Regioselective formation of medium-ring heterocycles of biological relevance by intramolecular cyclization
K. C.
Majumdar
*ab
aDepartment of Chemistry, University of Kalyani, Kalyani, 741235, W.B., India
bDepartment of Chemical Sciences, Tezpur University, Napaam, Tezpur, 784 028, Assam, India
First published on 13th October 2011
Abstract
This brief account describes our progress on the regioselective synthesis of medium-sized heterocycles. Formation of the medium-ring (seven-, eight-, nine-membered) oxa-, aza-, thia, oxa-thia- and aza-thia-heterocycles has been achieved by the intramolecular metal-mediated cyclization (especially Pd-catalyzed), diene and enyne metathesis, thiol-mediated cyclization, iodocyclization and azide-alkyne cycloaddition.
1. Introduction
Medium-ring carbo- and heterocycles are quite significant in organic chemistry as these are the structural core of a large number of biologically important natural products.1 These also serve as the target molecules for a number of synthetic studies.2 Some medium-ring heterocycles form part of the structure of a range of natural products and medicinally important compounds. 1,3,5,7-Tetranitro-octahydro-s-tetrazocine is a high melting explosive (HMX). My past experience with the synthesis of medium-ring heterocycles dates back to the early 1970's involving the condensation of urethanes with formaldehyde under appropriate conditions to give octahydro-1,3,5,7-tetracarbalkoxy-tetrazocine derivatives and also the conversion of 1,3,5-tricarbalkoxyhexahydro-s-triazines to 1,3,5,7-tetracarbalkoxy-octahydro-s-tetrazocines.3 A number of methodologies have been developed4 over the last several years for the synthesis of medium-ring systems. The synthetic protocols for five- and six-membered ring systems are common via the different types of cyclizations, but the formation of seven-, eight- and nine-membered ring heterocycles are not as abundant. Because of enthalpic and entropic factors medium-rings are difficult to prepare. Cyclization strategies are often hampered by entropic factors and transannular interactions.5 Therefore, relatively fewer methods based common general cyclization or cycloaddition reactions have been utilized6 for the preparation of medium-ring heterocycles from acyclic precursors.2. Construction of medium-heterocyclic ring by Palladium-catalyzed intramolecular cyclization
The intramolecular Heck reaction of appropriately tethered substrates has been extensively used for the construction of medium-ring heterocycles. A substrate having an activated Michael type olefinic fragment smoothly undergoes the intramolecular cyclization via the endo-trig mode.7 This is perhaps due to the minimization of entropy factor and transannular interaction.8 However, the case of unactivated olefinic system is totally different. The cyclization generally favors endo-trig mode with minor amount of exo-trig mode. Therefore, there is always a chance to produce a mixture of products giving the endo-Heck product as the major one.2.1 Construction of medium-ring oxa-heterocycles
Benzofused oxepine- and oxocine- derivatives belong to a class of naturally occurring important compounds.9 Despite their broad spectrum of biological activity, benzo-and dibenzo-fused oxepine- and oxocine derivatives are not sufficiently investigated. The Heck reaction has been extensively studied in our laboratory during the last few years.10 We, therefore, aimed at achieving a regioselective synthesis of naphthoxepine- and naphthoxocine derivatives by palladium-catalyzed intramolecular Heck cyclization. The tailored substrates 1 were prepared from o-hydroxybenzaldehyde, 2-hydroxy naphthalene-1-aldehyde via alkylation with o-bromobenzyl bromide followed by Wittig olefination. The o-bromobenzyl 2-(1-vinylnaphthalene) ethers 1a,b when subjected to Heck reaction condition [Pd(OAc)2, KOAc, TBAB, DMF, 100 °C, 4 h] afforded only the naphthoxocine derivatives 2a,b in 86% and 67% yields without any contamination from the exo-cyclization product (Scheme 1). Other substrates 1c–j when treated under the optimized reaction condition [(Pd(OAc)2/TBAB/KOAc/DMF/100 °C] also afforded exclusively the corresponding eight-membered cyclized oxocine-derivatives 2c–j without any contamination of the oxepine derivatives.11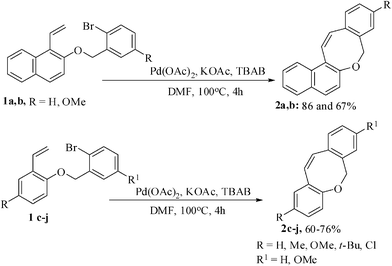 | ||
| Scheme 1 Regioselective synthesis of benzoxocine derivatives. | ||
As no exo-cyclization was observed with Pd(OAc)2 the reaction condition was further manipulated by changing the palladium catalyst and other variable parameters. When the Heck precursors 1a,b were allowed to react with 20 mol% Pd(PPh3)4 as catalyst, and dry Et3N as base in refluxing acetonitrile for 24 h, the required seven-membered naphthoxepines 3a,b were isolated as the major products along with naphthoxocine derivatives 2a,b as the minor products (Scheme 2).
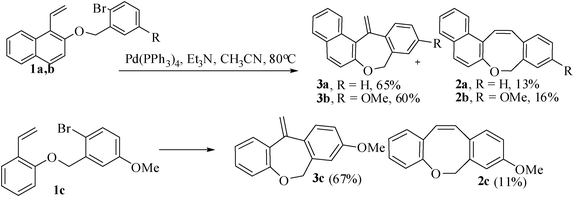 | ||
| Scheme 2 Synthesis of benzoxepine and benzoxocine derivatives. | ||
We have also investigated the synthesis of naphthoxocine derivatives based on the combined Claisen rearrangement and phosphine-free12 palladium-catalyzed intramolecular Heck reaction. Thus the appropriate substrates 4a–d with flexible tethers were prepared by the alkylation of 1-allyl-2-naphthol and 2-allyl-1-naphthols with either 2-bromobenzyl bromide or 2-bromo-5-methoxybenzyl bromide. When the intramolecular Heck reaction was carried out with 4a in the presence of 10 mol% of Pd(OAc)2 as catalyst and KOAc (2.75 equiv.) as base and tetrabutylammonium bromide as promoter13 in DMF at 120 °C for 4 h, the 8-exo cyclized product 6a was obtained as the major product in 78% yield, along with the 9-endo cyclized 7 as a minor product in 20% yield. The use of Pd(PPh3)2Cl2 as catalyst was also found to be effective to give the product 6a in low yield (49%) but the 9-membered endo-Heck product was completely absent. Substrates 4b–d were reacted under the same condition to afford the 8-membered exo-cyclic ring products 6b–d in 72–81% yields. The bis-allyl-bis-ether 5 derived from resorcinol on similar treatment gave mono-8-exo-trig product 8 in 69% yield along with 21% uncyclized product. Further manipulation of the reaction conditions did not improve the yield or give any indication of the formation of the desired bis-benzoxocine derivatives by the occurrence of another 8-exo-trig cyclization of 8 (Scheme 3).14
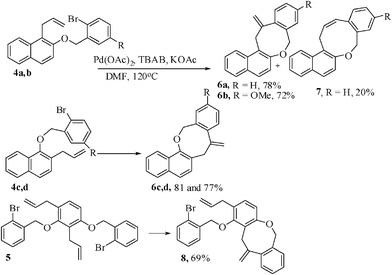 | ||
| Scheme 3 Synthesis of naphthoxocine derivatives. | ||
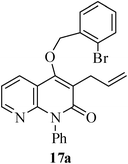
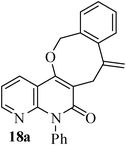
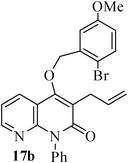
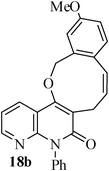
The same study was also extended to heterocyclic systems with appropriate tethers. We have prepared substrates 9 and 19,15,17 from 3-allyl-4-hydroxy-1-methylquinolin-2(1H)-one, 3-allyl-1,6-dimethyl-4-hydroxy-pyridin-2(1H)-one, 3-allyl-4-hydroxy-1-phenyl-1,8-naphthyridin-2(1H)-one respectively. When the Heck reaction was carried out with the substrates 9a,b,15a,b and 17a, each precursors smoothly underwent cyclization leading to the formation of the corresponding exo-Heck (kinetically controlled) product in good to excellent yields except for 17b (Scheme 4).15
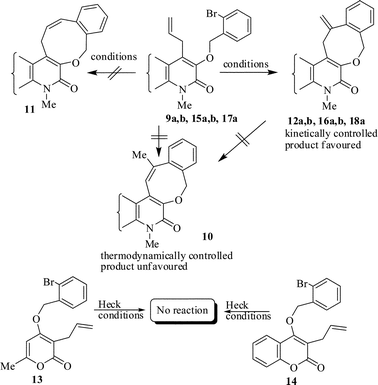 | ||
| Scheme 4 Formation of kinetically controlled vs. thermodynamically controlled products. | ||
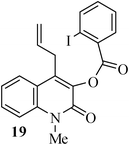
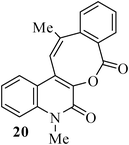
In the case of substrate 17b, the Heck reaction performed under the condition afforded exclusively the endo-Heck cyclization product 18b. Similar substrates 13 and 14 remained unchanged under the condition. No rationalization could be provided for this observation. Recently, Guy et al.16 attempted the synthesis of medium-sized lactone derivatives from the highly activated Heck precursors. However, their attempts to synthesize medium-sized lactone derivatives by palladium-catalyzed Heck reaction failed. We, therefore, prepared the tailored substrate 19 having appropriate tether and subjected this to intramolecular Pd-catalyzed cyclization. Our initial effort to cyclize the substrate 19 to the lactone also failed. However, when the reaction was carried out with Pd(PPh3)2Cl2, TBAB, NaOAc, DMA, 130 °C for 2 h to our pleasant surprise the corresponding lactone derivative 20 was obtained in 43% yield. The formation of lactone 20 from the substrate 19 has been explained via 8-exo-trig mode of cyclization followed by a double bond isomerization (thermodynamically controlled product). Here it is noteworthy that we have succeeded in preparing the medium 8-membered lactone by the implementation of the intramolecular Pd-catalyzed cyclization. The results are summarized in Table 1.
| Entry | Substrate | Optimized conditions | Products | Yield (%) |
|---|---|---|---|---|
| 1 |
|
Pd(PPh3)4, TBAB, KOAc, DMF, 95 °C, 2 h |
|
71 |
| 2 |
|
Pd(OAC)2, TBAB, KOAc, DMF, 120 °C, 6 h |
|
87 |
| 3 |
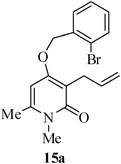
|
Pd(OAC)2, TBAB, KOAc, DMF, 120 °C, 5 h |
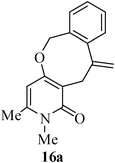
|
69 |
| 4 |
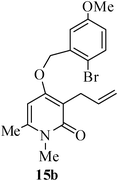
|
Pd(OAC)2, TBAB, KOAc, DMF, 95 °C, 3.5 h |
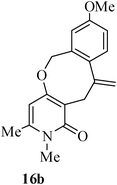
|
59 |
| 5 |
|
Pd(OAC)2, TBAB, KOAc, DMF, 95 °C, 3.5 h |
|
73 |
| 6 |
|
Pd(OAC)2, TBAB, KOAc, DMF, 120 °C, 5 h |
|
51 |
| 7 |
|
Pd(PPh3)2Cl2, TBAB, NaOAc, DMA, 130 °C, 2 h |
|
43 |
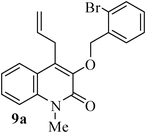
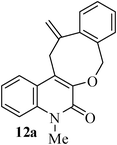
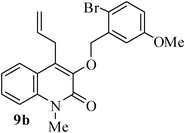
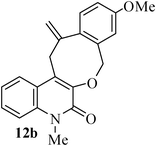
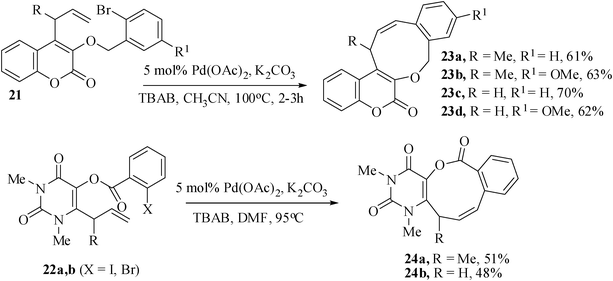
We have further extended our studies on the regioselective synthesis of coumarin- and uracil-annulated medium-ring oxa-heterocycles. For this purpose we have prepared the appropriately tethered substrates 21 from 4-(2’-methylbut-3’-enyl)-3-hydroxy coumarin, 4-allyl-3-hydroxy coumarin by alkylation with either 2-bromobenzyl bromide or 2-bromo-5-methoxybenzyl bromide, 22a,b from 6-(2’-methyl-but-3-enyl)-1,3-dimethyl-5-hydroxy uracil and 6-allyl-1,3-dimethyl-5-hydroxy uracil respectively by esterification with 2-iodobezoyl chloride and 25a–f from 6-(appropriately substituted allyl/vinyl)-1,3-dimethyl-5-hydroxy uracil by alkylation with either 2-bromobenzyl bromide or 2-bromo-5-methoxybenzyl bromide. When attempted under normal Heck reaction condition [(Pd(OAc)2/TBAB/KOAc/DMF] the substrate 21a failed to give any nine-membered product instead afforded the allylic double bond isomerized product. Thus the reaction condition was optimized. When the reaction of 21a–d were carried out using 5 mol% of Pd(OAc)2 as catalyst, K2CO3 as base, TBAB as additive in CH3CN at 80 °C for 2 h, the nine-membered cyclized product 23a–d were obtained in 61-70% yields (Scheme 5).17
 | ||
| Scheme 5 Unusual nine-endo mode of cyclization. | ||
The corresponding 2-iodo-benzoate-tethered substrates 22a,b failed to react under the above optimized conditions. When the reaction was carried out using modified optimized conditions [Pd(OAc)2, K2CO3, TBAB, DMF at 95 °C] the nine-membered lactone 24a,b were obtained in 51% and 48% yields respectively.
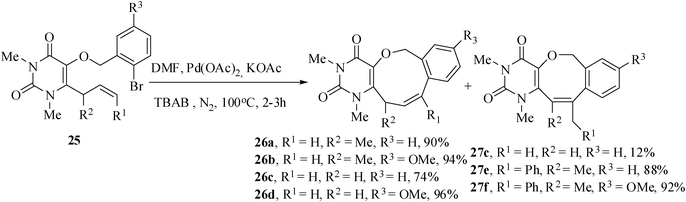
The intramolecular Heck reaction of the tailored substrates 25a–f were carried out with Pd(OAc)2 as catalyst, KOAc as base, TBAB as additive in CH3CN at 100 °C for 2–3.5 h. All the reactions gave exclusively endo-Heck heterocyclic rings 26a–d fused with aryl ring and uracil moiety by the unusual 9-endo-trig in 76–96% yields and 27e,f by 8-endo-trig cyclization mode in 88 and 91% yields. The substrate 25c gave the 9-endo product 26c (74%) along with the formation of eight-membered ring compound 27c (12%) by 8-exo-trig cyclization (Scheme 6).
 | ||
| Scheme 6 Synthesis of uracil-annulated eight-and nine-membered heterocycle. | ||
2.2 Construction of medium-ring aza-heterocycles
Other groups18 also reported the formation of medium-ring heterocyclization by palladium-catalyzed Heck reaction, but none has reported the synthesis of medium-ring heterocycles starting from unactivated allylic C–H activation method. Moreover, though the formation of medium-sized oxa-heterocycles are relatively abundant,19,20 the medium-sized nitrogen heterocycles by the implementation of intramolecular Heck reactions are rare. It is reported21 that the palladium-catalyzed cyclization by the application of intramolecular Heck reaction requires harsh reaction condition where a nitrogen-containing compound is used as the starting material. Recently, Guy et al.16 reported the eight-membered azocines by the intramolecular Heck reaction via 8-endo-trig mode of cyclization starting from highly activated precursors. We have recently achieved the synthesis of biologically interesting azocine intermediates by the application of combined aza-Claisen rearrangement and palladium-mediated intramolecular Heck reaction via 8-exo-trig mode of cyclization starting from the unactivated allylic systems. The tailored substrates 28a–f were prepared from the appropriate N-tosyl anilines by BF3.Et2O catalyzed aza-Claisen rearrangement, tosylation and alkylation with 2-bromobenzyl bromides. When the intramolecular Heck reactions were conducted with the substrates 28a–f applying the concept of Jeffrey's two-phase protocol in the presence of Pd(OAc)2, KOAc and TBAB in dry DMF under a nitrogen atmosphere for 6 h, the eight-membered exo-Heck products 29a–f were obtained in 72–79% yields (Scheme 7).22 Expectedly the tosyl deprotected amine substrates did not react under the optimized reaction conditions.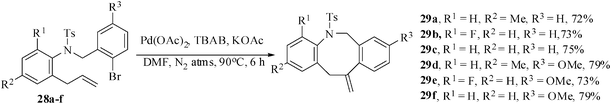 | ||
| Scheme 7 Formation of benzazocine derivatives. | ||
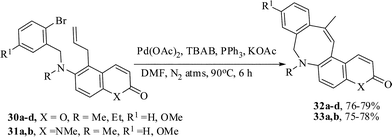
The aforesaid strategy was also applied to the synthesis of other heterocycles-annulated benzazocine derivatives. The appropriate precursors 30a–d,31a,b for this purpose were prepared from 6-amino coumarin and 6-amino quinolin-2(1H)-one. The intramolecular Heck reactions were performed in anhydrous N,N-dimethylformamide in the presence of potassium acetate as a base, tetrabutylammonium bromide as a promoter, triphenylphosphine as ligand, and palladium(II) acetate as catalyst under a nitrogen atmosphere at 90 °C for about 6–7.5 h. All the reactions afforded the eight-membered heterocyclic compounds 32a–d,33a,b in 75–79% yields (Scheme 8). In this case also kinetically controlled 8-exo-trig cyclized products perhaps underwent double-bond isomerization to give the thermodynamically stable endo-cyclic products or else might have been formed by prior allylic double bond isomerization followed by 8-endo-trig cyclization. Another point to note that in the present instance the phosphine ligand is required. In the absence of PPh3 ligand the reaction gives a lower yield of the product.23
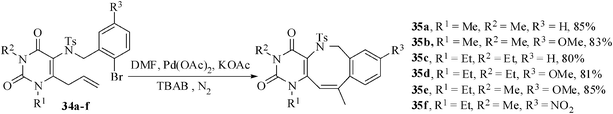
The same combined aza-Claisen rearrangement and palladium-mediated intramolecular Heck reaction has also been successfully applied to the synthesis of uracil-annulated benzazocine derivatives. The appropriate substrates 34a–f were prepared from 5-bromo-1,3-dialkyl uracils by reaction with allyl amine, aza-Claisen rearrangement, tosylation followed by alkylation with 2-bromobenzyl bromides. When the precursors 34a–e were heated at 90 °C in anhydrous N,N-dimethylformamide using potassium acetate (2.75 equiv) as a base, tetrabutylammonium bromide (1.2 equiv) as a promoter, and palladium(II) acetate (10 mol%) as the catalyst under a nitrogen atmosphere, the reaction proceeded smoothly and was complete within one hour affording the pyrimidoazocine derivatives 35a–e in 80-85% yields. However, the substrate 34f containing the 5-nitro-2-iodobenzyl moiety afforded an intractable complex mixture from which no cyclized product could be isolated (Scheme 9). The initially formed 8-exo cyclized products possessing an exo-methylene (kinetically controlled product) may undergo double bond isomerization to give the thermodynamically stable products 35. The other possibility is that a double bond isomerization prior to the Heck reaction may occur followed by 8-endo Heck cyclization to give the products 35.24
 | ||
| Scheme 9 Uracil-annulated benzazocine derivative formation. | ||
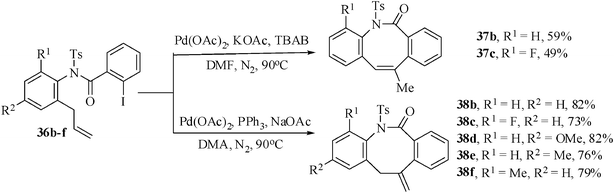
The combined aza-Claisen rearrangement and palladium-mediated intramolecular Heck reaction has also been successfully exploited for the synthesis of dibenzoazocinone framework. The appropriately tethered substrates were prepared by aza-Claisen rearrangement, tosylation followed by amidation with 2-iodobenzoyl chloride. The substrate 36a with free NH (i.e.; without Ts protection) failed to undergo any reaction under ligand free condition [Pd(OAc)2/KOAc/TBAB/DMF/N2] and also under phosphine-assisted standard condition [Pd(OAc)2/KOAc/PPh3/TBAB/DMA//N2]. The reason for the failure may be due to the fact that the palladium catalyst loses its catalytic activity by the formation of the chelate-complex25 (36A and 36B) between the palladium metal and the amide carbonyl oxygen atom (Fig. 1).
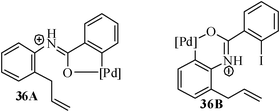 | ||
| Fig. 1 Formation of the chelate-complex between Pd and heteroatom. | ||
Therefore, we have conducted the Heck reaction with the substrate 36b,c under the aforesaid two reaction conditions [Method A and Method B]. The results are depicted in Scheme 10. From the results it is clear that the Jeffrey's two phase protocol (Method A) would be applicable for the construction of endo-cyclic product and the phosphine-assisted standard condition (Method B) will be suitable for the synthesis of exo-cyclic products. Considering further application potential of the exo-cyclic product we have treated other substrates 36 under the optimized reaction condition to afford the corresponding exo-Heck cyclized products dibenzoazocinones derivatives 38b–f in 73–82% yields (Scheme 10).
 | ||
| Scheme 10 Product formation in presence and absence of ligand PPh3. | ||
The synthesis of the isomeric products 37 and 38 from the same substrate 36 provided us an unique opportunity to get more insight into the mechanism of the formation of products. The exo-cyclic product 38 was subjected to the reaction under the condition of Method A. However, no reaction occurred and the unchanged 38 was recovered. As the exo-cyclic product 38 is not convertible to endo-cyclic product 37 under the condition of the method A, the question of double bond isomerization after the occurrence of 8-exo-trig cyclization is ruled out. Therefore, the formation of 37 should have occurred with prior allylic double bond isomerization of 36 followed by 8-endo-trig mode of cyclization.26
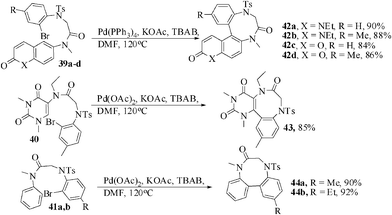
Buchwald and coworkers developed27 a procedure for the synthesis of benzodiazocine derivatives utilizing sequential Cu-catalyzed coupling of a β-lactam with bromo or iodo aryl amine. However, to our knowledge there is no report on the synthesis of heterocycles-annulated diazocines except for pyrrole.28 Therefore, we have recently reported a procedure for the regioselective synthesis of benzodiazocinone-annulated heterocycles by the implementation of palladium catalyzed Heck reaction. The precursors for this purpose, 39a,b were accessed from 6-(N-methylamino) coumarin, 39c,d from 6-(N-methylamino) quinolin-2-(1H)-one, 40 from 1-ethyl-6-(N-methylamino)-1,3-dimethyl-5-(N-ethylamino)uracil and 41a,b from N-methylaniline by the reaction of chloroacetyl chloride followed by further reaction with tosylated o-bromoanilines. The optimal reaction condition for the Heck reaction was established [10 mol% Pd(PPh3)4, KOAc, TBAB, DMF, 120 °C, 4–6 h]. Under the above optimized conditions all the substrates underwent intramolecular Heck reaction smoothly to give the benzodiazocine derivatives in 84–92% yield (Scheme 11).29
 | ||
| Scheme 11 Construction of benz-annulated diazocinones. | ||
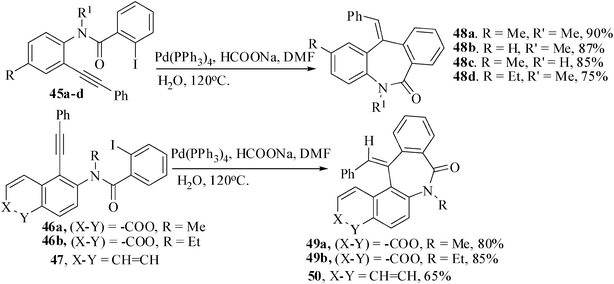
Dibenzoazepinone framework30 is widespread among natural compounds and important pharmaceuticals. Most often the construction of the medium-sized ring of the dibenzoazepinone framework is achieved via intramolecular Friedel–Crafts alkylation. However, this method demands an excess of Lewis acid and lacks flexibility for variation of substituents. Alternatively, ring closure is accomplished upon formation of the C–N bond via intramolecular reductive amination. We have recently achieved a new route to the synthesis of Dibenzoazepinones based on palladium-catalyzed reductive Heck cyclization. The required precursors for the reductive Heck cyclization were synthesized in three steps: bromination of the amines, Sonogashira coupling of the bromo derivatives with phenylacetylene, and subsequent amide-formation reaction with 2-iodobenzoyl chloride. The reductive Heck cyclization of 45a–d were carried out by heating (120 °C) in the presence of 3 mol% of Pd(PPh3)4 as the catalyst and sodium formate as the reducing agent in DMF–water (7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3) mixture for 4.2 h to give dibenzoazepinone derivatives 48a–d in 75–90% yields (Scheme 12).31 The corresponding substrates 46a,b derived from 3-(N-methylamino)coumarin and 47 from N-ethylnaphthyl amine also afforded the desired products 49a,b and 50. The structure of the compound 49b was confirmed from its X-ray crystal analysis.
:3) mixture for 4.2 h to give dibenzoazepinone derivatives 48a–d in 75–90% yields (Scheme 12).31 The corresponding substrates 46a,b derived from 3-(N-methylamino)coumarin and 47 from N-ethylnaphthyl amine also afforded the desired products 49a,b and 50. The structure of the compound 49b was confirmed from its X-ray crystal analysis.
 | ||
| Scheme 12 Synthesis of benz-azepinone derivatives by reductive Heck cyclization. | ||
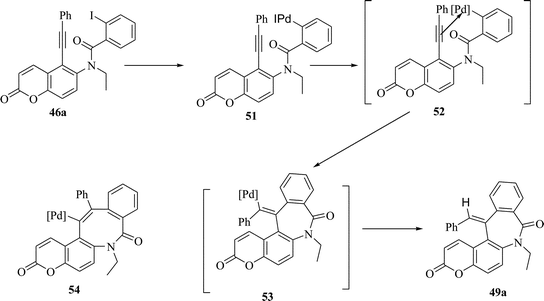
The regioselective formation of exo-cyclized products during reductive Heck cyclization can be rationalized via Initial formation of aryl palladium π-complex 52 which may get transformed into σ-vinyl palladium complex 53via simultaneous syn addition to the triple bond. The endo cyclization via a hypothetical intermediate 54 is fairly unlikely due to high strain exerted by the trans geometry around the double bond in the eight-membered ring. The Pd(0) catalyst is regenerated by the reducing agent present in the reaction mixture (Scheme 13)
 | ||
| Scheme 13 Probable mechanism of formation of benzazepinones. | ||
Larock et al. reported the synthesis of tetrahydrobenzo[d]azocine via palladium-catalyzed heteroannulation of allenes.32 However, this procedure gives the products as mixtures of E/Z isomers of the exo-cyclic double bond. Donets and Eycken33 recently developed a microwave-assisted cyclization procedure to synthesize extended alkyl chain containing dibenzoazocinone framework. However, the procedure is low yielding and applied for only two-electron-donating substrates. We have recently achieved the regioselective synthesis of dibenzoazocinone framework through palladium-mediated reductive Mizoroki–Heck cyclization. The substrate 55a,b for this synthesis were prepared by the reaction of N-ethylamino-2-phenyleth-2-ynyl)benzene/5-(phenyleth-2-ynyl)-6-(N-alkylamino)coumarin/1-methyl-5-(phenyleth-2-ynyl)-6-[N alkyl(methyl/ethyl)amino]quinolone with 2’-iodophenylacetyl chloride. The optimal reaction condition for the Heck reaction was established [Pd(PPh3)4/HCOONa/DMF–H2O (7:3), 100 °C, 0.5 h]. Under the optimized reaction conditions all the other substrates 55b and 56a–d were similarly treated to produce the 8-exo-cyclized products in 42–60% yields (Scheme 14). The cyclization process is extremely time dependent. The increase in the reaction time resulted in the decomposition of both the product and the unreacted starting material.34
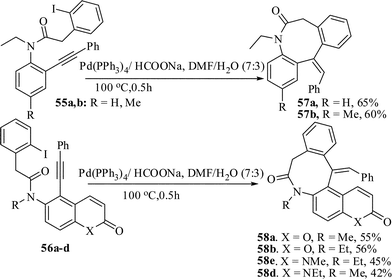 | ||
| Scheme 14 Synthesis of benzoxocine derivatives by reductive Heck reaction. | ||
The regioselective formation of 8-exo-cyclized product during the course of the reaction can be rationalized by a mechanism similar to the one proposed by Donets and Eycken.33
2.3 Construction of medium-ring oxa-thia-heterocycles
There are fewer examples of palladium-catalyzed Heck reaction of sulfur compounds35 in the literature and to our knowledge synthesis of medium-sized ring containing sulfur atom at appropriate position has been rare. Probably there is only one example to construct eight-membered sultones by Heck reaction conditions starting from highly activated vinylic system.14 But, there is no such example available in literature for the synthesis of sultone derivatives from the unactivated allylic systems. Recently we have reported interesting oxathiocine derivatives starting from unactivated vinylic systems with the help of palladium-catalyzed intramolecular Heck reaction. The intramolecular Heck reaction of the tailored substrates 2′-vinylphenyl-2-bromobenzene sulfonates 59 were carried out using the Jeffrey's two-phase protocol in the presence of Pd(OAc)2 as catalyst, KOAc as base, TBAB as promoter in dry DMF at 80 °C under nitrogen atmosphere to afford the 8-membered endo Heck products 60 in 79–91% yields (Scheme 15). 2′-(1′-vinylnaphthyl)-2-bromobenzene sulfonate also yielded the corresponding oxothiocine derivative in 90% yield.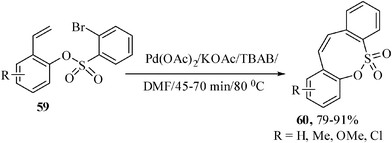 | ||
| Scheme 15 Pd-catalyzed synthesis of benzoxathiocine derivatives. | ||
The reaction is equally successful with substrates where R is a 3,4-phenylene residue. The substrate 61 derived from naphthylene-2,7-diol also provides a 26% yield of the bis-oxathiocine derivative 62 by exclusive 8-endo-trig cyclization along with unidentified products when reacted under the optimized reaction condition (Scheme 16).36a
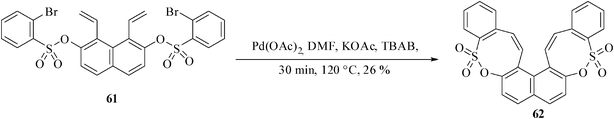 | ||
| Scheme 16 Palladium-catalyzed synthesis of bis-benzoxathiocines. | ||
The regioselectivity in the formation of oxathiocine ring was also investigated with a different set of substrates 2′-allyl-1′-naphthyl 2-bromobenzenesulfonate. The substrates 63a,b were subjected to Heck reaction under optimized condition [Pd(OAc)2, DMF, KOAc, TBAB, 100 °C] to give exclusively the 8-membered sultones 65 in 84–97% yields by 8-exo-trig mode of cyclization (Scheme 17). The (1′-allylnaphthalene)-2-bromobenzene sulfonates 64a,b also afforded the corresponding eight-membered sultones 66a,b in 95 and 84% yields respectively.36b
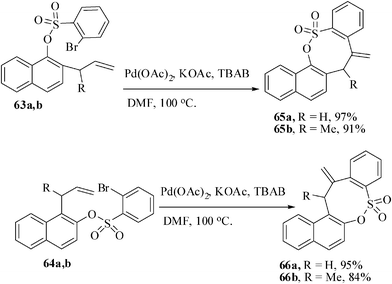 | ||
| Scheme 17 8-exo-trig mode of cyclization for the synthesis of benzoxathiocines. | ||
The cyclization of substrates 59 bearing the sulfonate ester tether may occur via two alternative pathways—either a 8-endo-trig or a 7-exo-trig cyclization. Denieul and Skrydstrup have reported37 that the palladium-catalyzed cyclization of a similar substrate with an ester tether gave a mixture of three products: the 7-exo trig cyclization product (i.e. seven-membered lactone, major product), the 8-endo-trig cyclization product (eight membered lactone, minor product) and the biaryl coupling product (trace). The exclusive formation of benzoxathiocine derivatives 60via the 8-endo-trig mode of cyclization is quite unusual. Beletskaya and Cheprakov reported38 that the endo-Heck cyclization can occur when the Heck precursor possesses a Michael-type olefinic fragment,39 otherwise exclusive exo-cyclization occurs.
The exclusive formation of naphthoxothiocines 65 and 66via 8-exo-trig mode of cyclization is also unusual in view of the report of Beletskaya and Cheprakov38 in which it was claimed that the exo-Heck cyclization can occur only in case of the substrate which generates small to common ring system, otherwise mixture of products of endo-cyclization and exo-cyclization is formed.
2.4 Construction of medium-ring aza-thia-heterocycles
Sultams are useful heterocycles in medicinal chemistry.40 Sultams have been synthesized by different methods. Recently transition metal-catalyzed syntheses of sultams including the use of copper,41 silver,42 rhodium,43 ruthenium44 and a very few examples by palladium-catalyzed cyclization45 have been reported. We have recently reported the regioselective synthesis of tricyclic and pentacyclic sultams by palladium-catalyzed ligand-free intramolecular cyclization. The cyclization of tailored substrates 67 and 68 were carried out using Jeffrey's two-phase protocol in the presence of Pd(PPh3)4 for tricyclic sultams and Pd(OAc)2 for pentacyclic sultams in anhyd. DMF with an organic base at 120 °C for 10 h under a nitrogen atmosphere to afford the corresponding tricyclic sultams 69 in 62–85% yields and the pentacyclic sultams 70 in 80–92% yields respectively (Scheme 18). The presence of electron withdrawing nitro group on the aryl ring bearing the halogen gave a lower yield of the tricyclic sultam (60%) compared to all other substrates (above 80%).46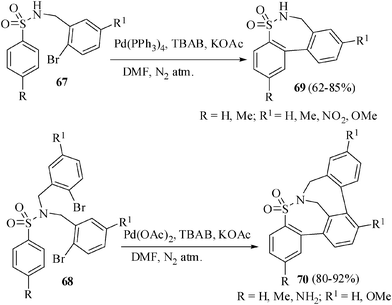 | ||
| Scheme 18 Synthesis of tri- and pentacyclic sultams. | ||
The use of PdCl2 or Pd(PPh3)2Cl2 as a catalyst reduces the yield dramatically. The use of DMSO or CH3CN also resulted in lower yield of the product. The reaction may probably proceed by an initial oxidative addition of the aryl bromide 68 to Pd(0) to form an aryl palladium bromide intermediate 71. The cyclization may then occur by an electrophilic attack of the aryl palladium bromide 71 at the electron-rich aromatic ring to give the mono-cyclized intermediate 73via the intermediate palladacycle 72. The mono-cyclized intermediate 73 may repeat one more catalytic cycle to finally afford the bis-cyclized product 70via intermediate 74 (Scheme 19).
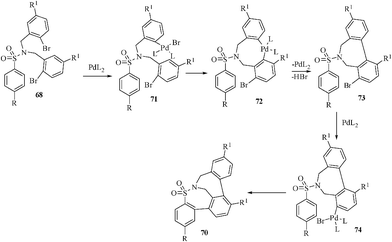 | ||
| Scheme 19 Probable mechanism of formation of tri- and pentacyclic sultams. | ||
3. Construction of medium-ring heterocycles by sulfanyl radical addition-cyclization
Radical chemistry is an established tool in preparative organic synthesis. Recently a cost effective tin-free protocol has been developed for the formation of carbon-carbon bonds by radical reaction based on sulfanyl radical addition-cyclization.47 These radical reaction occurs via the generation of carbon-centred radical species by the addition of a sulfanyl radical to an unsaturated bond. The resulting carbon-centered radical undergoes intramolecular endo-addition to another multiple bond followed by the abstraction of a hydrogen atom from the thiol to afford the product (Fig. 2).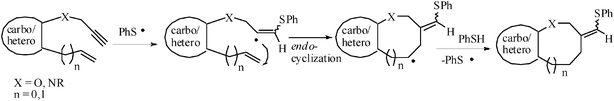 | ||
| Fig. 2 Endo-trig mode of cyclizaion of sulphanyl radical addition intermediate. | ||
Otherwise, the sulfanyl radical may add to the terminus of the triple bond to generate an alkenyl radical, which may undergo intramolecular exo-addition to another multiple bond followed by the abstraction of a hydrogen atom from the thiol to afford the product (Fig. 3).
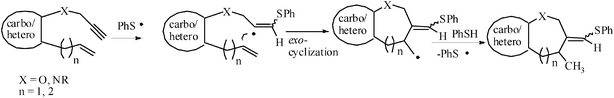 | ||
| Fig. 3 Exo-trig mode of cyclizaion of sulphanyl radical addition intermediate. | ||
This tin-free protocol offered easy work-up procedure for isolation of the product free from bi-product contamination. The cyclized products are often highly functionalized compounds and are useful intermediates for target molecules.48 The formation of different heterocyclic rings by thiol-mediated radical cyclization has been reported. The reaction tolerates a wide range of substrates.
3.1 Synthesis of medium-ring oxa-heterocycles
We have recently utilized the sulfanyl radical addition-cyclization for the regioselective synthesis of medium-ring heterocycles. The synthesis of medium-ring cyclic ethers annulated with aromatic ring is an active area of current research interest due to their presence in many bioactive natural products.49The benzoxepine moiety is present in many natural products50 and justifiably encouraged many research groups to synthesize these compounds. To the best of our knowledge, there are only few examples in the literature51 for the construction of seven-membered cyclic ethers by radical cyclization. Thus the tailored-substrates 76 were subjected to radical cyclization under optimized reaction condition (established through a series of experiments): 1.5 equiv. PhSH, 1.5 equiv. AIBN, benzene, 80 °C, 2 h to give regioselectively the benzoxepine derivatives in 72–88% yields (Scheme 20). The substrate 2-propargyloxy-1-vinylnaphthalene also gave the corresponding naphthoxepine derivative in 86% yields.52
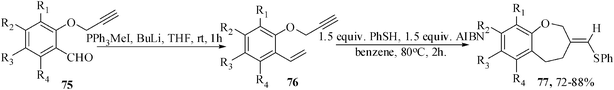 | ||
| Scheme 20 Thiophenol-mediated formation of benzoxepine derivative. | ||
The mechanistic rationalization for the formation of product 77 by sulfanyl radical addition-cyclization is depicted in Scheme 21. Initially, the phenyl sulfanyl radical generated from thiophenol and AIBN adds to the terminal alkyne moiety to form the corresponding vinyl radical intermediate 78. This vinyl radical 78 may undergo a 7-endo-trig intramolecular cyclization with the adjacent alkene to form the radical intermediate 81 which, on abstraction of a proton from thiophenol, leads to the formation of product 77. Both the 6-exo-trig and 7-endo-trig modes of cyclization are favorable, according to Baldwins rule53 to give the radicals 81 and 89. It is remarkable to note that here only 7-endo-mode of cyclization has occurred to afford the oxepine derivatives 77. The predominant formation of the products 77 may be explained not only by the stability of the secondary alkyl radical 81 as opposed to that of the primary alkyl radical 89 but also by the stability of the benzylic radical (extended conjugation) which is perhaps the driving force that allowed the reaction to follow pathway a. Alternatively, pathway b, a 6-exo-trig cyclization process followed by 1,2-alkenyl migration via a cyclopropyl methyl radical 82 (neophyl rearrangement), would also lead to the same product.
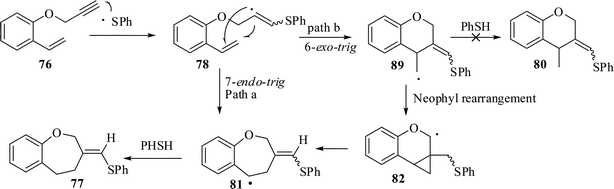 | ||
| Scheme 21 Probable mechanism of formation of benzoxepine ring by thiol-mediated cyclization. | ||
Thiol-mediated 8-endo-trig sulfanyl radical addition-cyclization has been utilized for the regioselective synthesis of aromatic ring/heterocycle-annulated eight-membered ring cyclic ethers. Some examples of natural products containing the 3,4,5,6- tetrahydro-2H-benz(b) oxocine core structure include helianane54, heliannuols A and K55 and protosappanine B56. The characteristic phytotoxic activity and the unique structural features enshrined in these compounds have made them attractive targets for synthesis.57 To the best of our knowledge, only one example of thiophenol-mediated 8-endo-trig radical cyclization has been reported.55b The tailored substrates o-allylphenyl-prop-2-ynyl ethers 83 were accessed from allyl phenyl ethers in two steps.
The optimized condition for the sulfanyl radical addition-cyclization was established. When a 0.1 M solution of 83 was refluxed with PhSH (2 equiv.) and AIBN (2 equiv.) in dry t-butanol for 4 h under a nitrogen atmosphere, afforded the benzoxocine derivatives 84 in 72–85% yields (Scheme 22).58 Out of the seven cases studied five cases gave the benzoxocine with 100% diastereoselectivity. Only in two cases having an ortho-substituent with respect to allyl group on the benzene ring of the substrate under similar condition resulted in a diastereomeric mixture of benzoxocine in 72 and 78% yields. The products were isolated as diastereomeric mixtures in the ratios 3:2, which were not separable by column chromatography. Similarly, the naphthyl substrate, 1-allyl-2-propargyloxy naphthalene when reacted in refluxing toluene also afforded the corresponding naphthoxocine derivative in 88% yield.
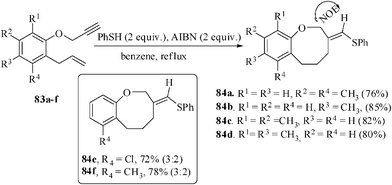 | ||
| Scheme 22 Formation of benzoxocine derivatives 8-endo mode of cyclization. | ||
The product formation may be rationalized by an 8-endo-trig sulfanyl radical cyclization and similar argument given earlier in case of the formation of benzoxepine derivatives 77. The stereochemistry of the exocyclic double bond was found to be exclusively E on the basis of NOE correlation.
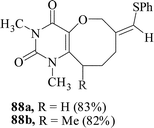
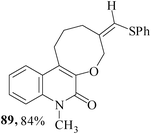
We have also extended59 the regioselective formation of oxocine ring for the synthesis of heterocycle-annulated oxocine compounds via 8-endo-trig cyclization. By the implementation of the same protocol, the uracil-, quinolone- and 1,8-naphthyridone-annulated oxocine derivatives were synthesized in 82–85% yields (Table 2). The stereochemistry of the exocyclic double bond was determined by single crystal XRD.
| Starting material | Product |
|---|---|
|
|
|
|
|
|
3.2 Synthesis of medium-ring aza-heterocycles
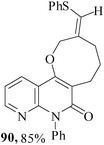
The same methodology has also been successful for the synthesis of uracil-annulated azocine derivatives.60 Azocine ring system is found in various structurally remarkable natural products.61 Although oxocines have been synthesized by different methods, so far no radical-mediated cyclization has been explored. The substrates 91 were treated with PhSH and benzoyl peroxide in refluxing dry t-butanol for 2 h under a nitrogen atmosphere to give uracil-annulated azocine derivatives in 70–85% yields. The usual radical initiator AIBN in place of benzoyl peroxide resulted in a relatively lower yield of the azocines (Scheme 23). The stereochemistry of the exocyclic double bond was determined by single crystal XRD.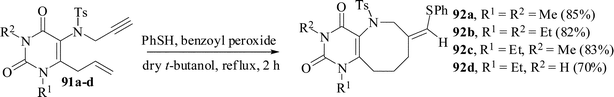 | ||
| Scheme 23 Thiophenol-mediated formation of benzazocine derivatives. | ||
4. Construction of medium-ring heterocycles by diene and enyne metathesis
The discovery and development of olefin metathesis over the past several years have revolutionized the area of medium-ring syntheses. The synthesis of medium-ring heterocycles by RCM methodology62 seem to work best when some conformational constrains favor the ring formation. Medium-ring oxa heterocycles are easily synthesized from appropriately oxygen-tethered substrates by RCM. We have explored the RCM and RCYEM strategy using first and second generation Grubbs' catalysts 1 and 2 (Fig. 4) for the regioselective synthesis of the heterocycles-annulated and carbocycle-annulated medium-ring heterocycles.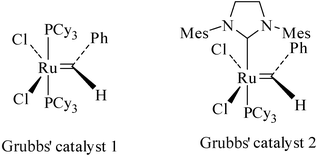 | ||
| Fig. 4 Grubb’s catalysts 1 & 2. | ||
4.1 Construction of oxa-heterocyclic medium-ring
We have reported the regioselective synthesis of pyranoxepine63 and pyridoxepine64 derivatives by tandem Claisen rearrangement and RCYEM protocol. When a dichloromethane solution of the enynes 93 and the Grubbs' catalyst 1 were stirred at rt, the corresponding oxepine derivatives 95 were obtained in 80–90% yields. The Diels–Alder reaction of the product 95 with dimethyl fumarate proceeded smoothly to afford the tricyclic compound 97 in excellent yield. The pyridone substrates 94 also behaved similarly to give the corresponding pyridoxepine derivatives 96 in 88–95% yields (Scheme 24).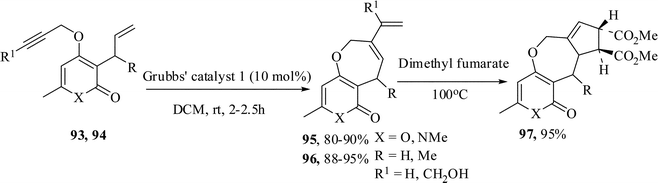
The study was also extended to the synthesis of oxepine- and oxocine-annulated pyrimidine derivatives. Thus 6-allyl-5-allyloxy/homoallyloxy-1,3-dimethyl pyrimidine-2,4-diones 98 on treatment with Grubbs' 1 catalyst in DCM at rt under nitrogen atmosphere furnished the oxepine- and oxocine-annulated pyrimidine derivatives in 75–77% yields while the 6-allyl-5-propargyloxy-1,3-dimethyl pyrimidine-2,4-diones 88a,b gave the oxepine-annulated pyrimidine derivatives 100a,b in 73–78% yields (Scheme 25).65
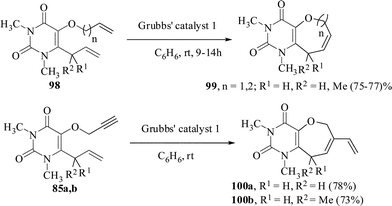 | ||
| Scheme 25 Construction of oxocine derivatives by diene and enyne metathesis reaction. | ||
The combined tandem Claisen rearrangement and the diene and enyne metathesis has also been successfully applied to the synthesis of oxepine- and oxocine-annulated quinolin-2(1H)-ones,66 oxepine- and oxocine-annulated quinoline derivatives67 (Scheme 26).
 | ||
| Scheme 26 Syntheis of quinolone-annulated oxepines. | ||
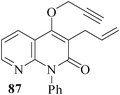
Substituted 1,8-naphthyridine derivatives are used for the diagnostic therapy of the human diseases including AIDS and for combating exo- and endo- parasites in agriculture.68 We have utilized the tandem Claisen rearrangement and RCYEM protocol for an efficient synthesis of the potentially bioactive oxepine-annulated naphthyridone compounds.69 When a dichloromethane solution of the enynes 87a–d and the Grubbs' catalyst 1 was stirred at rt, the medium-ring oxaheterocycles 104a–d were obtained in 90–95% yields (Scheme 27). The corresponding diene, 3-allyl-4-allyloxy-1-phenyl-1,8-naphthyridin-2(1H)-one 103 afforded the oxepine 105 in 97% yield.
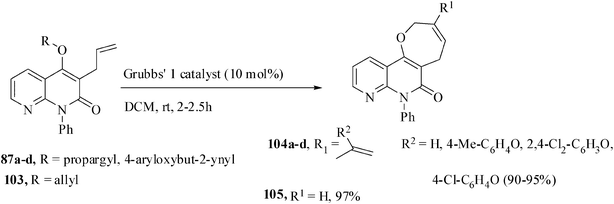 | ||
| Scheme 27 Formation of oxepines by RCM and RCYEM. | ||
Recently we have applied the combined Claisen rearrangement and RCM reaction as a new route to regioselectively synthesize a number of medium-ring heterocyclic compounds.70 The bis-benzoxepines and bis-benzoxocines remained largely unexplored till date. This may be due to lack of general methods of their synthesis, except a few.71 Thus the appropriately tethered substrates 106 and 107 were prepared from naphthalene-1,5-diol and naphthalene-1,6-diol by alkylation with allyl halides followed by 3,3-sigmatropic rearrangement. When a dichloromethane solution of 106 and commercially available Grubbs' catalyst 1 was stirred at rt under nitrogen atmosphere the reaction proceeded smoothly to give the bis-benzoxepine in 90% yield. The tailored-substrates 107a–c also reacted accordingly to give the corresponding products 109a–c in 60–85% yields (Scheme 28).
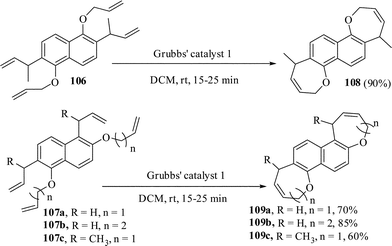 | ||
| Scheme 28 Synthesis of bis-fused heterocycles by RCM reaction. | ||
4.2 Construction of aza-heterocyclic medium-ring
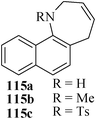
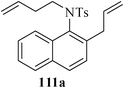
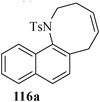
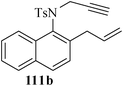
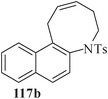
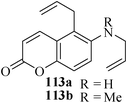
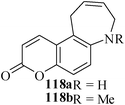
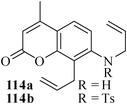
Various nitrogen heterocyclic systems (common to larger rings) have been conveniently synthesized using RCM protocol. We have also made some progress in the use of N-tethered dienes and enyns for the synthesis of azepine- and azocine-annulated carbocycles as well as heterocycles. The substituted N-tethered dienes and enyns were accessed by the use of aza-Claisen rearrangement as a key step. The RCM reactions were carried out using Grubbs' catalyst 1 in DCM at rt under nitrogen atmosphere. Toluene as solvent was required for the enyne substrate. The results are summarized in Table 2. Reaction of free amine substrate under RCM condition, expectedly, gave no cyclized product (Table 3, entry 6). The modified substrate 113b (by replacing NH with NMe, electron releasing) was also found to be not very effective. The azepine derivative 118b was obtained in only 35% yield. However, the substrate 114b containing a N-tosyl group afforded the RCM product 119b in excellent yield (89%).72| Entry | Starting materials | Conditions | Products | Yielda |
|---|---|---|---|---|
| a Isolated yield. b NR: No reaction. | ||||
| 1 |
|
dry CH2Cl2/r. t./30 min |
|
NRb |
| NR | ||||
| 96 | ||||
| 2 |
|
dry CH2Cl2/r. t./45 min |
|
94 |
| 3 |
|
dry toluene/110 °C/10 h |
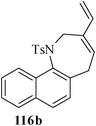
|
61 |
| 4 |
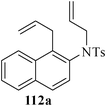
|
dry CH2Cl2/r. t./35 min |
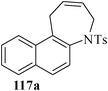
|
95 |
| 5 |
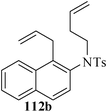
|
dry CH2Cl2/r. t./50 min |
|
89 |
| 6 |
|
dry CH2Cl2/r. t./10 h |
|
NR |
| 35 | ||||
| 7 |
|
dry CH2Cl2/r. t./40 min |
|
NR |
| 89 |
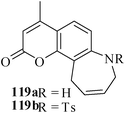
Palladium-catalyzed intramolecular Heck coupling reaction usually require harsh reaction conditions where a nitrogen containing substrate is used as a starting material.73 The other problem is the two different possible modes of cyclization, endo-trig and exo-trig, often compete with each other.74 Under the circumstances we have achieved the regioselective synthesis of biologically interesting pyrimidine-annulated azepine- and azocine- derivatives by tandem aza-Claisen rearrangement and intramolecular RCM reactions. Thus N-tethered dienes 120 and 121 were stirred with Grubbs' catalyst 1 in DCM at rt under nitrogen atmosphere to furnish the pyrimidine-annulated azepines 122 and pyrimidine-annulated azocines 123 in 87–90% and 85–89% yields respectively (Scheme 29). This sequence presents a very short, mild and efficient synthesis of 7-and 8-membered ring nitrogen heterocycles and provides an easy access to libraries for medicinal and pharmaceutical applications.75
 | ||
| Scheme 29 Formation of aza-heterocyclic compounds by RCM. | ||
4.3 Construction of medium-ring thia-heterocycles
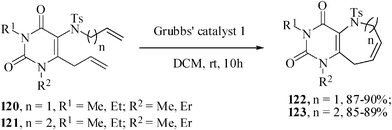
Many examples of the synthesis of oxa- and aza-heretocycles by RCM are known. However, the application of RCM to the synthesis of sulfur-containing medium-ring heterocycles remains very limited. Only tungsten,76 molybdenum77 and titanium78 catalysts were the three efficient catalyst for metathesis reaction of compounds bearing sulfur atom. Thus the S-tethered precursors 128,129 and 130 were prepared from 3-allyl-4-hydroxy coumarin 124 and 3-allyl-4-hydroxy-6-methyl pyran-2-one 125.Attempts to perform the metathesis reaction of the substrates 128–130 with Grubbs' catalyst 1 proved unsuccessful. However, the use of Grubbs' catalyst 2 in the enyne metathesis was successful and the substrates 128a–d afforded the products 131a–d in 65–72% yields. Substrates 129 also reacted with the Grubbs' catalyst 2 in refluxing benzene to give the pyrone-annulated thiepine derivatives 132 in 72–77% yields. The sulfonyl group is less likely to induce poisoning of the ruthenium catalyst 1.21 With this presumption the sulfides 130a,b were oxidized to their corresponding sulfones and then reacted in the presence of Grubbs' catalyst 1 in DCM at rt under nitrogen atmosphere for 7 h to smoothly afford the 7- and 8-membered cyclic sulfones 133 in 74 and 76% yields respectively (Scheme 30).79
 | ||
| Scheme 30 Synthesis is of thia heterocyclic compounds by diene and enyne metathesis. | ||
5. Miscellaneous
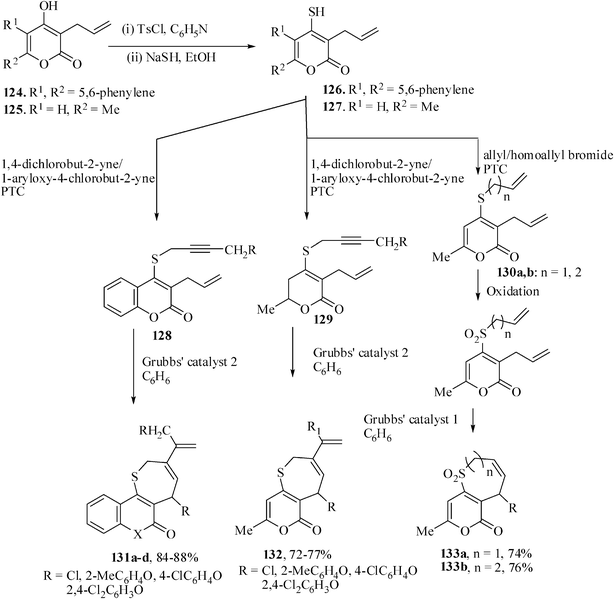
5.1 Synthesis of medium-ring oxa-heterocycles by iodocyclization
Recently iodine has found wide utility because it is an inexpensive, non-toxic and readily available electrophilic reagent for the iodocyclization to afford compounds with synthetic and biological applications.80 Although many examples are available to construct five- and six-membered heterocycles utilizing iodine-mediated cyclization, to our knowledge, the synthesis of medium-ring heterocycles has not been reported. With a view to synthesize some medium-ring oxa-heterocycles we have treated the appropriately tethered substrates 134 under the optimized reaction conditions with molecular iodine in CH3CN-MeOH in the presence of NaHCO3 at room temperature for 5–8 h. Pleasingly the seven-membered cyclic benzoxepines 135 were formed exclusively by 7-exo-trig cyclization to afford benzoxepine derivatives 7 in 48–55% yields (Scheme 31).81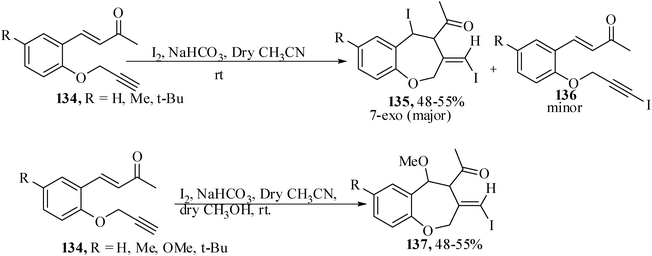 | ||
| Scheme 31 Molecular iodine-mediated synthesis of benzoxepine derivatives. | ||
It was also observed that in case of salicyladehyde derivatives containing an electron-withdrawing group, e.g., p-Cl and m-Cl, only uncyclized iodo derivatives 136 were isolated without any cyclized products. In the absence of MeOH the reaction gives a mixture of diiodo product 135 and iodo derivatives 136. Cyclization might occur either through ‘path a’ or ‘path b’. In case of the former, cyclization is assumed to proceed via activation of the carbon–carbon triple bond of 134 by coordination to I+ and, subsequently, the remaining I− ion may attack the double bond of 138 in a Michael fashion to form carbanions 140. Subsequent attack of the carbanions at the iodonium moiety in a 7-exo-mode may afford the seven-membered oxepine derivatives 135; but the formation of products 136 could be explained by considering an alternative pathway (pathway b). The cyclization is supposed to proceed through the formation of 1,2-diiodoalkenyl intermediates 142. Initially formed diiodoalkenyl intermediates 142 may subsequently undergo dehydroiodination under basic conditions to afford the minor products 136. A less probable explanation for the formation of products 135 proceeding through nucleophilic displacement of iodine from the envisioned diiodoalkenyl intermediates 142 by nucleophilic α-carbon (relative to the carbonyl group) cannot be ruled out (Scheme 32).
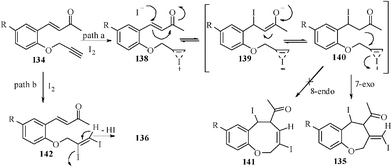 | ||
| Scheme 32 Probable mechanistic pathway for the formation of benzoxeine derivatives. | ||
5.2 Synthesis of medium-ring aza-heterocycles by azide-alkyne cycloaddition
There are some reports82 of the synthesis of 1,4-benzodiazepinones but triazole-fused 1,4-benzodiazepinones are still lacking in the literature. Owing to the importance of both 1,2,3-triazoles and benzodiazepinones, triazole-fused 1,4-benzodiazepinones83 may attract much attention from both biological and medicinal interests. The appropriately-tethered precursors 143 and 144 were accessed either from o-bromo-N-alkyl aniline or 5-bromo-6-alkyl amino coumarin by Sonogashira coupling with phenyl acetylene followed by PTC alkylation of the resulting products with chloroacetyl chloride. Treatment of the substrates 143 and 144 with an excess of NaN3 in DMF at 100 °C for 1 h gave the corresponding azide which were isolated and then heated in DMF at 110 °C for 4 h to give the desired benzodiazepinones 146 and 147 in 86–92% and 87–89% yields respectively by a catalyst-free 1,3-dipolar cycloaddition. The same sequence of two steps has been conducted in one step to give the 1,2,3-triazole-fused 1,4-benzodiazepinones without affecting the yield of the cycloaddition reaction. (Scheme 33).84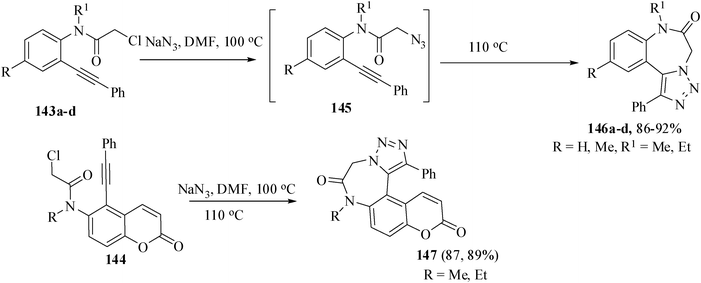 | ||
| Scheme 33 Formation of triazole-fused 1,4-benzo diazep in ones. | ||
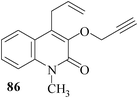
There are several routes available to access benzodiazocinone.85 However, there is no report on the synthesis of triazole-fused benzodiazocinone. We have recently reported a versatile route for the regioselective synthesis of 1,2,3-triazole fused- dibenzo[1,5]diazocine derivatives by intramolecular azide-alkyn[3+2]cycloaddition. The tailor-tethered substrates 148 were heated in DMF at 120 °C for 5 h to afford the desired 1,2,3-triazole fused dibenzo[1,5]diazocinones 151a–d exclusively in 93–97% yields using Huisgen 1,3-dipolar cycloaddition reaction. The corresponding substrates 149 derived from 6-(N-methylamino) coumarin and 150 from 6-(N-methylamino) quinolone gave the diazocinones 152 and 153 respectively, also in excellent yields (Scheme 34).86
![Construction of 1,2,3-triazole fused dibenzo [1,5] diazep in ones by Huisgen 1,3-dipolar cycloaddition reaction.](/image/article/2011/RA/c1ra00494h/c1ra00494h-s34.gif) | ||
| Scheme 34 Construction of 1,2,3-triazole fused dibenzo [1,5] diazep in ones by Huisgen 1,3-dipolar cycloaddition reaction. | ||
5.3 Synthesis of medium-ring aza-heterocycles by Cu-catalyzed intramolecular cyclization
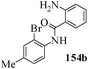
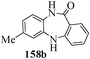
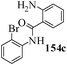
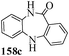
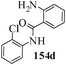
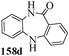
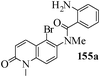
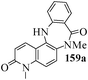
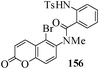
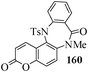
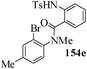
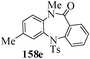
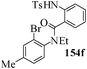
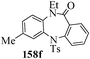
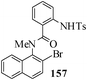
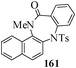
Several strategies to access 1,4-benzodiazepinones are known.87–88 Buchwald et al. reported89 a landmark development in the copper-catalyzed reactions which are found to be very effective for the aryl-amination reactions. Ma et al. introduced90 the synthesis of oxazepine ring system by CuI/L-proline catalyzed C–N bond formation.91 We have recently achieved regioselective synthesis of 1,4-benzodiazepinones by CuI/L-proline-catalyzed intramolecular aryl amination. The precursors 154a–d for this study were prepared from the reaction of o-haloanilines/o-halo-N-alkylanilines and o-nitrobenzyl chloride followed by reduction of the nitro group with SnCl2. Similar sequence of reactions of 5-bromo-1-methyl-6-[N-methylamino]quinilone afforded the substrates 155a. The other substrates were prepared using similar sequence of reactions followed by tosylation of the amino group with tosyl chloride-pyridine. Initially we have established the optimal condition for the reaction of free amino substrates and subjected the substrates 154a–d and 155a under the optimized conditions: Condition A [10 mol% CuI, 20 mol% L-proline, 2 equiv. Cs2CO3 in 5 ml DMSO at 120 °C] for 7–10 h to give the intramolecular aryl-amination products 158a–d and 159a in 58–72% and 66% yields respectively.92As the yields of the products were not high (58 to 72%), we have examined the effect of chelation owing to the presence of the free amine function in the substrate by introducing a tosyl group. The optimal condition for the intramolecular aryl-amination of the tosyl derivatives was also established: Condition B [10 mol% CuI, 20 mol% L-proline, 6 equiv. DABCO in 4 ml DMSO and 1 ml H2O at 120 °C]. Under this optimized condition the tosyl substrates 154e,f,155b,c,156 and 157 gave the 1,4-benzodiazepinones 158e,f,159b,c,160 and 161 in 77–92% yields (Table 4). The tosyl derivatives of the benzodiazepinones, can also be detosylated for further substitution at nitrogen atom. Beccalli and co-workers93 also reported the synthesis of [1,4] diazepinones using palladium-catalyzed reaction conditions. This methodology may suffer from some limitations particularly in case of industrial application of the methodology due to the air and moisture sensitivity of the palladium catalyst94 as well as the prohibiting cost.
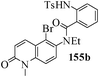
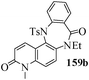
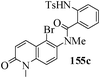
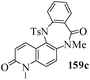
6. Conclusion
The concept of tethering of two reaction components to make a reaction intramolecular is a well-known synthetic strategy. The proximity effects of two reacting partners result in considerable enhancement of rate of an intramolecular reaction compared to the intermolecular reaction by decreasing the entropic demand on the activation energy and thereby allowing the reaction to be carried out at a lower temperature. For the formation of medium-rings, it is beneficial to use the appropriately tethered substrates causing inevitable increase in conformational restrictions of the reaction transition state to achieve greater regio- and stereoselectivity. We have made applications of this concept in the synthesis of various oxygen, nitrogen and sulfur heterocycles. The examples described above illustrate the broad applicability of the palladium-catalyzed intramolecular cyclization, intramolecular cyclization of sulfanyl radical and ring closing diene and enyne metathesis for the synthesis of medium-ring heterocycles of various importances. It is evident that these reactions have been useful in view of their ability to form medium- heterocyclic rings. Though considerable progress has been made, there is still enormous scope to develop this area especially in the synthesis of useful complex synthetic and natural molecules. Further applications are expected to emerge along this line.Acknowledgements
I thank the CSIR (New Delhi) and DST (New Delhi) for financial assistance. I also thank University of Kalyani and Tezpur University for infrastructural facilities. My special thanks are due to Mr. Biswajit Sinha (Senior Research Fellow) for his help during the preparation of this manuscript.References
- (a) T. K. Devon and A. I. Scott, Handbook of Naturally Occurring Compounds, Vol. 2; Academic Press: New York and London, 1972 Search PubMed; (b) D. J. Faulkner, Nat. Prod. Rep., 1984, 1, 251 RSC.
- K. Carl L. Joseph, U. S. Patent, 4,073,912, 1972; Chem. Abstr. 1978, 89, p. 2456 Search PubMed.
- K. C. Majumdar and B. S. Thyagarajan, J. Heterocycl. Chem., 1974, 11, 937 CrossRef.
- (a) L. Yet, Chem. Rev., 2000, 100, 2963 CrossRef CAS; (b) J. O. Hoberg, Tetrahedron, 1998, 54, 12631 CrossRef CAS.
- E. L. Eliel and S. H. Wilen, Stereochemistry of Organic Compounds, Wiley, New York, 1994 Search PubMed.
- P. A. Evans and A. B. Holmes, Tetrahedron, 1991, 47, 9191 CrossRef.
- (a) V. H. Rawal and C. Michovd, J. Org. Chem., 1993, 58, 5583 CrossRef CAS; (b) Z. Owczarczyk, F. Lamaty, E. J. Vawter and E. I. Negishi, J. Am. Chem. Soc., 1992, 114, 10091 CrossRef CAS; (c) A. C. Albeniz, P. Espinet and Y. S. Lin, J. Am. Chem. Soc., 1996, 118, 7145 CrossRef CAS; (d) S. E. Gibson, N. Guillo, R. J. Middleton, A. Thuilliez and M. J. Tozer, J. Chem. Soc., Perkin Trans. 1, 1997, 447 RSC.
- G. Illuminati and L. Mandolini, Acc. Chem. Res., 1981, 14, 95 CrossRef CAS.
- (a) R. E. TenBrink, J. M. McCall and H. G. Johnson, J. Med. Chem., 1980, 23, 1058 CrossRef CAS; (b) R. E. TenBrink, J. M. McCall, D. T. Pals, R. B. McCall, J. Orley, S. J. Humphrey and M. G. Wendling, J. Med. Chem., 1981, 24, 64 CrossRef CAS; (c) D. F. O'Shea and J. T. Sharp, J. Chem. Soc., Perkin Trans. 1, 1995, 515 Search PubMed; (d) J. R. Tretter, US Patent 3514449, 1970. Chem. Abstr. 1970, 73, 35404u Search PubMed; (e) J. Elks, C. R. Ganellin, ed., “Dictionary of Drugs,” 1st ed., Chapman and Hall, London, 1990, p. 984 Search PubMed; (f) M. Stefinovic and V. Snieckus, J. Org. Chem., 1998, 63, 2808 CrossRef CAS; (g) H. Kishuku, M. Shindo and K. Shishido, Chem. Commun., 2003, 350 RSC; (h) Y. Satoh, A. H. Libby, C. Powers, T. Kowalski, D. H. White and E. F. Kimble, Bioorg. Med. Chem. Lett., 1994, 4, 549 CrossRef CAS; (i) R. Kiyama, T. Honma, K. Hayashi, M. Ogawa, M. Hara, M. Fujimoto and T. Fujishita, J. Med. Chem., 1995, 38, 2728 CrossRef CAS.
- (a) K. C. Majumdar, S. Nath, B. Chattopadhyay and B. Sinha, Synthesis, 2010, 3918 CrossRef CAS; (b) K. C. Majumdar, K. Ray and S. Ganai, Tetrahedron Lett., 2010, 51, 1736 CrossRef CAS; (c) K. C. Majumdar, T. Ghosh and S. Chakravorty, Tetrahedron Lett., 2010, 51, 3372 CrossRef CAS; (d) K. C. Majumdar, S. Chakravorty, T. Ghosh and B. Sridar, Synlett, 2009, 3137 Search PubMed; (e) K. C. Majumdar, B. Sinha, P. K. Maji and S. K. Chattopadhyay, Tetrahedron, 2009, 65, 2751 CrossRef CAS; (f) K. C. Majumdar, A. Taher and P. Debnath, Synthesis, 2009, 793 CrossRef CAS; (g) K. C. Majumdar, S. Chakravorty, P. K. Shyam and A. Taher, Synthesis, 2009, 403 CrossRef CAS; (h) K. C. Majumdar, S. Chakravorty and N. De, Tetrahedron Lett., 2008, 49, 3419 CrossRef CAS; (i) K. C. Majumdar, S. Chakravorty and K. Ray, Synthesis, 2008, 2991 CrossRef CAS.
- K. C. Majumdar, I. Ansary, B. Sinha and B. Chattopadhyay, Synthesis, 2009, 3593 CrossRef CAS.
- (a) C. Gutler and S. L. Buchwald, Chem.–Eur. J., 1999, 5, 3107 CrossRef; (b) N. A. Bumagin, V. V. Bykov, L. I. Sukhomlinova, T. P. Tolstaya and I. P. Beletskaya, J. Organomet. Chem., 1995, 486, 259 CrossRef CAS; (c) N. A. Bumagin, P. G. More and I. P. Beletskaya, J. Organomet. Chem., 1989, 371, 397 CrossRef CAS; (d) M. T. Reetz, E. Westermann, R. Lohmer and G. Lohmer, Tetrahedron Lett., 1998, 39, 8449 CrossRef CAS; (e) M. Moreno-Manas, R. Pleixats and A. Roglans, Synlett, 1997, 1157 CrossRef CAS.
- T. Jeffery, Tetrahedron, 1996, 52, 10113 CrossRef CAS.
- K. C. Majumdar, B. Chattopadhyay and K. Ray, Tetrahedron Lett., 2007, 48, 7633 CrossRef CAS.
- K. C. Majumdar, B. Chattopadhyay, R. N. De and B. Roy, Synlett, 2009, 2083 CrossRef CAS.
- L. A. Arnold, W. Luo and R. K. Guy, Org. Lett., 2004, 6, 3005 CrossRef CAS.
- (a) K. C. Majumdar and B. Chattopadhyay, Synthesis, 2009, 2385 CrossRef CAS; (b) K. C. Majumdar and B. Chattopadhyay, Synlett, 2008, 0979 CrossRef CAS.
- (a) S. E. Gibson, N. Guillo, J. O. Jones, I. M. Buck, S. B. Kalindjian, S. Roberts and M. J. Tozer, Eur. J. Med. Chem., 2002, 37, 379 CrossRef CAS; (b) S. E. Gibson, J. O. Jones, R. McCague, M. J. Tozer and N. J. Whitcombe, Synlett, 1999, 954 CAS.
- (a) K. C. Majumdar, B. Chattopadhyay and B. Sinha, Tetrahedron Lett., 2008, 49, 1319 CrossRef CAS; (b) K. C. Majumdar, B. Sinha, B. Chattopadhyay and K. Ray, Tetrahedron Lett., 2008, 49, 4405 CrossRef CAS.
- (a) B. Alcaide, P. Almendros and R. Rodriguez-Acebes, J. Org. Chem., 2005, 70, 2713 CrossRef CAS; (b) G. A. Molander and F. Dehmel, J. Am. Chem. Soc., 2004, 126, 10313 CrossRef CAS.
- (a) C.-H. Park, V. Ryabova, I. V. Seregin, A. W. Sromek and V. Gevorgyan, Org. Lett., 2004, 6, 1159 CrossRef CAS; (b) E. David, J. Perrin, S. Pellet-Rostaing, J. F. Chabert and M. Lemaire, J. Org. Chem., 2005, 70, 3569 CrossRef CAS; (c) B. Glover, K. A. Harvey, B. Liu, M. J. Sharp and M. F. Tymoschenko, Org. Lett., 2003, 5, 301 CrossRef CAS; (d) B. Sezen and D. Sames, J. Am. Chem. Soc., 2003, 125, 5274 CrossRef CAS; (e) T. Okazawa, T. Satoh, M. Miura and M. Nomura, J. Am. Chem. Soc., 2002, 124, 5286 CrossRef CAS; (f) A. Yokooji, T. Okazawa, T. Satoh, M. Miura and M. Nomura, Tetrahedron, 2003, 59, 5685 CrossRef CAS; (g) D. Kalyani, N. R. Deprez, L. V. Desai and M. S. Sanford, J. Am. Chem. Soc., 2005, 127, 7330 CrossRef CAS; (h) C. A. Zificsak and D. J. Hlasta, Tetrahedron, 2004, 60, 8991 CrossRef CAS.
- K. C. Majumdar, B. Chattopadhyay and S. Samanta, Tetrahedron Lett., 2009, 50, 3178 CrossRef CAS.
- K. C. Majumdar, R. K. Nandi, S. Samanta and B. Chattopadhyay, Synthesis, 2010, 985 CrossRef CAS.
- K. C. Majumdar, S. Mondal and D. Ghosh, Synthesis, 2010, 1315 CrossRef CAS.
- (a) K. C. Majumdar, B. Chattopadhyay and S. Nath, Tetrahedron Lett., 2008, 48, 1609 CrossRef; (b) J. Tsuji Palladium Reagents and Catalysts. In New Perspective for the 21st Century; Wiley, 2004, 2nd Chapter Search PubMed.
- K. C. Majumdar, S. Samanta and B. Chattopadhyay, Tetrahedron Lett., 2009, 50, 4866 CrossRef CAS.
- A. Klapars, S. Parris, K. W. Anderson and S. L. Buchwald, J. Am. Chem. Soc., 2004, 126, 3529 CrossRef CAS.
- (a) K. Koriatopoulou, N. Karousis and G. Varvounis, Tetrahedron, 2008, 64, 10009 CrossRef CAS; (b) V. V. Potapov, N. A. Fetisova, A. V. Nikitin and A. V. Ivachtchenko, Tetrahedron Lett., 2009, 50, 2790 CrossRef CAS; (c) S. BouzBouz and M. Sanselme, Tetrahedron Lett., 2009, 50, 5884 CrossRef CAS.
- K. C. Majumdar, K. Ray and S. Ganai, Tetrahedron Lett., 2010, 51, 1736 CrossRef CAS.
- (a) D. Kaufmann, P. C. Fünfschilling, U. Beutler, P. Hoehn, O. Lohse and W. Zaugg, Tetrahedron Lett., 2004, 45, 5275 CrossRef CAS; (b) A. H. Fauq, K. Simpson, G. M. Maharvi, T. Golde and P. Das, Bioorg. Med. Chem. Lett., 2007, 17, 6392 CrossRef CAS; (c) A. Kling, G. Backfisch, J. Delzer, H. Geneste, C. Graef, U. Holzenkamp, W. Hornberger, U. E. W. Lange, A. Lauterbach, H. Mack, W. Seitz and T. Subkowski, Bioorg. Med. Chem. Lett., 2002, 12, 441 CrossRef CAS.
- K. C. Majumdar, S. Chakravorty, T. Ghosh and B. Sridhar, Synlett, 2009, 3127 CrossRef.
- R. C. Larock, C. Tu and P. Pace, J. Org. Chem., 1998, 63, 6859 CrossRef CAS.
- P. A. Donets and E. V. V. Eycken, Org. Lett., 2007, 9, 3017 CrossRef CAS.
- K. C. Majumdar, T. Ghosh and S. Chakravorty, Tetrahedron Lett., 2010, 51, 3372 CrossRef CAS.
- (a) Z. Jin and P. L. Fuchs, Tetrahedron Lett., 1993, 34, 5205 CrossRef CAS; (b) C. Fu and S. Ma, Org. Lett., 2005, 7, 1605 CrossRef CAS.
- (a) K. C. Majumdar, B. Chattopadhyay and B. Sinha, Synthesis, 2008, 3857 CrossRef CAS; (b) K. C. Majumdar, B. Chattopadhyay and B. Sinha, Lett. Org. Chem., 2009, 6, 453 CrossRef CAS.
- M.-P. Denieul and T. Skrydstrup, Tetrahedron Lett., 1999, 40, 4901 CrossRef CAS.
- I. P. Beletskaya and A. V. Cheprakov, Chem. Rev., 2000, 100, 3009 CrossRef CAS.
- (a) S. E. Gibson and R. J. Middleton, J. Chem. Soc., Chem. Commun., 1995, 1743 RSC; (b) S. E. Gibson, N. Guillo, S. B. Kalindjian and N. J. Tozer, Bioorg. Med. Chem. Lett., 1997, 7, 1289 CrossRef CAS.
- (a) S. Hanessian, H. Sailes and E. Therrien, Tetrahedron, 2003, 59, 7047 CrossRef CAS; (b) L. Zhuang, J. S. Wai, M. W. Embrey, T. E. Fisher, M. S. Egbertson, L. S. Payne, J. P. Guare, J. P. Jr. Vacca, D. J. Hazuda, P. J. Felock, A. L. Wolfe, K. A. Stillmock, M. V. Witmer, G. Moyer, W. A. Schleif, L. J. Gabryelski, Y. M. Leonard, J. J. Lynch, S. R. Jr. Michelson and S. D. Young, J. Med. Chem., 2003, 46, 453 CrossRef CAS; (c) R. A. Miller, G. R. Humphrey, D. R. Lieberman, S. S. Ceglia, D. J. Kennedy, E. J. J. Grabowski and P. J. Reider, J. Org. Chem., 2000, 65, 1399 CrossRef CAS.
- (a) P. Dauban and R. H. Dodd, Org. Lett., 2000, 2, 2327 CrossRef CAS; (b) P. Dauban, L. Saniere, T. Aurelie and R. H. Dodd, J. Am. Chem. Soc., 2001, 123, 7707 CrossRef CAS; (c) E. S. Sherman, S. R. Chemler, T. B. Tan and O. Gerlits, Org. Lett., 2004, 6, 1573 CrossRef CAS.
- X.-Y. Liu, C.-H. Li and C.-M. Che, Org. Lett., 2006, 8, 2707 CrossRef CAS.
- (a) J.-L. Liang, S.-X. Yuan, P. W. H. Chan and C.-M. Che, Org. Lett., 2002, 4, 4507 CrossRef CAS; (b) A. Padwa, A. C. Flick, C. A. Leverett and T. Stengel, J. Org. Chem., 2004, 69, 6377 CrossRef CAS; (c) M. J. Hopkins and P. R. Hanson, Org. Lett., 2008, 10, 2223 CrossRef.
- P. R. Hanson, D. A. Probst, R. E. Robinson and M. Yau, Tetrahedron Lett., 1999, 40, 4761 CrossRef CAS.
- (a) S. Merten, R. Frohlich and O. Kataeva, Adv. Synth. Catal., 2005, 347, 754 CrossRef CAS; (b) A. Vasudevan, P.-S. Tseng and S. W. Djuric, Tetrahedron Lett., 2006, 47, 8591 CrossRef CAS; (c) D. K. Rayabarapu, A. Zhou, K. O. Jeon, T. Samarakoon, A. Rolfe, H. Siddiqui and P. R. Hanson, Tetrahedron, 2009, 65, 3180 CrossRef CAS.
- (a) K. C. Majumdar, S. Mondal and N. De, Synlett, 2008, 2851 CrossRef CAS; (b) K. C. Majumdar, S. Mondal and N. De, Synthesis, 2009, 3127 CrossRef.
- (a) O. Miyata and T. C. Naito, R. Acad. Sci., 2001, 4, 401 CAS; (b) K. C. Majumdar and P. Debnath, Tetrahedron Report No 848, 64, 2008, 9799 CAS.
- (a) A. F. Barrero, S. Arseniyadia, M. M. Herrador, J. F. Q. de Moral and J. F. Arteaga, Synlett, 2005, 591 CrossRef CAS; (b) G. E. Keck, T. T. Wager and J. F. D. Rodriguez, J. Am. Chem. Soc., 1999, 121, 5176 CrossRef CAS.
- (a) Y. Asakawa, E. Kusube, T. Takemoto and C. Suire, Phytochemistry, 1978, 17, 2115 CrossRef CAS; (b) M. Murata and T. Yusumoto, Nat. Prod. Rep., 2000, 17, 293 RSC; (c) F. A. Macias, J. M. G. Molinillo, R. M. Varela, A. Torres and F. R. Fronczek, Tetrahedron Lett., 1993, 34, 1999 CrossRef CAS.
- (a) F. A. Macias, J. M. G. Molinillo, R. M. Varela, A. Torres and F. R. Fronczek, J. Org. Chem., 1994, 59, 8261 CrossRef CAS; (b) B. Dinda, S. K. Das, A. K. Hajra, A. Bhattacharya, K. De, G. Chel and B. Achari, Indian J. Chem., Sect. B, 1999, 38, 577 Search PubMed; (c) M. Engler, T. Anke and Z. O. Sterner, Natur forsch., 1998, 53c, 318 Search PubMed; (d) Y. Asakawa, R. Takeda, M. Toyota and T. Tsunematsu, Phytochemistry, 1981, 20, 858 CrossRef CAS; (e) Y. Asakawa, T. Hashimoto, K. Takikawa, M. Tori and S. Ogawa, Phytochemistry, 1991, 30, 235 CrossRef CAS; (f) Y. Asakawa, M. Toyota and T. Takemoto, Phytochemistry, 1978, 17, 2005 CrossRef CAS; (g) S. Yamaguchi, K. Furihata, M. Miyazawa, H. Yokoyama and Y. Hirai, Tetrahedron Lett., 2000, 41, 4787 CrossRef CAS; (h) S. McCormick, K. Robson and B. Bohm, Phytochemistry, 1986, 25, 1723 CrossRef CAS; (i) M. Breuer, G. Leeder, P. Proksch and H. Budzikiewicz, Phytochemistry, 1986, 17, 495 CrossRef.
- (a) S. K. Mandal and S. C. Roy, Tetrahedron Lett., 2006, 47, 1599 CrossRef CAS; (b) S. Wu, M. Journet and M. Malacria, Tetrahedron Lett., 1994, 35, 8601 CrossRef CAS; (c) A. Neogi, T. P. Majhi, N. Ghosal and P. Chattopadhyay, Tetrahedron, 2005, 61, 9368 CrossRef CAS; (d) H. Magono and S. Hara, Tetrahedron Lett., 2004, 45, 4329 CrossRef.
- K. C. Majumdar and A. Taher, Tetrahedron Lett., 2009, 50, 228 CrossRef CAS.
- J. E. Baldwin, J. Chem. Soc., Chem. Commun., 1976, 734 RSC.
- P. Crews and B. Harrison, J. Org. Chem., 1997, 62, 2646 CrossRef.
- (a) F. A. Macias, J. M. G. Molinillo, R. M. Varela, A. Torres and F. R. Fronczek, J. Org. Chem., 1994, 59, 8261 CrossRef CAS; (b) F. A. Macias, R. M. Varela, A. Torres and J. M. G. Molinillo, J. Nat. Prod., 1999, 62, 1636 CrossRef CAS; (c) F. A. Macias, R. M. Varela, A. Torres, J. M. G. Molinillo and F. R. Fronczek, Tetrahedron Lett., 1993, 34, 1999 CrossRef CAS; (d) K. Takabatake, I. Nishi, M. Shindo and K. Shishido, J. Chem. Soc., Perkin Trans. 1, 2000, 1807 RSC.
- (a) S. R. Oh, D. S. Kim, I. S. Lee, K. Y. Jung, J. J. Lee and H. –K. Lee, Planta Med., 1998, 64, 456 CrossRef CAS; (b) M. Nagai and S. Nagumo, Chem. Pharm. Bull., 1987, 35, 3002 CAS.
- (a) B. Biswas, P. K. Sen and R. V. Venkateswaran, Tetrahedron, 2007, 63, 12026 CrossRef CAS; (b) Z. Liang, S. Ma, J. Yu and R. Xu, Tetrahedron, 2007, 63, 977 CrossRef CAS; (c) K. Sato, T. Yoshimura, M. Shindo and K. Shishido, J. Org. Chem., 2001, 66, 309 CrossRef CAS; (d) D. Fuminao, O. Takahisa, S. Takeshi and N. Shigeru, Tetrahedron Lett., 2003, 44, 4877 CrossRef; (e) E. L. Grimm, S. Levac and L. A. Trimble, Tetrahedron Lett., 1994, 35, 6847 CrossRef CAS.
- K. C. Majumdar, K. Ray, P. Debnath, P. K. Maji and N. Kundu, Tetrahedron Lett., 2008, 49, 5597 CrossRef CAS.
- K. C. Majumdar, P. K. Maji, K. Ray and P. Debnath, Tetrahedron Lett., 2007, 48, 9124 CrossRef CAS.
- K. C. Majumdar, S. Mondal and D. Ghosh, Tetrahedron Lett., 2010, 51, 348 CrossRef CAS.
- (a) E. Lunt, CompreIn hensive Organic Chemistry; Barton, D., Ollis, W. D., ed.; Pergamon Press, 1974; Vol. 4, pp 493 Search PubMed; (b) J. D. Brown, In Comprehensive Heterocyclic Chemistry; Katrizky, A. R., Rees, C. W., ed.; Pergamon Press: Oxford, 1984; Vol. 3, pp 57 Search PubMed; (c) E. D. Clercq and R. J. Beraaerts, Biol. Chem., 1987, 262, 14905 Search PubMed.
- S. K. Chattopadhyay, S. Karmakar, T. Biswas, K. C. Majumdar, H. Rahman and B. Roy, Tetrahedron report 796, Tetrahedron, 2007, 63, 3919 CAS.
- K. C. Majumdar, H. Rahaman, S. Muhuri and B. Roy, Synlett, 2006, 466 CrossRef CAS.
- K. C. Majumdar, H. Rahaman and B. Roy, Lett. Org. Chem., 2006, 3, 526 CrossRef CAS.
- K. C. Majumdar, P. K. Maji, H. Rahaman and B. Roy, Lett. Org. Chem., 2006, 3, 845 CrossRef CAS.
- K. C. Majumdar, P. Debnath and S. Samanta, Lett. Org. Chem., 2007, 4, 309 CrossRef CAS.
- K. C. Majumdar, P. Debnath and A. Taher, Lett. Org. Chem., 2008, 5, 169 CrossRef CAS.
- V. P. Litvinov, S. V. Roman and V. D. Dyachenko, Russ. Chem. Rev., 2000, 69, 201 CrossRef CAS.
- K. C. Majumdar, H. Rahaman, R. Islam and B. Roy, Tetrahedron Lett., 2006, 47, 2111 CrossRef CAS.
- K. C. Majumdar, B. Chattopadhyay and S. Chakravorty, Synthesis, 2009, 674 CrossRef CAS.
- S. K. Chattopadhyay, T. Biswas and S. Maity, Synlett, 2006, 2211 CrossRef CAS.
- K. C. Majumdar, S. Samanta, B. Chattopadhyay and R. K. Nandi, Synthesis, 2010, 863 CrossRef CAS.
- K. C. Majumdar and B. Chattopadhyay, Curr. Org. Chem., 2009, 13, 731 CrossRef CAS.
- M. Quadir, J. Cobb, P. W. Sheldrahe, N. Whittal, A. J. P. White, K. K. M. Hii, P. N. Horton and M. B. Hursthouse, J. Org. Chem., 2005, 70, 1545 CrossRef.
- K. C. Majumdar, S. Mondal and D. Ghosh, Synthesis, 2010, 1176 CrossRef CAS.
- (a) J. –L. Couturier, C. Paillet, M. Leconte, J. –M. Basset and K. Weiss, Angew. Chem., Int. Ed. Engl., 1992, 31, 628 CrossRef; (b) J.-L. Couturier, K. Tanaka, M. Leconte, J.-M. Basset and J. Pllivier, Angew. Chem., Int. Ed. Engl., 1993, 32, 112 CrossRef.
- S. K Armstrong and B. A. Christie, Tetrahedron Lett., 1996, 37, 9373 CrossRef.
- T. Fujiwara, Y. Kato and T. Tekeda, Tetrahedron, 2000, 56, 4859 CrossRef CAS.
- K. C. Majumdar, S. Muhuri, H. Rahaman, R. Islam and B. Roy, Chem. Lett., 2006, 35, 1430 CrossRef CAS.
- (a) M. J. Mphahlele, Molecules, 2009, 14, 4814 CrossRef CAS; (b) D. Yue and R. C. Larock, Org. Lett., 2004, 6, 1037 CrossRef CAS; (c) R. Halim, P. J. Scammells and B. L. Flynn, Org. Lett., 2008, 10, 1967 CrossRef CAS; (d) X. Zhang, M. A. Campo, T. Yao and R. C. Larock, Org. Lett., 2005, 7, 763 CrossRef CAS; (e) G. Alvaro, R. D. Fabio, A. Gualandi, C. Fiorelli, M. Monari, D. Savoia and L. Zoli, Tetrahedron, 2007, 63, 12446 CrossRef CAS; (f) M. Amjad and D. W. Knight, Tetrahedron Lett., 2004, 45, 539 CrossRef CAS; (g) I. Aillaud, E. Bossharth, D. Conreaux, P. Desbordes, N. Monteiro and G. Balme, Org. Lett., 2006, 8, 1113 CrossRef CAS; (h) D. Yue, T. Yao and R. C. Larock, J. Org. Chem., 2006, 71, 62 CrossRef CAS.
- K. C. Majumdar, B. Sinha, I. Ansary and S. Chakravorty, Synlett, 2010, 1407 CrossRef CAS.
- (a) K. Kim, S. K. Volkman and J. A. Ellman, J. Braz. Chem. Soc., 1998, 9, 375 CAS; (b) A. Correa, M. T. Herrero, I. Tellitu, E. Domínguez, I. Moreno and R. SanMartin, Tetrahedron, 2003, 59, 7103 CrossRef CAS; (c) J. Dourlat, W. Q. Liu, N. Gresh and C. Garbay, Bioorg. Med. Chem. Lett., 2007, 17, 2527 CrossRef CAS.
- (a) R. Alvarez, S. Valaquez, F. San, S. Aquaro, C. De, C. F. Penro, A. Karlsson, J. Balzarini and M. J. Camarasa, J. Med. Chem., 1994, 37, 4185 CrossRef CAS; (b) S. Valquez, R. Alvarez, C. Perez, F. Cago, C. De, J. Balzarini and M. J. Camarasa, Antivir. Chem. Chemother., 1998, 9, 481 Search PubMed; (c) D. R. Buckle, C. J. M. Rockell, H. Smith and B. A. Spicer, J. Med. Chem., 1986, 29, 2262 CrossRef CAS; (d) A. A. Patchett and R. P. Nargund, Annu. Rep. Med. Chem., 2000, 35, 289 CrossRef CAS.
- K. C. Majumdar, K. Ray, S. Ganai and T. Ghosh, Synthesis, 2010, 858 CrossRef CAS.
- (a) N. Corres, J. J. Delgado, M. D. Valverde, S. Marcaccini, T. Rodriguez, J. Rojo and T. Torroba, Tetrahedron, 2008, 64, 2225 CrossRef CAS; (b) Tetrahedron Lett 2009, 50, p. 5805 Search PubMed.
- K. C. Majumdar, K. Ray and S. Ganai, Synthesis, 2010, 2101 CrossRef CAS.
- (a) E. M. Beccalli, G. Broggini, G. Paladino, T. Pilati and G. Pontremoli, Tetrahedron: Asymmetry, 2004, 15, 687 CrossRef CAS; (b) M. D. Surman, M. J. Mulvihill and M. Miller, Org. Lett., 2002, 4, 139 CrossRef CAS; (c) G. Broggini, G. Molteni, A. Terraneo and G. Zecchi, Tetrahedron, 1999, 55, 14803 CrossRef CAS; (d) R. M. Keenan, J. F. Callahan, J. M. Samanen, W. E. Bondinell, R. R. Calvo, L. Chen, C. DeBrosse, D. S. Eggleston, R. C. Haltiwanger, S. M. Hwang, D. R. Jakas, T. W. Ku, W. H. Miller, K. A. Newlander, A. Nichols, M. F. Parker, L. S. Southhall, I. Uzinskas, J. A. Vasko-Moser, J. W. Venslavsky, A. S. Wong and W. F. Huffman, J. Med. Chem., 1999, 42, 545 CrossRef CAS; (e) T. Sugimori, T. Okawa, S. Eguchi, A. Kakehi, E. Yashima and Y. Okamoto, Tetrahedron, 1998, 54, 7997 CrossRef CAS; (f) A. Walser, R. I. Fryer, In Bicyclic Diazepines; Wiley: New York, 1991; Ch. VII Search PubMed.
- (a) J. M. Matthews, A. B. Dyatkin, M. Evangelisto, D. A. Gauthier, L. R. Hecker, W. J. Hoekstra, F. Liu, B. Poulter, K. L. Sorgi and B. E. Maryanoff, Tetrahedron: Asymmetry, 2004, 15, 1259 CrossRef CAS; (b) K. Hemming and C. Loukou, Tetrahedron, 2004, 60, 3349 CrossRef CAS; (c) S. Herrero, M. T. GarcÌa-López and R. Herranz, J. Org. Chem., 2003, 68, 4582 CrossRef CAS; (d) L. Abrous, J. Hynes, S. R. Jr. Friederich, A. B. Smith and R. Hirschmann, III, Org. Lett., 2001, 3, 1089 CrossRef CAS; (e) F. Ammadi, S. Boukhris, A. Souizi and G. Coudert, Tetrahedron Lett., 1999, 40, 6517 CrossRef CAS; (f) G. L. Grunewald, V. H. Dahanukar, P. Ching and K. R. Criscione, J. Med. Chem., 1996, 39, 3539 CrossRef CAS.
- (a) A. Klapars, J. C. Antilla, X. Huang and S. L. Buchwald, J. Am. Chem. Soc., 2001, 123, 7727 CrossRef CAS; (b) M. Wolter, A. Klapars and S. L. Buchwald, Org. Lett., 2001, 3, 23 CrossRef; (c) F. Y. Kwong, A. Klapars and S. L. Buchwald, Org. Lett., 2002, 4, 4 Search PubMed; (d) R. Martin, H. C. Larsen, A. Ana Cuenca and S. L. Buchwald, Org. Lett., 2007, 9, 17 CrossRef.
- (a) L. Guo, B. Li, W. Huang, G. Pei and D. Ma, Synlett, 2008, 12, 1833 Search PubMed; (b) D. Ma and Q. Cai, Org. Lett., 2003, 5, 21 Search PubMed.
- (a) I. P. Beletskaya and A. V. Cheprakov, Coord. Chem. Rev., 2004, 248, 2337 CrossRef CAS; (b) S. V. Ley and A. W. Thomas, Angew. Chem., Int. Ed., 2003, 42, 5400 CrossRef CAS; (c) K. Kunz, U. Scholz and D. Ganzer, Synlett, 2003, 2428 CrossRef CAS.
- K. C. Majumdar and S. Ganai, Synlett, 2011 Search PubMed (accepted).
- E. M. Beccalli, G. Broggini, G. Paladino and C. Zoni, Tetrahedron, 2005, 61, 61 CrossRef CAS.
- H. Zhang, Q. Cai, D. Ma and J. Org, Chem., 2005, 70, 5164 CAS.
| This journal is © The Royal Society of Chemistry 2011 |