Fe3O4@SiO2@Pd-Au: a highly efficient and magnetically separable catalyst for liquid-phase hydrodechlorination of 4-chlorophenol†
Zhijie
Wu
,
Chongxia
Sun
,
Yan
Chai
and
Minghui
Zhang
*
Institute of New Catalytic Materials Science, Department of Material Chemistry, College of Chemistry, and Key Laboratory of Advanced Energy Materials Chemistry (MOE), Nankai University, Tianjin, 300071, China. E-mail: zhangmh@nankai.edu.cn; Fax: +86 22-2350-7730; Tel: +86 22-2350-7730
First published on 7th September 2011
Abstract
A magnetic core–shell nanocomposite, Fe3O4@SiO2@Pd-Au, was synthesized by reducing palladium and gold cations previously bound to the amine ligand-modified surface of silica-encapsulated magnetic iron oxide (Fe3O4) nanoparticles, and served as a highly efficient and easily-recyclable catalyst for liquid-phase hydrodechlorination of 4-chlorophenol under mild conditions.
Chlorophenols (CPs) are widely known carcinogenic, teratogenic and mutagenic pollutants, which are toxic to the environment and are resistant to biodegradation.1,2 Much attention has been paid to finding sustainable methods for the disposal of CP waste which do not pose an environmental hazard.1 Hydrodechlorination (HDC) is regarded as the most effective method for their elimination, because the only organic product (phenol) can be subsequently recycled or safely incinerated.1–5Palladium is unique among the catalytic metals for its activity and selectivity in the HDC process, and as a consequence large numbers of fundamental and practical studies have been done with this metal.2–14 Many reports mentioned a beneficial effect of Au metal on palladium catalysts for chlorinated paraffin HDC, claiming its improvement on either the stability of metal dispersion, or the activity, or the selectivity for partial HDC.13–16 Since those alloy catalysts were mostly prepared by impregnation or adsorption methods, evolution of this research had to consider other catalyst's preparation techniques which would lead to better Pd-Au anchoring and recycling. Moreover, there is no report on the synthesis of Pd-Au alloy catalysts for the hydrodechlorination of CPs. Here, we propose a simple route to synthesize an easily-separable and highly active supported Pd-Au alloy catalyst for the HDC of 4-chlorophenol (4-CP). A silica-encapsulated magnetic carrier (Fe3O4) modified by amine was used as the support for Pd-Au nanoparticles, in which the amine acts as a bridge for anchoring highly dispersed Pd-Au nanoparticles onto the supports, and the presence of Fe3O4 enables the catalyst to be facilely separated from the reaction solution under an external magnetic field, then reused.
The synthesis of the magnetic nanoparticle-supported Pd-Au catalyst followed the procedure illustrated in Scheme 1. The surface of Fe3O4 nanoparticles was covered by a SiO2 shell (SiO2 encapsulated Fe3O4 particles, Fe3O4@SiO2) viahydrolysis of tetraethyl orthosilicate (TEOS) in cyclohexane solution,17 and then the resulting Fe3O4@SiO2 support was modified by a bifunctional (3-aminopropyl)triethoxysilane (APTS) ligand.18,19 The amine (–NH) group of the APTS ligand interacts with cationic Pd and Au, while the alkoxysilane moiety forms covalent Si–O–Si linkages and anchors on the surface of Fe3O4@SiO2. Thus, highly dispersed cationic Pd and Au were anchored on the surface of magnetic nanoparticles by the homogeneously distributed APTS,18–20 leading to small and highly dispersed Pd-Au clusters after reduction by KBH4 (potassium borohydride).
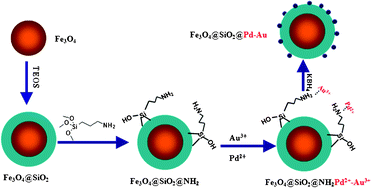 | ||
| Scheme 1 Synthesis procedure of Fe3O4@SiO2@Pd-Au catalyst. | ||
The XRD patterns indicate that the crystal structure of magnetic Fe3O4 is indexed as a face centered cubic (fcc) (JCPDS card no. 19-629) structure (Fig. 1). No other characteristic peaks corresponding to SiO2, or Pd and Au metal/oxides were observed, suggesting the high dispersion of surface species with low weight loading on Fe3O4@SiO2. TEM images of the magnetic samples are shown in Fig. 2. The Fe3O4@SiO2 nanocomposites with 30–45 nm diameter possess white patches on the surface of the magnetic Fe3O4 core, indicating the surface covering layer of SiO2. 2 nm Pd particles and 3 nm Pd64Au36 alloy particles are located on the surface of Fe3O4@SiO2@Pd and Fe3O4@SiO2@Pd-Au samples, respectively. However, some small metal or alloy particles are difficult to be distinguished in our TEM experiments. In order to obtain the average particle size of active metal sites, Pd dispersions on Fe3O4@SiO2@Pd and Fe3O4@SiO2@Pd-Au were measured as 0.64 and 0.47 (corresponding to smaller particle sizes: 1.8 and 2.3 nm), respectively.
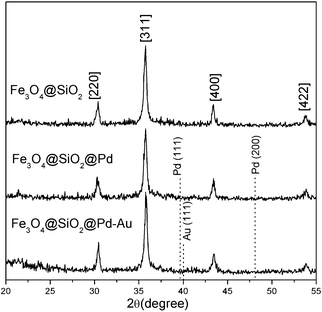 | ||
| Fig. 1 XRD patterns of samples. | ||
 | ||
| Fig. 2 TEM and HRTEM images of samples (∼3 nm Pd-Au nanoparticles in Fe3O4@SiO2@Pd-Au and ∼2 nm Pd nanoparticles in Fe3O4@SiO2@Pd). | ||
The FT-IR spectra of CO adsorption were recorded to confirm the formation of Pd-Au alloy. For Pd-Au alloy samples, a CO vibrational frequency shift can be used to identify the changes in the interactions between the substrate and CO molecules, which reflect the electronic modifications of the substrate surface induced by Au metal (the ligand effect).21d As shown in Fig. 3, two bands at 1965 (CO mainly on Pd bridging sites)21 and 2088 cm−1 (CO occupying Pd atop sites)21 appear for the Fe3O4@SiO2@Pd, and one band at 2110 cm−1 (CO adsorption on Au atop sites)21 occurs for Fe3O4@SiO2@Au. For Fe3O4@SiO2@Pd-Au, the band at 2109 cm−1 should be due to CO adsorption on Au, and the band corresponding to CO adsorption on Pd atop sites has shifted from 2088 cm−1 to 2068 cm−1, which indicates the influence of a ligand effect via the changing chemical environment around Pd atoms.21c Since the frequency shift is largely affected by the chemical interaction between the substrate and CO (mainly the back-donation of electrons from the metal d bands to CO 2π* anti-bonding orbital), the addition of Au to the Pd surface alters the width and center of the Pd d bands and thus shifts the CO internal frequency.21e With the presence of Au, the alloy particles also caused the band initially at 1965 cm−1 to shift toward higher frequency, indicating the decrease of adsorbate–adsorbate interactions on the Pd surface after Au dilution.21 Another obvious feature of Fig. 3 is that the CO-IR intensity for the Au atop feature drops dramatically on the Pd-Au sample, suggesting that CO adsorption on isolated Pd sites is enhanced, while that on Au is attenuated. These results reflect the intimate contact and interaction between surface Au and Pd atoms in Pd-Au alloy nanoparticles, leading to the change of the chemical environment around the Pd surface when mixing with Au, which is in agreement with the UV-vis spectra results (Fig. S1, see ESI†).
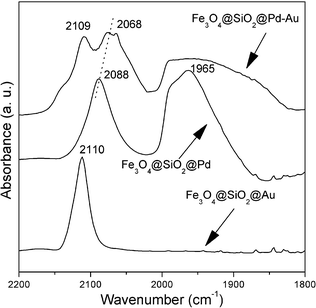 | ||
| Fig. 3 FT-IR spectra of CO adsorption on catalysts. | ||
Pd nanoparticles were highly active in HDC of 4-CP,1,22 while Au nanoparticles were almost inert towards the reaction (Fig. 4). Here, the formation of Pd-Au (Pd64Au36) alloy led to a dramatic enhancement in conversion (Fig. 4), and a complete HDC conversion was realized in ∼20 min much faster than Pd metal catalysts (∼3 h). In our previous work, the HDC of 4-CP followed a first-order reaction based on the conversion of 4-CP under the current reaction conditions.22 Here, we used HDC turnover rates (TORs) based on the exposed Pd metal surface to represent the activity of Pd and Pd-Au catalysts. The reaction rate in terms of initial turnover rate on Fe3O4@SiO2@Pd-Au is 486.2 mol/(h mol surface Pd), which is 12-fold higher than that of Fe3O4@SiO2@Pd catalyst (37.3 mol/(h mol surface Pd)).
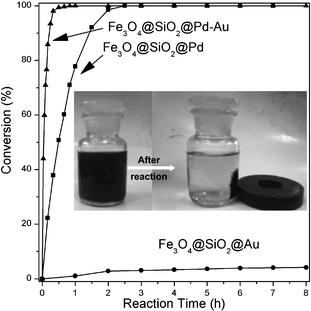 | ||
| Fig. 4 4-CP HDC conversions on different catalysts. | ||
For HDC reactions, large Pd particles possess high TORs because the nature of structure-sensitive reactions.1–9 Thus, Fe3O4@SiO2@Pd-Au catalyst should possess a lower TOR, because Au serves as a dilution element for the Pd-Au nanoparticles,23 resulting in surface enrichment of Au atoms and low surface coverage of Pd metal. However, the dramatic enhancement of TOR on Pd-Au particles indeed occurs, suggesting a key role of Au in promoting HDC activity. For HDC reactions catalyzed by Pd-Au, Au promotes catalysis through geometric and electronic effects.24 Two-dimensional ensembles or islands of Pd atoms are more active sites than pure Pd surface,24b and they are formed on the Au surface defects. On the other hand, the Pd-Au interface provides another set of active sites for HDC based on similar work on Pd-Au and Pd-Cu alloy catalysts, in which the Au induces the adsorption of chlorinated compounds and Pd serves to activate H2 and catalyze HDC.24 Additionally, the presence of the Au can influence the adsorption of reactants or desorption of products.25 For instance, chlorine atoms can bind to the Au surface,26 which would enhance the desorption of products and inhibit the poisoning of the Pd surface.
Magnetic separation is an attractive alternative for filtration or centrifugation as it prevents the loss of catalyst and increases the reusability of the catalyst for liquid-phase reactions. Fe3O4 particles are known for their paramagnetic properties.17 Here, Fe3O4@SiO2@Pd-Au or Fe3O4@SiO2@Pd nanocomposites are superparamagnetic in nature, which makes them amenable to magnetic separation, ensuring that the separation of these nanocomposites from reactants and products is very easy and efficient. The catalyst was recovered by simple decantation of liquid products in the presence of an external magnet as depicted in the insert of Fig. 4. The catalyst was then washed with ethanol and then water, and used directly for the next cycle of the reaction without further purification. The recoverability and reusability were investigated by the HDC reaction of 4-CP and the results are summarized in Fig. 5. Fe3O4@SiO2@Pd-Au exhibits much better stability than Fe3O4@SiO2@Pd, and little decrease in conversion and initial HDC turnover rate occurs after 8 recycling times, suggesting the presence of Au could obviously improve the stability of the Pd metal catalyst.
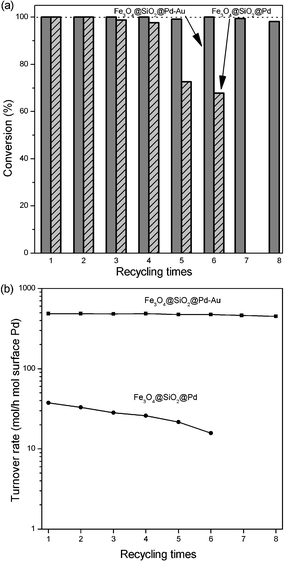 | ||
| Fig. 5 (a) Conversions and (b) initial HDC turnover rates of 4-CP HDC over recycled catalysts. | ||
Here, we report a general synthesis protocol for the design and synthesis of a highly dispersed and easily separated and recycled supported bimetallic or alloy cluster catalyst for catalytic reactions. The superparamagnetic nature of Fe3O4@SiO2@Pd-Au allows for easy separation and redispersion of the catalyst for its reuse. The formation of a Pd-Au alloy promotes the 4-CP HDC activity and stability.
Experimental section
Catalyst preparation
Analytical methods
The chemical composition of samples was analyzed by inductively coupled plasma atomic emission spectrometry (ICP-AES) on an IRIS intrepid spectrometer. Transmission electron microscopy (TEM) images were acquired using a FEI Tecnai G2 high-resolution transmission electron microscope operating at 200 kV. Powder X-ray diffraction (XRD) patterns of the catalysts were obtained on a Rigaku D/max 2500 X-ray diffractometer (Cu Kα, λ = 1.54178 Å) operated at 40 kV and 40 mA in the 2θ range of 10–50° and with a scanning speed of 2° min−1. The dispersion of Pd metal crystallites was measured using CO chemisorption (ASAP2020, Micromeritics) at 40 °C. Mean cluster sizes were estimated from these dispersions by assuming spherical structures and the atomic density of bulk Pd (14.70 × 10−3 nm3). Infrared measurements were made with a Nicolet NEXUS 670 infrared spectrometer. The samples used for infrared study were ground in an agate mortar to a fine powder, diluted with SiO2 power (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 weight ratio of catalyst/SiO2), and compressed into thin wafers under 1 × 104 lb in−2 pressure. All the samples were reduced at 200 °C with flowing H2 (40 ml min−1) for 1 h and evacuated for 1 h at the same temperature prior to cooling to 0 °C for IR measurements.
:2 weight ratio of catalyst/SiO2), and compressed into thin wafers under 1 × 104 lb in−2 pressure. All the samples were reduced at 200 °C with flowing H2 (40 ml min−1) for 1 h and evacuated for 1 h at the same temperature prior to cooling to 0 °C for IR measurements.
Activity testing
The batch experiments for 4-chlorophenol HDC were performed in a 500 mL three-necked flask at 25 °C and atmospheric pressure,22 to which 0.5 g catalyst, 0.05 g NaOH, deionized water and hydrogen flow were added. To evaluate the catalyst activity under catalytic control, reaction conditions were first established. The effect of increasing H2 flow rate and stirring speed on hydrodechlorination performance was investigated, and a hydrogen flow rate of 60 mL min−1 and a stirring speed of 600 rpm were set to exclude the limit of H2 diffusion. After stirring for 10 min, a certain amount of 4-chlorophenol was introduced and the reaction was timed immediately. To evaluate the conversion of the reaction, the sample was collected with a glass syringe at an interval of 5 min, and then passed through a 0.45 μm membrane filter. In many cases, the reactions were tested for the repeatability under identical conditions, and the reaction reproducibility was better than ±5%. A carbon balance was performed by comparing the amount of carbon entering per unit time and the number of products exiting per unit time. Carbon mass balance was complete to better than ±1%.Acknowledgements
The present work was supported by the Natural Sciences Foundation of Tianjin (10JCYBJC04400) and MOE (IRT-0927).References
- M. A. Keane, ChemCatChem, 2011, 3, 800 CrossRef CAS.
- J. B. Hoke, G. A. Gramiccioni and E. N. Balko, Appl. Catal., B, 1992, 1, 285 CrossRef CAS.
- V. V. Lunin and E. S. Lokteva, Russ. Chem. Bull., 1996, 45, 1519 CrossRef.
- F. J. Urbano and J. M. Marinas, J. Mol. Catal. A: Chem., 2001, 173, 329 CrossRef CAS.
- F. Alonso, I. P. Beletskaya and M. Yus, Chem. Rev., 2002, 102, 4009 CrossRef CAS.
- M. A. Keane, J. Chem. Technol. Biotechnol., 2005, 80, 1211 CrossRef CAS.
- G. Yuan and M. A. Keane, Catal. Commun., 2003, 4, 195 CrossRef CAS.
- H. M. Roy, C. M. Wai, T. Yuan, J.-K. Kim and W. D. Marshall, Appl. Catal., A, 2004, 271, 137 CrossRef CAS.
- E. Diaz, J. A. Casas, A. F. Mohedano, L. Calvo, M. A. Gilarranz and J. J. Rodríguez, Ind. Eng. Chem. Res., 2008, 47, 3840 CrossRef CAS.
- Z. Jin, C. Yu, X. Wang, Y. Wan, D. Li and G. Lu, Chem. Commun., 2009, 4438 RSC.
- Z. M. de Pedro, E. Diaz, A. F. Mohedano, J. A. Casas and J. J. Rodriguez, Appl. Catal., B, 2011, 103, 128 CrossRef CAS.
- Z. Jin, C. Yu, X. Wang, Y. Wan, D. Li and G. Lu, J. Hazard. Mater., 2011, 186, 1726 CrossRef CAS.
- B. Coq, S. Hub, F. Figueras and D. Tournigant, Appl. Catal., A, 1993, 101, 41 CrossRef CAS.
- M. Bonarowska, J. Pielaszek, W. Juszczyk and Z. Karpinski, J. Catal., 2000, 195, 304 CrossRef CAS.
- M. Bonarowska, A. Malinowski, W. Juszczyk and Z. Karpinski, Appl. Catal., B, 2001, 30, 187 CrossRef CAS.
- M. Bonarowska, B. Burda, W. Juszczyk, J. Pielaszek, Z. Kowalczyk and Z. Karpinski, Appl. Catal., B, 2001, 35, 13 CrossRef CAS.
- J. Yu, L. Zhao and B. Cheng, J. Solid State Chem., 2006, 179, 226 CrossRef CAS.
- (a) L. M. Rossi, I. M. Nangoi and N. J. S. Costa, Inorg. Chem., 2009, 48, 4640 CrossRef CAS; (b) S. Devaraian, P. Bera and S. Sampath, J. Colloid Interface Sci., 2005, 290, 117 CrossRef.
- R. Arundhathi, D. Damodara, P. R. Likhar, M. L. Kantam, P. Saravanan, T. Magdaleno and S. H. Kwon, Adv. Synth. Catal., 2011, 353, 1591 CrossRef CAS.
- Y. X. Li, H. Y. Xian and Y. Zhou, Appl. Catal., A, 2011, 401, 226 CrossRef CAS.
- (a) M. Primet, M. V. Mathieu and W. M. H. Sachtler, J. Catal., 1976, 44, 114 CrossRef; (b) Y. L. Lam and M. Boudart, J. Catal., 1977, 50, 530 CrossRef CAS; (c) E. L. Kugler and M. Boudart, J. Catal., 1979, 59, 201 CrossRef CAS; (d) T. Wei, J. H. Wang and D. W. Goodman, J. Phys. Chem. C, 2007, 111, 8781 CrossRef CAS; (e) F. M. Hoffmann, Surf. Sci. Rep., 1983, 59, 201 Search PubMed.
- C. X. Sun, Z. J. Wu, Y. Z. Mao, X. Q. Yin, L. Y. Ma, D. H. Wang and M. H. Zhang, Catal. Lett., 2011, 141, 792 CrossRef CAS.
- D. D. Eley and J. Res. Inst. Catalysis, Hokkaido Univ., 1968, 16, 101 CAS.
- (a) M. O. Nutt, J. B. Hughes and M. S. Wong, Environ. Sci. Technol., 2005, 39, 1346 CrossRef CAS; (b) M. O. Nutt, K. N. Heck, P. Alvarez and M. S. Wong, Appl. Catal., B, 2006, 69, 115 CrossRef CAS; (c) Y. L. Fang, J. T. Miller, N. Guo, K. N. Heck, P. J. J. Alvarez and M. S. Wong, Catal. Today, 2011, 160, 96 CrossRef CAS; (d) Y. L. Fang, N. Guo, K. N. Heck, P. J. J. Alvarez and M. S. Wong, ACS Catal., 2011, 1, 128 CrossRef CAS.
- (a) Z. J. Wu, M. H. Zhang, Z. F. Zhao, W. Li and K. Y. Tao, J. Catal., 2008, 256, 323 CrossRef CAS; (b) Z. J. Wu, Z. F. Zhao and M. H. Zhang, ChemCatChem, 2010, 2, 1606 CrossRef CAS.
- G.N. Kastanas and B.E. Koel, Appl. Surf. Sci., 1993, 64, 235 CrossRef CAS.
- Z. L. Mo, C. Zhang, R. B. Guo, S. J. Meng and J. X. Zhang, Ind. Eng. Chem. Res., 2011, 50, 3534 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available: UV-vis spectra of alloy samples. See DOI: 10.1039/c1ra00491c/ |
| This journal is © The Royal Society of Chemistry 2011 |