Electrochemical capacitors utilising transition metal oxides: an update of recent developments
Wentao
Deng
a,
Xiaobo
Ji
*a,
Qiyuan
Chen
a and
Craig E.
Banks
*b
aInstitute of Metallurgical Physicochemistry, College of Chemistry and Chemical Engineering, Central South University, Changsha, 410083, China. E-mail: xji.csu.edu@gmail.com; Fax: +86-731 88879616; Tel: +86-731 88879616
bFaculty of Science and Engineering, School of Science and the Environment, Division of Chemistry and Environmental Science, Manchester Metropolitan University, Chester Street, Manchester M1 5GD, Lancs, UK. E-mail: c.banks@mmu.ac.uk; Fax: +44 (0) 1612476831; Tel: +44 (0) 1612471196
First published on 17th October 2011
Abstract
Transition metal oxides receive considerable attention in the area of electrochemistry not only due to their beneficial reported structural, mechanical or electronic properties, but because of their capacitive properties ascribed to their multiple oxide states they exhibit pseudo capacitances which carbon counterparts generally cannot. Typically transition metal oxides may be classified as noble transition metal oxides which exhibit excellent capacitive properties but have the drawback of generally being relatively expensive. Alternatively base metal oxides may also be utilised which are considerably cheaper and more environment friendly than noble transition metals as well as exhibiting good capacitive properties. In considering that nanostructured materials can help ameliorate the electrochemical performances of transition metal oxides, this review summarizes the recent investigations of fundamental advances in understanding the electrochemical reactivity of transition metal oxides, thus leading to an improved capacitive performance, which is essential for their continual use in a plethora of supercapacitor applications.
Introduction
The rapidly growing commercial markets providing portable electronic devices and other related electronics such as electric vehicles (EV) requires high-performance energy-storage systems.1,2 There is of course global issues threatening the environmental resulting in academics and industry exploring clean alternative energies where electricity storage needs to be either stored as electricity or in other forms of energy to meet growing consumers needs.3 Electrochemical supercapacitors (ECs), also called supercapacitors or ultracapacitors, are electronic components that can be rapidly charged and discharged and relied upon to store energy reliably for long periods.4 Emerging as an ideal model, they have been touted as a solution to the mismatch between the fast growth in power required by devices and the inability of batteries in various applications which require transient but high/peak power pulses for the time-dependent usage.5ECs were first reported in 1957 Becker's patent6 and then appeared on the commercial market in 1978 (Gold Capacitors from Panasonic/Matsushita) and in 1980 (Supercap from NEC/Tokin).7 They are often used as uninterruptible power supplies and load-leveling, such as backup sources for memories and microcomputers. They also have potential applications in electric vehicles and hybrid electric vehicles. It has been reported that the Airbus A380 has applied electrochemical double-layer capacitors (EDLCs) within its emergency doors.1
Note that ECs possess high power density, fast power delivery or uptake, excellent reversibility, low equivalent series resistance (ESR) and long cycle life when compared with batteries. A direct comparison between batteries and ECs is shown in Table 1.8 It can be found that the Ragone plot in Fig.1 (specific power density against specific energy density) shows that ECs shorten the gap between conventional capacitors and batteries,1 exhibiting much higher specific capacitances than conventional capacitors owing to their high specific area. It has been revealed that as for electrode-active materials, specific surface area and pore size are two of the crucial factors that can affect the specific capacitance of capacitors. It was once believed by scientists that it was advantageous to increase the pore size allowing for greater ion adsorption leading to higher capacitances. Interestingly, Gogotsi et al. demonstrated that the capacitance can be greatly enhanced with pore size down to the solvated ion size.9
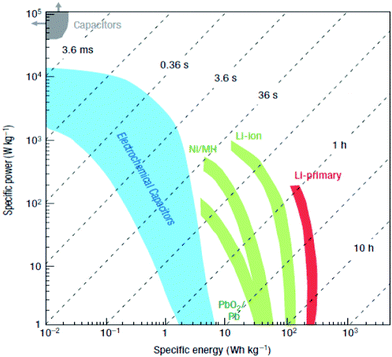 | ||
| Fig. 1 Ragone Plot. Reproduced from ref. 6 by permission of Nature Publishing Group. | ||
| Battery | Supercapacitor |
|---|---|
| Ideally single-valued free energies of components | Continuous variation of free energy with degree of conversion of materials |
| Not capacitative, except in very general sense. | Capacitative Behaviour |
| Usual irreversible behaviour | High degree of reversibility is common |
| Response to linear modulation of potential gives irreversible i vs. V profile with nonconstant current. | Response to linear modulation of potential gives more or less constant charging current |
| Discharge at constant current arises at a more or less constant potential | Discharge at constant current gives linear decline of potential with times |
According to charge storage mechanisms, ECs may be placed into two distinctive classes: electrochemical double-layer capacitors (EDLCs) and pseudo-capacitors.1,5 Since the 1990s, electrochemical supercapacitors ushered in its development with materials mostly used as EDLCs based on carbonaceous materials such as activated carbon,9–12carbide-derived carbon,9,12–15graphene16–19 and carbon nanotubes20–23 with a high surface area essential for the charge to reside. Alternatively pseudo-capacitors combine electrosorption with fast surface redox reactions to store energy, where the amount of charge held is proportional to the applied voltage.7 Transition metal oxides are often utilised or alternatively electronically conductive polymers may be used as electrode materials.2,24–27 Impressively, their specific capacitance can be 10–100 times higher that those obtained from EDLCs.8
Researchers had no clear concept regarding pseudo-capacitance until Trasatti and Buzzanca discovered the capacitive properties of RuO2.28 While supercapacitors benefit from the highest known power capability (10 kW kg−1), high current capability, ease of maintenance and long-term cycling stability (>106 cycles),29 their moderate energy density for the widespread deployment in everyday technology as clean and renewable energy media are still far away to meet the performances and cost requirements, in particular providing power boosts to start the engine or to assist acceleration.
In order to make capacitors commercially viable and become extensively used, the energy that they can store needs to increase significantly and to achieve supercapacitors with sufficient energy for higher requirements in the future is still a challenge. Extensive efforts have been devoted to improve the specific capacitance of supercapacitors by introducing pseudo-capacitive metal oxides. Compared with EDLC-based capacitors, pseudocapacitors based on transition metal oxide electrodes have attracted a large amount of attention since they can produce higher capacitances than double-layer carbonaceous materials. Despite their importance, efforts are still needed to further improve the practical use of metal oxide supercapacitors.
Transition metal oxides used as electrode active materials for ECs have developed at a high rate over the past decades and may be classed as either noble or based metal oxides. Noble metal oxides, for instance, RuO2, IrO2, etc exhibit good conductivities and excellent power densities. However, their relatively high cost and potential (reported) harmful nature to the environment have limited their widespread application in supercapacitors. In terms of base metal oxides such as MnO2, NiO, Fe3O4, etc, their outstanding pseudo-capacitive behaviour, practical availability, environmental compatibility and lower cost have been demonstrated when compared to the state-of-the-art supercapacitor material RuO2. The detailed maximum specific capacitance of these materials and energy density in aqueous solution is depicted in Table 2.
The aim of this review is to detail the recent investigations of using metal oxides as electrode materials for utilisation in electrochemical capacitors as well as reporting on the fundamental advances in understanding the electrochemical reactivity of metal oxides.
1. Noble metal oxides
Noble metal oxides such as RuO2, IrO2 and so forth were first found to exhibit good capacitive properties with rich electrochemical performances. Iridium dioxide is one kind of transition metal oxide with rutile structure.30 It can be used as optical switching layers in electrochromic displays,31 ferroelectric capacitors for non-volatile memories32 and supercapacitors.30,33,34 The fact that IrO2 did not draw much attention in comparison to RuO2 for use in supercapacitors can mostly be attributed to its relatively small value of mass-specific capacitance caused by its heavy relative atomic mass.30 For example, Chen et al. fabricated a composite of IrO2 and multiwalled carbon nanotubes, which showed a low specific capacitance of 69 F/g.35 After that, the authors prepared IrOx nanofoils with high surface area and sputtered them on multi-wall carbon nanotubes for supercapacitor application.30 The active materials exhibited an improved specific capacitance of 370 F/g, which is still poor when compared with RuO2. Hence, we will focus on summarizing the electrochemical properties of Ruthenium oxide.1.1 Synthesis methods of RuO2
Trasatti and Buzzanca first reported that RuO2 films could form a rectangular shaped cyclic voltammogram which closely resembled the carbon-based electric double-layer capacitor.28 Since this pioneering report, researchers have explored the capacitive properties of RuO2 as its theoretical value of specific capacitance is estimated to range from 1300–2200 F/g36 and is now consequently playing an important role in the application of electro-catalysis37–40 and power sources36,41,42.Nowadays nanostructured materials are used to help ameliorate the electrochemical performances of transition metal oxides. Transforming bulk transition metal oxides into nano-porous structures was thought to have the advantage of effectively alleviating the strain generated during the ion insertion/desertion process and leading to improved capacitive performance.43 A key methodology to boost the specific capacitance is their morphological and/or chemical composition design at the nanometre-scale, because high surface-to-volume ratio with suitable pore sizes is desirable for the penetration of electrolytes and reactants into the whole electrode matrix, and can promote the electric double-layer capacitances and accommodate a large amount of superficial electroactive species to participate in Faradaic redox reactions.
Generally there are two common methods to prepare ruthenium oxides.44 The first, chemical deposition, is to synthesize the materials by thermal decomposition and then oxidize the precursor at high temperatures. The other is a sol–gel synthesis methodology which firstly produces a sol–gel precursor which is then annealed at low temperatures. It was found that amorphous ruthenium oxides can be obtained at low temperatures while the crystalline phase were observed with higher temperatures; the resultant capacitance was found to be as high as 768 F/g.7
Zhang et al. synthesized nanotubular ruthenium oxides by using manganite nanorods as a morphology sacrificial template and obtained a high specific capacitance of 860 F/g with a good capacitive retention.45 Extensive research has been undertaken by researchers in this field with the specific capacitance reported to reach more than 1300F/g.36,43,46–57 Hu et al. used the membrane-templated synthesis route to obtain hydrous RuO2 (RuO2·xH2O) nanotubular structures by means of an anodic deposition technique.36 They obtained ruthenium oxide which exhibited a ultrahigh specific capacitance (1300 F g−1), excellent charge/discharge behaviour at 1000 mV s−1, and high-frequency (4.0–7.8 kHz) capacitive responses, which were much higher than any value ever reported before. Some typical synthesis methods and synthesising processes of RuO2 are given in Table 3 and Fig. 2.
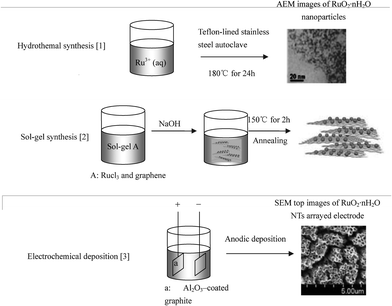 | ||
| Fig. 2 Typical synthesis processes of RuO2. Reproduced from ref. 24, 73, 74 by permission of the American Chemical Society, the Electrochemical Society and Wiley. | ||
| Method | Precursor | Solution | Specific capacitance |
|---|---|---|---|
| Chemical deposition | RuCl3 | Water | 210 F/g 134 |
| Sol–gel synthesis | RuCl3 | Water and ethanol | 570 F/g (38.3 wt% Ru loading)47 |
| Electrochemical deposition | RuCl3 | Water | 1300 F/g 36 |
1.2 Energy storage mechanism of RuO2
As typical pseudo-capacitor electrode materials, ruthenium oxides have a completely different mechanism with that of electrochemical double-layer capacitors and have multiple redox states while demonstrating good electrical conductivity. Note that RuO2 has three distinct oxidation states accessible within 1.2V, and its pseudo-capacitive behaviour in acidic solution can be described by the following equations:1,43| RuO2 + xH+ + xe+ ↔ RuO2−x(OH)x 0 ≤ x ≤ 2 | (1) |
| RuOx(OH)y + δH+ + δe− ↔ RuOx−δ(OH)y+δ | (2) |
As described in the equations above, protons can participate in the redox reaction, where RuO2 acts as a proton condenser and the reaction is electrochemically reversible.28,50,51 It should be noted that amorphous structures are the key parameter in determining the value of specific capacitances, as the proton can be easily intercalated into the bulk of RuO2·xH2O but not into the crystalline phase.43,58 It is also proposed that hydrous ruthenium dioxide has improved capacitive properties than those observed from anhydrous structures as hydrous regions are more permeable to protons and proton conduction inside ruthenium dioxide is dominant when compared with the electron conduction.59
2. Base transition metal oxides
Though RuO2, IrO2 and similar structures exhibit excellent capacitive performances, the drawback is their relative high cost and consequently scientists are exploring other low cost alternatives which still exhibit good capacitive properties. Attention has been focused on base transition metal oxides such as MnO2, NiO and Fe3O4 for electrode active materials of ECs in recent years.2.1 NiO
Nickel is cheap and abundant in the earth, and it is often used as a catalyst60–62 and the active material of batteries,63–65 especially in the form of nickel hydroxide. It is a very versatile material and has shown to exhibit good electrochemical activity based on the transformation of Ni(OH)2/NiOOH.53,55 Hierarchical micro-flowers, hollow microspheres and nanosheets have all been successfully prepared with various methodologies, followed by the paramount important steps of dehydration and re-crystallization of Ni(OH)2 in preparing NiO.66,67Sol–gel process is another efficient way to obtain the desired nanostructures. Typically the first step is to prepare a sol–gel and the second step is to apply heat treatment. Though the sol–gel progress is very simple, it is difficult to control, especially when very fine nanostructures are required.69–73 Yang et al. obtained hexagonal and single crystalline NiO nanowires by a sol–gel process and subsequent calcination, with the lateral size of the nanowires found to be ∼20–60 nm.70
In terms of electrochemical deposition, a thin Ni film is deposited electrochemically onto a substrate. In comparison with the methods identified above, it is more controllable and relatively easy to control the mass and thickness of films by simply controlling the current, bath chemistry and temperature.74–76 Porous nickel oxide films are directly deposited onto conducting indium tin oxide coated glass substrates by cyclic voltammetric (CV), galvanostatic, and potentiostatic strategies.77 It was shown that the deposition potential influences the pore distribution of the film and with an optimized deposited potential at + 0.9 V, the obtained sample showed a high specific capacitance as 320 F/g.
There are many other methods such as chemical bath deposition53,57,78 and the spray pyrolysis method;79 typical methodologies and their processes to synthesize Ni(OH)2 or NiO are reported in Table 4 and Fig. 3.
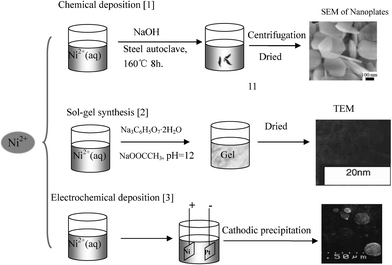 | ||
| Fig. 3 Typical synthesis processes of Ni(OH)2 or NiO. Reproduced from ref. 26, 50, 70 by permission of Springer, the Electrochemical Society, and Elsevier. | ||
| Method | Precursor | Solution | Specific capacitance |
|---|---|---|---|
| Chemical deposition | NiCl2·6H2O | Water | 390 F/g (NiO)44 |
| Sol–gel synthesis | NiSO4·6H2O | Water | 698 F/g 137 |
| Electrochemical deposition | Ni(NO3)2 | Water | 277 F/g (NiOx)80 |
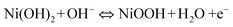 | (3) |
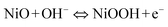 | (4) |
2.2 MnO2
Manganese oxide is a material that possesses the advantages of low cost, no toxicity, easy to obtain and high specific capacitances and consequently has been considered as the most promising material in application of ECs. | ||
| Fig. 4 SEM images of nanostructure MnO2 with different morphologies: (a) nanowire, (b) nanoflower, (c) hollow urchin, and (d) clews. Reproduced from Ref. 54 by permission of the American Chemical Society. | ||
The active MnO2 used in supercapacitors can also be prepared via a sol–gel synthesis. For example, Pang et al. fabricated a MnO2 film viadip-coating or drop-coating with manganese dioxide suspensions (sols) onto nickel foils.90 The resultant samples were found to exhibit good capacitive behaviours with specific capacitance values as high as 698 F/g. Electrochemical deposition is another effective methodology to prepare active MnO2. Adelkahani et al. synthesized large-scale oriented nano-sheets of MnO2 with thicknesses of less than 30 nm by pulse current electro-deposition.93 The resultant MnO2 nano-sheets exhibited high porosity, cation vacancy and water content producing a capacitance of 280 F/g at a scan rate of 5 mV s−1 in a Na2SO4 electrolyte. Hu et al. designed composite nanowires consisting of MnO2, Mn3O4 nanocrystallites and single-crystalline MnOOHvia a simple two-step electrochemical deposition process,94 which exhibited a specific capacitance of 470 F/g in CaCl2 and demonstrated excellent reversibility as well as outstanding stability. Extensive work is on-going on to prepare a better modality of MnO2 in order to achieve higher specific capacitances; common methodologies are depicted in Table 5 and a typical synthesis process of each method is highlighted in Fig. 5.
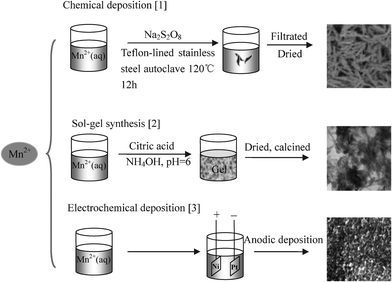 | ||
| Fig. 5 Typical synthesis processes of MnO2. Reproduced from ref. 13, 61,75 by permission of the American Chemical Society, the Electrochemical Society, and Elsevier. | ||
| Method | Precursor | Solution | Specific capacitance |
|---|---|---|---|
| Hydrothermal synthesis 20,87–89 | MnAc2·4H2O | Water | 168F/g 20 |
| Sol–gel synthesis90,135,136 | Mn(ClO4)2 | Water | 698F/g 90 |
| Electrochemical deposition 94,95 | MnSO4·H2O | Water | 310 F/g 95 |
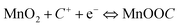 | (5) |
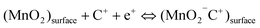 | (6) |
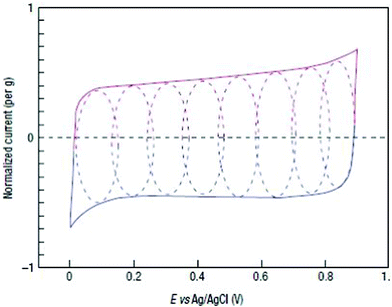 | ||
| Fig. 6 CV of a single MnO2 electrode Reproduced from ref. 6 by permission of Nature Publishing Group. | ||
Though two points of view have some differences, they both indicate a redox reaction between the III and IV oxidation states of Manganese.
2.3 Other base metal oxides and composite materials
Other base metal oxides such as iron oxide,100,101indium oxide,102,103tin oxide,104–106 and vanadium oxide107–109 also have their applications in ECs. Transition metal oxides may be prepared as a powder and used to prepare composite electrodes or can be directly deposited onto a conducting substrate to overcome their poor conductive performances.110–112 For example, Du et al. prepared nanoparticle Fe3O4/active carbon supercapacitors by a microwave fabrication methodology producing a resultant nano structure Fe3O4 with average particle sizes of 35 nm.100 This AC–Fe3O4 hybrid supercapacitor was found to deliver a specific capacitance of 37.9 F/g and yet was reported to still keep 82% of initial capacity over 500 cycles in characteristic cyclic tests.3. Mixed metal oxides
Mixed metal oxides are used as active materials of supercapacitor because the additive metal oxide can affect the structure and improve the electrochemical properties of the composites to a certain extent.113,114 Some work has been performed in pursuing better ingredients of mixed metal oxides, such as Ni–Co, V–Sn, Ru–Ti and Ru–Co composites, etc.113–117 It is reported that the presence of cobalt oxide can help to increase the electronic and redox properties of metal oxides composites, the additive of Co into the lattice of nickel hydroxide can not only efficiently increase its conductivity but shift the redox peaks of Ni(OH)2 to less positive potential.115,118,119 Electrochemical characterization of a SnO2–V2O5 composite indicates superior properties than the corresponding V2O5.117 The presence of SnO2 affects the structure of V2O5, which enhances the use ratio of V to a different dimension actually. Kim et al. also prepared a Ru–Co mixed oxide. The Ru–Co mixed oxide was co-deposition on single-walled carbon nanotube (SWNT)114 which exhibited a specific capacitance of 570 F/g at a scan rate 500 mV s−1, much better than the corresponding RuO2 electrode (475 F/g), owing to the enhanced electronic conduction for which Co provided structural support through the active Ru species.4. Asymmetrical supercapacitors
It is well known that the energy density of a supercapacitor is proportional to the potential window which can effectively enhance the energy density.120,121 Asymmetrical supercapacitors (ASC) can utilise ionic liquids or environmentally harmful organic reagents as electrolytes to improve cell voltage,20,29 and will often use appropriate electrode materials that allow working in wide potential windows which can significantly broaden the cell voltage of supercapacitor in order to achieve high energy density.121–127 In an ASC, the active material in one electrode is not the same as that used in the other electrode in a cell system. For instance, Quet al. fabricated an ASC in which active carbon (AC) was used as anode and MnO2 as cathode.124 It was reported to exhibit an energy density of 17 Wh/kg at 2 kW kg−1 with a voltage 1.8 V in 0.5 mol L−1K2SO4 aqueous solution, which was much higher than its symmetrical counterparts (AC/K2SO4/AC). Ganesh et al. obtained similar conclusion that ASC provide higher capacitance value than symmetrical one when comparing asymmetrical cell (NiO/KOH/AC) with their corresponding symmetrical cell (NiO/KOH/NiO).123Considering the compromises of power density causing by poor conductivity of metal oxides, scientists have mixed metal oxide with materials of good conductivity like active carbon, graphene, carbon nanotube to fabricate electrodes forward to an improvement of electrochemical properties. Reduced graphene oxide sheets (RGO) and its modified ruthenium oxide (RGO–RuO2) have been used as anode and cathode,127 leading to an energy density of 26.3 Wh/kg
5. Summary and outlook
We have summarized the current literature regarding the relatively new progress of transition metal oxides and explored their applications in electrochemical supercapacitors. Their development can be summed as:1. A continued exploration into new materials in order to enhance capacitance properties with a particular emphasis on finding materials those are relatively low in cost and environmentally friendly. Since RuO2 was first found to have a capacitive property, many transition metal oxides have been explored as electrode materials in ECs. The morphology of electrode materials exerts a wide influence over the speed of ion transfer and the ability to synthesize materials with excellent morphology (pore size, surface area and so on) in order to achieve good capacitive performances with adequate electrolyte wetting is crucial; to meet these criteria is a challenge for scientists to design better materials, or composite materials in order to further improve capacitances.
2. Improvement of the electrochemical properties of known oxides. It is known that ions transfer in the surface or bulk of transition metal oxides when redox reaction occurs and the majority of the transition metal oxides suffer a disadvantage of relatively bad conductivity.
Although the energy and power density of ECs can be enhanced to a level to achieve high energy densities, using hybrid systems might be a final solution via combining double layer and pseudo-behaviour electrodes. In summary, for ECs to become more commercially viable, the materials at the heart of the capacitor will need to become significantly cheaper and more efficient.
Acknowledgements
Financial support from the National Natural Science Foundation of China (No.21050110115), International Joint Project from The Royal Society (No. JP090644), and Hunan Province Foundation of Natural Science (10JJ6026) are greatly appreciated.References
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845 CrossRef CAS.
- Y. Zhang, H. Feng, X. Wu, L. Wang, A. Zhang, T. Xia, H. Dong, X. Li and L. Zhang, Int. J. Hydrogen Energy, 2009, 34, 4889 CrossRef CAS.
- P. Li, IEEE Nanotechnology Magazine, 2008, 13 Search PubMed.
- B. E. Conway, Electrochemical supercapacitors, Kluwer Academic Publishers, 1999 Search PubMed.
- B. E. Conway and W. G. Pell, J. Solid State Electrochem., 2003, 7, 637 CrossRef CAS.
- H. E.Becker, U.S. Patent, 2800616, 1957 Search PubMed.
- R. Kotz and M. Carlen, Electrochim. Acta, 2000, 45, 2483 CrossRef CAS.
- B. E. Conway, J. Electrochem. Soc., 1991, 138, 1539 CrossRef CAS.
- C. Largeot, C. Portet, J. Chmiola, P.-L. Taberna, Y. Gogotsi and P. Simon, J. Am. Chem. Soc., 2008, 130, 2730 CrossRef CAS.
- L. Demarconnay, E. Raymundo-Pinero and F. Béguin, Electrochem. Commun., 2010, 12, 1275 CrossRef CAS.
- K. Xia, Q. Gao, J. Jiang and J. Hu, Carbon, 2008, 46, 1718 CrossRef CAS.
- A. Balducci, R. Dugas, P. L. Taberna, P. Simon, D. Plée, M. Mastragostino and S. Passerini, J. Power Sources, 2007, 165, 922 CrossRef CAS.
- Y. Gogotsi, A. Nikitin, H. Ye, W. Zhou, J. E. Fischer, B. Yi, H. C. Foley and M. W. Barsoum, Nat. Mater., 2003, 2, 591 CrossRef CAS.
- J. Chmiola, C. Largeot, P. L. Taberna, P. Simon and Y. Gogotsi, Science, 2010, 328, 480 CrossRef CAS.
- J. Torop, V. Palmre, M. Arulepp, T. Sugino, K. Asaka and A. Aabloo, Carbon, 2011, 49, 3113 CrossRef CAS.
- S. Chen, J. Zhu, X. Wu, Q. Han and X. Wang, ACS Nano, 2010, 4, 2822 CrossRef CAS.
- Z. Chen, W. Ren, L. Gao, B. Liu, S. Pei and H.-M. Cheng, Nat. Mater., 2011, 10, 424 CrossRef CAS.
- J. R. Miller, R. Outlaw and B. Holloway, Science, 2010, 329, 1637 CrossRef CAS.
- Y. Wang, Z. Shi, Y. Huang, Y. Ma, C. Wang, M. Chen and Y. Chen, J. Phys. Chem. C, 2009, 113, 13103 CAS.
- P. C. Chen, G. Shen, Y. Shi, H. Chen and C. Zhou, ACS Nano, 2010, 4, 4403 CrossRef CAS.
- C. Yu, C. Masarapu, J. Rong, B. Wei and H. Jiang, Adv. Mater., 2009, 21, 4793 CrossRef CAS.
- D. N. Futaba, K. Hata, T. Yamada, T. Hiraoka, Y. Hayamizu, Y. Kakudate, O. Tanaike, H. Hatori, M. Yumura and S. Iijima, Nat. Mater., 2006, 5, 987 CrossRef CAS.
- F. D. S. Marquis, Journal of the Minerals, Metals and Materials Society, 2011, 63, 48 CAS.
- B. Conway, V. Birss and J. Wojtowicz, J. Power Sources, 1997, 66, 1 CrossRef CAS.
- C. Meng, C. Liu, L. Chen, C. Hu and S. Fan, Nano Lett., 2010, 10, 4025 CrossRef CAS.
- G. M. Suppes, B. A. Deore and M. S. Freund, Langmuir, 2008, 24, 1064 CrossRef CAS.
- C. D. Lokhande, D. P. Dubal and O.-S. Joo, Curr. Appl. Phys., 2011, 11, 255 CrossRef.
- S. Trasatti and G. Buzzanca, Journal of Electroanalytical Chemistry and Interfacial Electrochemistry, 1971, 29, A1 Search PubMed.
- L. L. Zhang and X. Zhao, Chem. Soc. Rev., 2009, 38, 2520 RSC.
- Y.-M. Chen, Nanotechnology, 2011, 22, 355708 CrossRef.
- K. Nishio, Y. Watanabe and T. Tsuchiya, Thin Solid Films, 1999, 350, 96 CrossRef CAS.
- T. Sakoda, T. S. Moise, Scott R. Summerfelt, L. Colombo, G. Xing, S. R. Gilbert, A. L. S. Loke, S. Ma, R. Kavari, L. A. Wills and J. Amano, Jpn. J. Appl. Phys., 2001, 40, 2911 CrossRef CAS.
- D.-Q. Liu, S.-H. Yu, S.-W. Son and S.-K. Joo, ECS Trans., 2008, 16, 103 CrossRef CAS.
- Y. Chen, Y. Huang, K. Lee, D. Tsai and K. Tiong, J. Mater. Sci.: Mater. Electron., 2011, 22, 890 CrossRef CAS.
- Y. M. Chen, Nanotechnology, 2011, 22, 115706 CrossRef CAS.
- C. C. Hu, K. H. Chang, M. C. Lin and Y. T. Wu, Nano Lett., 2006, 6, 2690 CrossRef CAS.
- L. C. Jiang and W. D. Zhang, Electroanalysis, 2009, 21, 1811 CrossRef CAS.
- D. Pletcher, J. Appl. Electrochem., 1984, 14, 403 CrossRef CAS.
- S. Zweidinger, J. Hofmann, O. Balmes, E. Lundgren and H. Over, J. Catal., 2010, 272, 169 CrossRef CAS.
- H. Over, Y. Kim, A. Seitsonen, S. Wendt, E. Lundgren, M. Schmid, P. Varga, A. Morgante and G. Ertl, Science, 2000, 287, 1474 CrossRef CAS.
- T. Gujar, V. Shinde, C. Lokhande, W. Y. Kim, K. D. Jung and O. S. Joo, Electrochem. Commun., 2007, 9, 504 CrossRef CAS.
- P. Balaya, H. Li, L. Kienle and J. Maier, Adv. Funct. Mater., 2003, 13, 621 CrossRef CAS.
- J. Zheng, P. Cygan and T. Jow, J. Electrochem. Soc., 1995, 142, 2699 CrossRef CAS.
- X. Zhang, W. Shi, J. Zhu, W. Zhao, J. Ma, S. Mhaisalkar, T. L. Maria, Y. Yang, H. Zhang and H. H. Hng, Nano Res., 2010, 3, 643 CrossRef CAS.
- J. Zhang, J. Ma, L. L. Zhang, P. Guo, J. Jiang and X. Zhao, The Journal of Physical Chemistry C Search PubMed.
- C. C. Hu, W. C. Chen and K. H. Chang, J. Electrochem. Soc., 2004, 151, A281 CrossRef CAS.
- K. H. Chang, C. C. Hu and C. Y. Chou, Electrochim. Acta, 2009, 54, 978 CrossRef CAS.
- C. C. Hu and W. C. Chen, Electrochim. Acta, 2004, 49, 3469 CrossRef CAS.
- W. Sugimoto, H. Iwata, Y. Yasunaga, Y. Murakami and Y. Takasu, Angew. Chem., Int. Ed., 2003, 42, 4092 CrossRef CAS.
- S. Trasatti, Electrochim. Acta, 1991, 36, 225 CrossRef CAS.
- D. Galizzioli, F. Tantardini and S. Trasatti, J. Appl. Electrochem., 1974, 4, 57 CrossRef CAS.
- J. Zheng and T. Jow, Journal of the Electrochemical Society;(United States), 1995, 142 Search PubMed.
- U. M. Patil, K. V. Gurav, V. J. Fulari, C. D. Lokhande and O. S. Joo, J. Power Sources, 2009, 188, 338 CrossRef CAS.
- W. Sugimoto, H. Iwata, K. Yokoshima, Y. Murakami and Y. Takasu, J. Phys. Chem. B, 2005, 109, 7330 CrossRef CAS.
- K.-C. Liu and M. A. Anderson, J. Electrochem. Soc., 1996, 143, 124 CrossRef CAS.
- L.-X. Yang, Y.-J. Zhu, H. Tong, Z.-H. Liang, L. Li and L. Zhang, J. Solid State Chem., 2007, 180, 2095 CrossRef CAS.
- M. Akinc, N. Jongen, J. Lemaìtre and H. Hofmann, J. Eur. Ceram. Soc., 1998, 18, 1559 CrossRef.
- J. P. Zheng and T. R. Jow, J. Electrochem. Soc., 1995, 142, L6 CrossRef CAS.
- W. Sugimoto, H. Iwata, K. Yokoshima, Y. Murakami and Y. Takasu, J. Phys. Chem. B, 2005, 109, 7330 CrossRef CAS.
- A. Kudo, A. Tanaka, K. Domen, K. Maruya, K. Aika and T. Onishi, J. Catal., 1988, 111, 67 CrossRef CAS.
- C. Bradu, L. Frunza, N. Mihalche, S. M. Avramescu, M. Neata and I. Udrea, Appl. Catal., B, 2010, 96, 548 CrossRef CAS.
- B. Q. Xu, J. M. Wei, H. Y. Wang, K. Q. Sun and Q. M. Zhu, Catal. Today, 2001, 68, 217 CrossRef CAS.
- A. Hammouche, E. Karden and R. W. De Doncker, J. Power Sources, 2004, 127, 105 CrossRef CAS.
- M. Cao, X. He, J. Chen and C. Hu, Cryst. Growth Des., 2007, 7, 170 CAS.
- J. M. Tarascon and M. Armand, Nature, 2001, 414, 359 CrossRef CAS.
- C. Y. Cao, W. Guo, Z. M. Cui, W. G. Song and W. Cai, Journal of Materials Chemistry, 2011, 21, 3204 RSC.
- C. Yuan, X. Zhang, L. Su, B. Gao and L. Shen, J. Mater. Chem., 2009, 19, 5772 RSC.
- L. Wang, Y. Hao, Y. Zhao, Q. Lai and X. Xu, Journal of Solid State Chemistry, 2010 Search PubMed.
- H. Jiang, T. Zhao, C. Li and J. Ma, Journal of Materials Chemistry, 2011 Search PubMed.
- Q. Yang, J. Sha, X. Ma and D. Yang, Mater. Lett., 2005, 59, 1967 CrossRef CAS.
- A. Šurca, B. Orel and B. Pihlar, J. Sol-Gel Sci. Technol., 1997, 8, 743 CrossRef.
- F. Jia, L. Zhang, X. Shang and Y. Yang, Adv. Mater., 2008, 20, 1050 CrossRef CAS.
- T. Y. Wei, C. H. Chen, H. C. Chien, S. Y. Lu and C. C. Hu, Adv. Mater., 2010, 22, 347 CrossRef CAS.
- K. R. Prasad and N. Miura, Appl. Phys. Lett., 2004, 85, 4199 CrossRef CAS.
- M.-S. Wu, C.-H. Yang and M.-J. Wang, Electrochim. Acta, 2008, 54, 155 CrossRef CAS.
- L. Berkat, L. Cattin, A. Reguig, M. Regragui and J. C. Bernède, Mater. Chem. Phys., 2005, 89, 11 CrossRef CAS.
- B. Pejova, T. Kocareva, M. Najdoski and I. Grozdanov, Appl. Surf. Sci., 2000, 165, 271 CrossRef CAS.
- Y. Tan, S. Srinivasan and K.-S. Choi, J. Am. Chem. Soc., 2005, 127, 3596 CrossRef CAS.
- V. Srinivasan and J. W. Weidner, J. Electrochem. Soc., 1997, 144, L210 CrossRef CAS.
- K. W. Nam and K. B. Kim, J. Electrochem. Soc., 2002, 149, A346 CrossRef CAS.
- R. Jayashree, P. V. Kamath and G. Subbanna, J. Electrochem. Soc., 2000, 147, 2029 CrossRef CAS.
- W. G. Pell and B. E. Conway, J. Power Sources, 2001, 96, 57 CrossRef CAS.
- R. Barnard, C. F. Randall and F. L. Tye, J. Appl. Electrochem., 1980, 10, 109 CrossRef.
- H. Liang, H. Yang, L. Liu, Z. Yang and Y. Yang, Superlattices Microstruct., 2010, 48, 569 CrossRef CAS.
- X. Wang and Y. Li, J. Am. Chem. Soc., 2002, 124, 2880 CrossRef CAS.
- R. N. Reddy and R. G. Reddy, J. Power Sources, 2003, 124, 330 CrossRef CAS.
- V. Subramanian, H. Zhu, R. Vajtai, P. M. Ajayan and B. Wei, J. Phys. Chem. B, 2005, 109, 20207 CrossRef CAS.
- P. Yu, X. Zhang, D. Wang, L. Wang and Y. Ma, Cryst. Growth Des., 2008, 9, 528 Search PubMed.
- M. Xu, L. Kong, W. Zhou and H. Li, J. Phys. Chem. C, 2007, 111, 19141 CAS.
- S. C. Pang, M. A. Anderson and T. W. Chapman, J. Electrochem. Soc., 2000, 147, 444 CrossRef CAS.
- X. Huang, D. Lv, H. Yue, A. Attia and Y. Yang, Nanotechnology, 2008, 19, 225 Search PubMed.
- Y. S. Ding, X. F. Shen, S. Gomez, H. Luo, M. Aindow and S. L. Suib, Adv. Funct. Mater., 2006, 16, 549 CrossRef CAS.
- H. Adelkhani and M. Ghaemi, J. Alloys Compd., 2010, 493, 175 CrossRef CAS.
- S. Devaraj and N. Munichandraiah, Electrochem. Solid-State Lett., 2005, 8, A373 CrossRef CAS.
- F. Ataherian, K.-T. Lee and N.-L. Wu, Electrochim. Acta, 2010, 55, 7429 CrossRef CAS.
- M. Toupin, T. Brousse and D. Bélanger, Chem. Mater., 2004, 16, 3184 CrossRef CAS.
- M. Toupin, T. Brousse and D. Bélanger, Chem. Mater., 2002, 14, 3946 CrossRef CAS.
- M. Ghaemi, F. Ataherian, A. Zolfaghari and S. Jafari, Electrochim. Acta, 2008, 53, 4607 CrossRef CAS.
- X. Du, C. Wang, M. Chen, Y. Jiao and J. Wang, J. Phys. Chem. C, 2009, 113, 2643 CAS.
- N.-L. Wu, Mater. Chem. Phys., 2002, 75, 6 CrossRef CAS.
- K. R. Prasad, K. Koga and N. Miura, Chem. Mater., 2004, 16, 1845 CrossRef CAS.
- K. Rajendra Prasad and N. Miura, Electrochem. Commun., 2004, 6, 1004 CrossRef CAS.
- M. Jayalakshmi, N. Venugopal, K. P. Raja and M. M. Rao, J. Power Sources, 2006, 158, 1538 CrossRef CAS.
- F. Li, J. Song, H. Yang, S. Gan, Q. Zhang, D. Han, A. Ivaska and L. Niu, Nanotechnology, 2009, 20, 455602 CrossRef.
- R. K. Selvan, I. Perelshtein, N. Perkas and A. Gedanken, J. Phys. Chem. C, 2008, 112, 1825 CAS.
- R. Demir-Cakan, Y. S. Hu, M. Antonietti, J. Maier and M. M. Titirici, Chem. Mater., 2008, 20, 1227 CrossRef CAS.
- H. Y. Lee and J. Goodenough, J. Solid State Chem., 1999, 148, 81 CrossRef CAS.
- G. Wee, H. Z. Soh, Y. L. Cheah, S. G. Mhaisalkar and M. Srinivasan, J. Mater. Chem., 2010, 20, 6720 RSC.
- Z. Chen, Y. Qin, D. Weng, Q. Xiao, Y. Peng, X. Wang, H. Li, F. Wei and Y. Lu, Adv. Funct. Mater., 2009, 19, 3420 CrossRef CAS.
- H. Wang, H. S. Casalongue, Y. Liang and H. Dai, J. Am. Chem. Soc., 2010, 132, 7472 CrossRef CAS.
- J. W. Lee, T. Ahn, D. Soundararajan, J. M. Ko and J. D. Kim, Chem. Commun., 2011, 47, 6305 RSC.
- Q. Huang, X. Wang, J. Li, C. Dai, S. Gamboa and P. Sebastian, J. Power Sources, 2007, 164, 425 CrossRef CAS.
- C.-C. Hu, H.-Y. Guo, K.-H. Chang and C.-C. Huang, Electrochem. Commun., 2009, 11, 1631 CrossRef CAS.
- B. C. Kim, G. G. Wallace, Y. I. Yoon, J. M. Ko and C. O. Too, Synth. Met., 2009, 159, 1389 CrossRef CAS.
- S. I. C. d. Torresi, K. Provazi, M. Malta and R. M. Torresi, J. Electrochem. Soc., 2001, 148, A1179 CrossRef.
- M. Vidotti, M. R. Silva, R. P. Salvador, S. I. C. d. Torresi and L. H. Dall'Antonia, Electrochim. Acta, 2008, 53, 4030 CrossRef CAS.
- M. Jayalakshmi, M. M. Rao, N. Venugopal and K.-B. Kim, J. Power Sources, 2007, 166, 578 CrossRef CAS.
- K. Provazi, M. J. Giz, L. H. Dall'Antonia and S. I. Córdoba de Torresi, J. Power Sources, 2001, 102, 224 CrossRef CAS.
- A. K. Sood, J. Appl. Electrochem., 1986, 16, 274 CrossRef CAS.
- M. D. Stoller and R. S. Ruoff, Energy Environ. Sci., 2010, 3, 1294 CAS.
- C. Peng, S. Zhang, X. Zhou and G. Z. Chen, Energy Environ. Sci., 2010, 3, 1499 Search PubMed.
- N. W. Duffy, W. Baldsing and A. G. Pandolfo, Electrochim. Acta, 2008, 54, 535 CrossRef CAS.
- V. Ganesh, S. Pitchumani and V. Lakshminarayanan, J. Power Sources, 2006, 158, 1523 CrossRef CAS.
- Q. Qu, P. Zhang, B. Wang, Y. Chen, S. Tian, Y. Wu and R. Holze, J. Phys. Chem. C, 2009, 113, 14020 CAS.
- H. Wang, Y. Liang, T. Mirfakhrai, Z. Chen, H. S. Casalongue and H. Dai, Nano Research, 1 Search PubMed.
- C. Xu, H. Du, B. Li, F. Kang and Y. Zeng, J. Electrochem. Soc., 2009, 156, A435 CrossRef CAS.
- J. Zhang, J. Jiang, H. Li and X. S. Zhao, Energy & Environmental Science, 2011 Search PubMed.
- C.-C. Hu and W.-C. Chen, Electrochim. Acta, 2004, 49, 3469 CrossRef CAS.
- P. Lin, Q. She, B. Hong, X. Liu, Y. Shi, Z. Shi, M. Zheng and Q. Dong, J. Electrochem. Soc., 2010, 157, A818 CrossRef CAS.
- G.-W. Yang, C.-L. Xu and H.-L. Li, Chem. Commun., 2008, 6537 RSC.
- L. Cao, F. Xu, Y. Y. Liang and H. L. Li, Adv. Mater., 2004, 16, 1853 CrossRef CAS.
- K.-H. Chang and C.-C. Hu, Electrochem. Solid-State Lett., 2004, 7, A466 CrossRef CAS.
- L. Cao, F. Xu, Y. Y. Liang and H. L. Li, Adv. Mater., 2004, 16, 1853 CrossRef CAS.
- Z.-S. Wu, D.-W. Wang, W. Ren, J. Zhao, G. Zhou, F. Li and H.-M. Cheng, Adv. Funct. Mater., 2010, 20, 3595 CrossRef CAS.
- C.-C. Hu, C.-Y. Hung, K.-H. Chang and Y.-L. Yang, J. Power Sources, 2011, 196, 847 CrossRef CAS.
- D.-L. Fang, B.-C. Wu, A.-Q. Mao, Y. Yan and C.-H. Zheng, J. Alloys Compd., 2010, 507, 526 CrossRef CAS.
- J. Cheng, G.-P. Cao and Y.-S. Yang, J. Power Sources, 2006, 159, 734 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |