Morphology and pore control in carbon materials via templating
M.
Inagaki
*a,
H.
Orikasa
b and
T.
Morishita
b
aProfessor Emeritus of Hokkaido University, Sapporo, 060-8628, Japan. E-mail: im-ii@xj.commufa,jp
bAdvanced Carbon Technology Center, Toyo Tanso Co., Ltd., Takeshima, Nishiyodogawa-ku, Osaka, 555-0011, Japan
First published on 27th October 2011
Abstract
Carbonization of the precursors by using various templates (template carbonization) was reviewed as the process to control morphology of the carbon and pore structure in the carbon. Prospects are discussed for the processes using various templates and comparison among the processes for pore structure control is presented.
1. Introduction
The carbonization process is the most important for the preparation of carbon materials because it governs the morphology and nanotexture of the resultant carbon materials. Carbonization of the precursors by using various templates (template carbonization) has attracted the attention of scientists and engineers since 1988 because of the paper by Kyotani et al. reporting the fabrication of flaky carbon particles by using a clay mineral as a template.1 Later, various templates, such as anodic aluminum oxide films, zeolite, silica, MgO, etc. were proposed and studied in detail. Template carbonization is the process to replicate the structure of the template into the carbon on the nanoscale, which has been successfully applied to prepare carbon flakes, carbon nanotubes and nanofibers, and nanoporous carbons. Modern science, engineering and technology demand a specific pore structure for a specific application.2–7 It has been pointed out in different literatures, therefore, that the exact control of pore structure in carbon materials is strongly required in different fields of their applications. In the applications of conventional activated carbons, mostly micropores with sizes less than 2 nm have been controlled in both their size and number. In some recent applications, however, even larger pores, mesopores (2–50 nm) and macropores (larger than 50 nm) are responsible for the functions of carbon materials used, for example, mesopores in electrode carbons of electric double-layer capacitors, macropores for heavy oil sorption, etc. In addition to the number and size of the pores, homogeneity in pore size and also in pore morphology must be controlled. In order to respond to these requirements, various processes and techniques were proposed to create pores in carbon materials and to control their number and size.8,9 Most of the newly proposed processes do not include the so-called activation process, which has been used for the industrial production of conventional activated carbons.Template carbonization was firstly proposed to control the morphology of carbon materials and then used to prepare nanoporous carbons with controlled size and alignment of the pores by employing various templates. Reviews on this technique have been published from different points of view,10–15 most of which are focused on pore control in carbon materials, and are also discussed in the reviews on specific applications.16–19 In the present review, template carbonization is divided into the processes aimed at controlling morphology and pore structure of carbon materials. Template carbonization processes were discussed with regards to their prospects for their development, including the possibility to apply these techniques to the production of carbon at a large scale. The comparison among the processes for pore structure control in carbons is also presented.
2 Template carbonization for morphology control
The template carbonization technique was firstly developed for the preparation of thin oriented graphite films using two-dimensional spaces in layered compounds, such as montmorillonite and taeniolite.1,20–25 In Fig. 1, a fundamental scheme of this template method is shown by using acrylonitrile (AN) as the carbon precursor and montmorillonite (MONT) as the template. Firstly, Na1+ ions in a commercially available MONT (Na[Al3.40Fe3+0.09Fe2+0.06Mg0.47][Si7.65Al0.35]O20(OH)4·nH2O) were exchanged for Ca2+ and then exposed to AN vapor for 24 h at room temperature. The mass gain and X-ray diffraction after this exposure confirmed the intercalation of the monolayer of AN into the gallery between the lamellae of the host MONT. The MONT–AN complex was irradiated by γ-ray at room temperature in order to polymerize the intercalated AN to poly(acrylonitrile) (PAN) and then heat-treated at 700 °C for 3 h under N2 flow to carbonize PAN. The heat-treated complex was put into 46% HF solution at 0 °C under rigorous stirring and then refluxed in 37% HCl solution in order to recover the resultant carbon. The resultant carbon contained relatively large amount of foreign atoms, 2.5 mass% H, 16 mass% N and 15.6 mass% O, but was composed of flaky particles.1,20 Its flaky morphology was maintained during the treatment at high temperatures, even though marked structural changes were observed, a decrease in layer spacing d002, a growth of crystallites along both a- and c-axes, and a decrease in intensity ratio of D-band to G-band in Raman spectrum, ID/IG, although the carbon precursor AN itself did not show appreciable growth of graphite structure by high temperature treatment.21 The flakes heat-treated up to 2800 °C showed the lattice fringe image under TEM similar to graphite and the negligibly small D-band in Raman spectrum. In Fig. 2, lattice fringe images are shown on the flake heat-treated at 1400, 2100 and 2800 °C. Many layers are stacked in parallel already after the treatment at 1400 °C, but regular graphitic stacking is realized only above 2500 °C.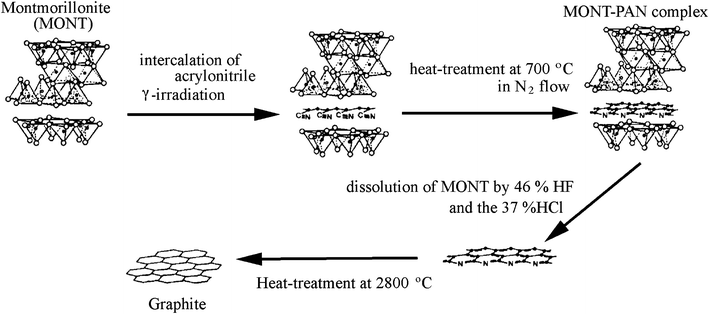 | ||
| Fig. 1 Scheme for the template carbonization using montmorillonite (courtesy of Prof. T. Kyotani of Tohoku University, Japan). | ||
 | ||
| Fig. 2 Lattice fringe images of carbon flakes prepared through templating and heat-treatment at different temperatures (courtesy of Prof. T. Kyotani of Tohoku University, Japan). | ||
Furfuryl alcohol (FA) and vinyl acetate (VAC) were successfully used as carbon precursor.22,23 For MONT–FA complex, MONT was stirred in 20% benzene solution of FA for 3 days at room temperature and the polymerization of FA in the MONT gallery was performed by heating up to 150 °C. Other layered compounds, saponite Na2Mg3Si4−xAlxO10(OH)2 (SAPO) and taeniolite LiMg2LiSi4O10F2 (TAEN), were also used as templates.24,25SAPO–FA and TAEN–FA complexes with FA content of 0.18 and 0.1 g g−1of clay, respectively, were prepared in 20% FA solution of benzene and their carbonization was performed at 700 and 900 °C, respectively.25 All of these complexes gave flakes with high graphitizability. Since the templates used were easily obtained as films, graphite films could be obtained from VAC.26Carbon flakes thus prepared had thicknesses of about 40 nm and could be formed into a pellet without any binder by simple compression.27 They reacted with potassium vapor to form first-stage intercalation compound.28
Tubular carbons were prepared by carbon deposition from propylene at 800 °C in a flow of a mixture of N2 and propylene (2.5%) on the inner wall of nano-sized channels in the anodic aluminum oxide (AAO) film,29,30 the process being a kind of chemical vapor infiltration (CVI). The preparation procedure is schematically shown in Fig. 3, consisting of carbon deposition on the pore wall, followed by the dissolution of aluminum oxide either with an excess of 46% HF at room temperature, or with a 10 mol L−1NaOH aqueous solution at 150 °C for 6 h in an autoclave. SEM and TEM images of carbon tubules, which were prepared by using a laboratory-made AAO film having 30 or 200 nm diameter channels, are shown in Fig. 4. In the as-prepared tubes, carbon layers are small and do not yet have a perfect orientation along the tube axis, as shown in Fig. 4c, but they grow markedly and aligned perfectly along the tube axis after heat treatment at a high temperature of 2800 °C, the resultant carbons being comparable to multi-walled carbon nanotubes synthesized by arc discharging. The control of channel size (diameter and length) of template oxide films have been well established by changing the electrolyte, current density, temperature and time for oxidation of aluminum metal plate. Therefore, the diameter and length of tubular carbons can be controlled by the diameter of the channels and thickness of the template film, respectively, and the wall thickness of tubular carbons by the deposition conditions. The prepared tubes have a very uniform diameter and length (Fig. 4a and 4b), and align along the channels of the AAO film, which are perpendicular to the film surface.
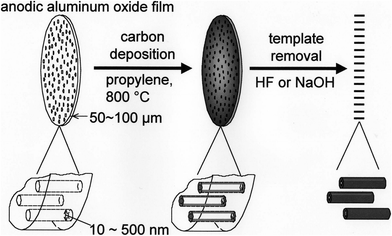 | ||
| Fig. 3 Scheme for the template carbonization using anodic aluminum oxide film (courtesy of Prof. T. Kyotani of Tohoku University, Japan). | ||
 | ||
| Fig. 4 SEM image (a) and TEM images (b and c) of carbon tubules prepared by template carbonization using anodic aluminum oxide film (courtesy of Prof. T. Kyotani of Tohoku University, Japan). | ||
Either Co or Fe was deposited at the bottom of the channels of the AAO template by AC electrolysis in a solution of either CoSO4 or FeSO4, with HBO3 and ascorbic acid, followed by the reduction in a flow of CO at 600 °C for 4–5 h. The deposited metals 100–200 nm in length worked as a catalyst for CVI to inner wall of the channels to form tubular carbons.30 By using the same template with the channel diameter of 200 nm and the thickness of 60 μm, tubular carbons were synthesized by CVI of ethylene gas and pyrene vapor with or without Ni, Co or Fe as a catalyst at 900 °C, with nanoparticles of the catalyst metal being included in the tubular carbons.31 The resultant tubular carbons were aligned by making their tubular axes parallel and so could be used as a membrane with a porosity of 60% and thickness of 60 μm.32,33 The membranes thus prepared were tested for Li intercalation/deintercalation, electro-catalyzed O2reduction and methanol oxidation after deposition of Pt, Pt/Ru and Fe.32 Highly-ordered arrays of tubular carbons were prepared by CVI at 650 °C in a flow of 10% acetylene in N2 using cobalt catalyst.33,34
By using AAO films with Y-branched channels with the channel diameter of 60 nm, tubular carbons having a Y-junction were prepared through CVI of acetylene at 650 °C.35 By repeating the anodization and CVI of acetylene, tube-in-tube and linearly joined tubular carbons were prepared.36 Well-aligned bundles of nanotubes were re-aligned to a monolayer film at the liquid–liquid interface by dispersing in aqueous ethanol/toluene solution, which was transferable to a solid substrate.37,38
This template carbonization technique has many advantages for the control of the structure of the tubular carbons, in addition to their high homogeneity in both the diameter and length of the tubes. One advantage is that the tubular carbons are fixed inside the channel of the template aluminum oxide where one of the ends is open (carbon nano-test tubes). It gives a possibility to fill relatively easily the inside of the tubes by either metals or metal oxides to prepare nanowires.
Filling of highly-crystallized Pt was carried out; a carbon-deposited AAO film was impregnated into an ethanol solution of H2PtCl6 at room temperature and then heat-treated at 500 °C in a H2 flow.39Pt and Pt/Ru alloys were deposited on the walls of the tubules as nanoparticles, about 7 and 1.6 nm, by immersing into an aqueous solution of H2PtCl6, and H2PtCl6 + RuCl3, respectively, followed by heating at 580 °C in a flow of H2.40 Crystalline Fe3O4 and metallic Ni were filled into the tubular carbons through metal-organic chemical vapor deposition (MOCVD) of ferrocene (Fe(C5H5)2) and nickelocene (Ni(C5H5)2).41,42Fe3O4 filled in the tubes composed from nanocrystals with an average size of 24 nm and filling efficiency was more than 20%.41Ni nanowire formed in the tube were supposed to be a single crystal with a uniform diameter of 4 nm, of which (111) planes are preferentially parallel to the tube axis.42NiO nanoribbons (4 × 20 × 800 nm)43 and also Ni(OH)2 single crystals44,45 were successfully filled in the tubes. Filling of permalloy (Fe–Ni alloys) being about 40 nm in diameter and 900 nm in length, was also carried out by electroplating in an electrolyte.46–48 In Fig. 5, a TEM image of permalloy-filled nanotubes and its selected electron diffraction pattern are shown.48 These permalloy-filled nanotubes thus prepared were shown to be effective for the suppression of electromagnetic noise emission.46,48
 | ||
| Fig. 5 TEM image of permalloy-filled nanotubes (a) and its selected-area electron diffraction pattern (b).48 | ||
The inner surface of these tubular carbons was also modified by fluorination49–51 and oxidation,52 because the inner surfaces of the tubes were exposed to the atmosphere but the outer surfaces were completely covered by the template Al2O3. Fluorination was performed by using elemental fluorine at a temperature of 50–200 °C for 5 days.49 After fluorination, tubular carbons were confirmed to retain their structural characteristics, even though fluorine atoms bonded covalently with carbon atoms on the inner surface of the tubules with the sp3 hybrid orbital. Oxidation of the inner surface of the tubular carbons was performed by immersing them into 20% HNO3 under refluxing conditions.52
The tubular carbons separated from AAO template (nano-test tubes) were found to be water-dispersible without any post-treatment, when their length was not more than 5 μm.53 However, nano-test tubes filled with permalloy could be dispersed in water, even when the length was around 1 μm, probably due to the magnetic interaction among the tubes. Upon the oxidation of the outer-surface of the permalloy-filled test tubes in H2O2, water dispersibility was highly improved, which might open a possibility to apply these tubes as carriers for magnetic drug delivery systems.54 It was experimentally confirmed that a dye, as a model drug, sealed into these tubes was able to dissolve out,55 and also that the adsorption capacity of the tubes for two proteins depends on both the nature of the inner surface and the size of the tubes.56 Aligned nano-test tubes with coaxially-doubled layers of N-doped and undoped carbons57,58 and also of N-doped and B-doped carbons59,60 were successfully synthesized by using propylene, acetonitrile and benzene with BCl3 as precursors.
Carbon-coated AAO films after fluorination could permeate water molecules preferentially from the water/ethanol mixture.61 Frictional properties were studied on the film in which tubular carbons were aligned.62
The synthesis of tubular carbons in the channels of AAO templates was reviewed by focusing on their formation and metal filling10 and also as one of techniques to synthesize the aligned carbon nanotubes.63
Instead of CVI of organic gases, carbon deposition inside the channels was carried out through the impregnation of furfuryl alcohol under vacuum, followed by polymerization and carbonization at 900 °C, but the resultant tubes contained many bubbles.64 By step-wise heating of an AAO film filled with furfuryl alcohol with ZnCl2 catalyst, glassy carbon nanopillar arrays were obtained.65 By liquid phase carbonization of a mesophase pitch,66,67hexa(4-dodecylphenyl)-peri-hexabenzocoronene,68 and poly(vinyl chloride) (PVC) and poly(vinyl alcohol)69 in the channels of AAO films, carbon nanofibers having platelet nanotexture were obtained. Nanofibers with three different nanotextures were prepared:70 nanofibers with platelet nanotexture by using PVC (sample A, Fig. 6a-1), nanotube formed by CVI of acetylene and then filling by PVC decomposition (sample B, Fig. 6a-2) and nanotubes filled by Ni (sample C, Fig. 6a-3), and their electrocatalytic activity for redox reaction of ferri/ferro hexacyanide was compared by plotting the difference in the peak position between the oxidation and reduction peaks ΔEp against the sweeping rate of cyclic voltammetry in Fig. 6b. Nanofibers named A with a platelet nanotexture after heat treatment at 1200 °C show an activity comparable with Pt, but samples B and C have lower reversibility, even after heat treatment, than Pt. The estimated electron transfer rate showed a large difference between samples A and B, which was supposed to be due to the difference in the nanotexture on the surface of the nanofibers, as schematically shown in Fig. 6a-1 and a-2.
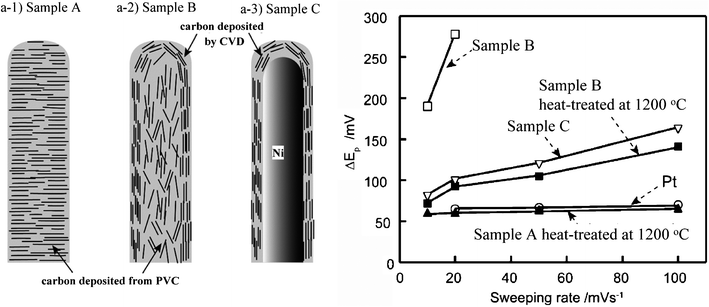 | ||
| Fig. 6 Carbon nanofibers with three different nanotextures prepared by using an AAO film as template (a) and comparison with Pt wire for electrocatalytic activity by plotting ΔEp against sweeping rate (b).70 | ||
Surfactants were used to control the morphology of various polymers, nanoparticles, nanofibers and hollow nanospheres.71 Some of these morphology-controlled polymers can be converted to carbon materials keeping their morphology.72–75 Monomeric acrylonitrile (AN) was added drop-wise to an aqueous solution of a surfactant, dodecyltrimethyleammonium bromide, to form micelles containing AN monomers inside, and then AN was polymerized, followed by conversion of PAN spherical micelles to cylindrical ones by adding FeCl3.72 The PAN nanofibers thus prepared had a uniform diameter of 25 nm and could be converted to carbon nanofibers at 900 °C, of which the diameter was about 20 nm. Microporous carbons were obtained by using cationic surfactants, cetyltrimethyl-, decyltrimethyl- and tetrapropyle-ammonium bromides, to synthesize resorcinol-formaldehyde (RF) resins.73 RF nanowires and nanospheres were fabricated by the NaOH-catalyzed polymerization confined to the vesicular assemblies of the surfactants, cetyltrimethylammonium bromide mixed with 1,3,5-trimethylbenzene and tert-butanol.74,75 RF nanowires and nanospheres, depending on the amount of butanol added, could be carbonized at 1000 °C to get carbon nanowires 45–240 nm in diameter and nanospheres 260–650 nm in diameter. The obtained carbons were microporous, had a BET surface area of 1777 m2 g−1 and total pore volume of 1.11 cm3 g−1.75
3 Template carbonization for pore structure control
Template carbonization also gives a high possibility to control pore structure in carbon materials. Various templates have been used, zeolite crystals, different mesoporous silicas, MgO and triblock surfactants. These template carbonization techniques have common advantages, giving a sharp distribution in pore size and no necessity of activation process, although activation is the most important process for the production of the so-called activated carbons. In this section, the template carbonization procedure and the pore structure in the resultant carbons are reviewed and classified by the templates used.3.1 Zeolite-templated carbons
Zeolites are aluminosilicate minerals with the framework consisting of tetrahedra of (AlO4) and (SiO4), which are microporous and so widely used as adsorbents. Three types of zeolites, zeolite Y, β and L, which have different frameworks and consequently different pore interconnections as schematically shown in Fig. 7, have been used for template carbonization.76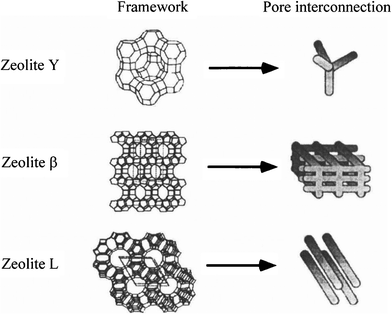 | ||
| Fig. 7 Relation of the framework in template zeolite to pore interconnection in the resultant carbon (courtesy of Prof. T. Kyotani of Tohoku University, Japan). | ||
Filling of carbon precursor into these micropores of Na-form zeolite Y through either the deposition of acrylonitrile (AN) vapor, or the liquid impregnation of furfuryl alcohol (FA) could not give highly porous carbons after carbonization at 700 °C and removing the template zeolite by washing with HF.77 However, chemical vapor infiltration (CVI) of propylene in the H-form zeolite Y at 700 °C resulted in a carbon with a high BET surface area SBET up to 2200 m2 g−1 after washing with HF and HCl solutions, of which pores consisted of both micropores and mesopores.77 Impregnation, polymerization, pyrolysis and carbonization in the pores of different zeolites had been reported on PAN78 and on phenol-formaldehyde,79 but high surface area was not obtained, except the sample obtained by using zeolite Y at 900 °C (SBET of 1580 m2 g−1 and total pore volume Vtotal of 0.83 cm3 g−1).79Carbon filling of the zeolite channel was also done by CVI of propylene,80,81 with the resultant carbon being porous with an SBET of 1380 m2 g−1 and Vtotal of 0.60 cm3 g−1.81
Coupling the impregnation of PFA with CVI of propylene into Na-form zeolite Y template was found to be effective to synthesize highly microporous carbons.82,83 The obtained carbon had an SBET of 3600 m2 g−1, micropore volume Vmicro of 1.5 cm3 g−1 and negligibly small mesopore volume Vmeso, and the micropores were supposed to be aligned regularly because it showed a sharp peak around 6° in 2θ in the XRD pattern and highly ordered lattice fringes in the TEM image, as shown in Fig. 8, revealing a structural regularity with the periodicity of about 1.4 nm, similar to the template zeolite.83 Detailed studies showed that the coupling of the impregnation of polymer, such as PFA, and the CVI of organic gas, such as propylene, (two-step filling) and the heat treatment at a high temperature such as 900 °C in the zeolite channels were essential to get highly microporous carbons.84 Through this two-step filling, almost complete filling of zeolite channels was possible, which seemed to result in the formation of the carbon framework rigid enough even after being removed from the zeolite channels. Since the zeolite-templated carbon thus obtained had a very high microporosity, its pore structure was studied by various analysis methods of N2 adsorption/desorption isotherm and its high microporous surface area Smicro of 3700 m2 g−1, high micropore volume Vmicro of 1.8 cm3 g−1 and sharp pore sizes in the range of 1.0–1.5 nm were confirmed.85
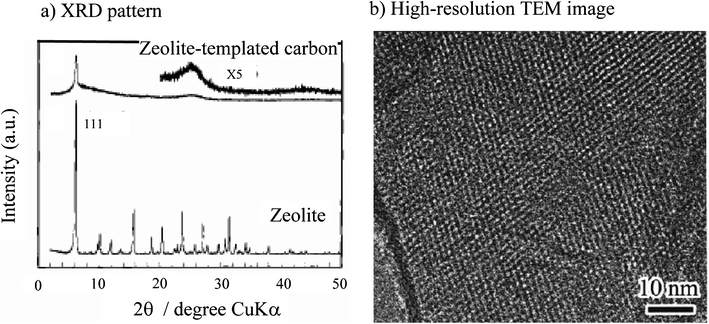 | ||
| Fig. 8 XRD pattern at low diffraction angle for the template zeolite and the resultant carbon (courtesy of Prof. T. Kyotani of Tohoku University, Japan). | ||
The two-step filling procedure associated with high temperature treatment at 700 and 800 °C was applied to other zeolite templates, zeolite β, ZSM-5, mordenite and zeolite L.86 A relatively high SBET of about 2000 m2 g−1 was obtained from zeolite β, but could not exceed that obtained from zeolite Y. By using the NH4-form of zeolite Y, microporous carbons having nitrogen-containing functional groups were obtained by the impregnation of phenol formaldehyde after carbonization above 900 °C.87SBET increased with an increase in the heat treatment temperature, from more than 1700 m2 g−1 after 900 °C treatment to 3700 m2 g−1 after 1100 °C. The particle size of the template zeolite was shown to have an influence on the pore structure of the resultant carbons: small zeolite particles produced carbon with a slightly higher SBET, higher Vtotal and higher carbonization yield than larger particles.88 A laboratory-prepared zeolite EMC-2, which had interconnected cages along the a-axis and straight channels along the c-axis, could be replicated by two-step filling with FA and propylene to result in the carbon with a high SBET of 4000 m2 g−1 and Vmicro of 1.8 cm3 g−1.89
In order to simplify the preparation procedure for zeolite-templated carbons, the liquid FA impregnation process was replaced by CVI of acetylene at 600 °C, which was followed by CVI of propylene at 700–800 °C and heat treatment at 900 °C.90 The resultant carbons are comparable in SBET and microporosity with the carbons prepared by the above-mentioned two-step filling process, but they seemed to have a better long-range regularity and uniformity of micropores. Two zeolite-templated carbon powders prepared by the acetylene CVI and by the two-step filling of PFA and propylene after the heat treatment at 900 °C could be pelletized without any binder at 300 °C and 147 MPa, their densities being 0.7–0.9 g cm−3, although no pellets could be obtained from commercially available activated carbons.91 By this densification, volumetric surface area increased from 1100 to 1300 m2 cm−3 and the average micropore size decreased with increasing pelletizing pressure, suggesting a possibility to be tuned by pressure.
The structural model of the carbon prepared at 900 °C by using furfuryl alcohol as a carbon precursor and zeolite Y as a template is shown in Fig. 9, on the basis of the experimental results by micro-Raman spectroscopy, powder X-ray diffraction, electron energy-loss spectroscopy, elemental composition, temperature programmed oxidation, temperature programmed desorption, and Fourier transform infrared spectroscopy.92 The zeolite-templated carbon is comprised of the assembly of single, non-stacked nanometre-sized graphene fragments and these graphene sheets are curved like buckybowls due to the steric hindrance of the template nanochannels. It contains some oxygen-containing functional groups bound to the edge sites of the buckybowl unit. The curvature of pore wall, pore size, pore volume estimated from this structural model containing oxygen at the edges agreed well with experimental results. These curved graphenes and periodical nanopore structure were confirmed to be preserved up to 380 °C by X-ray diffraction and Raman spectroscopy.93 The presence of curved graphene sheets in the zeolite-templated carbons was supposed to be responsible for their high-temperature ferromagnetism.94
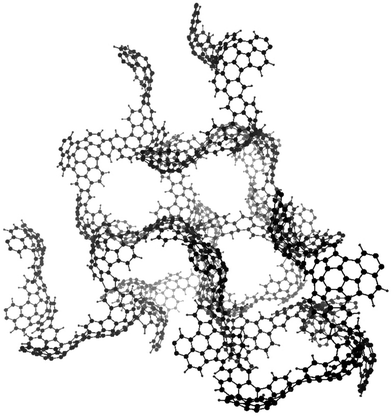 | ||
| Fig. 9 Structural model for zeolite-templated carbon (courtesy by Prof. T. Kyotani of Tohoku University, Japan). | ||
The adsorption behavior of various gases was studied on these microporous zeolite-templated carbons.95–100Nitrogen-doped carbon, which was prepared by using impregnation of furfuryl alcohol and following CVD of acetonitrile, showed higher affinity to H2O molecules than the nitrogen-free carbon.95 Adsorption of hydrogen was studied experimentally96,97 and theoretically,98 with the highest uptake of hydrogen being reported to be 6.9 mass% under 20 bar pressure at −196 °C.97 Their ordered micropore structure was preserved by potassium adsorption at 380 °C, although it was destructed by bromine adsorption, and their magnetic properties were found to be sensitive to the adsorption of these gases, including inert He.93,994He molecules adsorbed in the zeolite-templated carbon with the micropores having 1.2 nm width in a 1.4 nm periodicity formed a layer with about 1.4 atomic thickness.100 Pt-loaded zeolite-templated carbon was reported to have higher specific activity for room-temperature methanol oxidation than the commercial catalyst.101 Pt-loaded carbon was prepared by using Pt-loaded zeolite X as the template with the impregnation of furfuryl alcohol, in which Pt particles were 1.3–2.0 nm in size.102 Application of zeolite-templated carbons to the electrodes of electric double-layer capacitors was examined in many papers.103–107
3.2 Silica-templated carbons
Ordered mesoporous structures of silicas, which were formed by templating a self-assembly of surfactants and named as MCM-48, MCM-41, SBA-1 and SBA-15, etc., were successfully inherited into carbons to form ordered mesoporous carbons.108–131 In Table 1, pore structures in the carbons prepared by using various silica templates are summarized. Reviews focusing on silica-templated carbonization have also published.12,132,133| Silica template | Mesoporous carbon synthesized | Ref. | |||||
|---|---|---|---|---|---|---|---|
| Notation | Symmetry | Comments | Notation | Precursor (process) | Symmetry (XRD) | Pore parameters | |
| a a: unit cell parameter of ordered mesopores (nm), SBET: BET surface area (m2 g−1), Vt: total pore volume (mL g−1), Vmeso: mesopore volume (mL g−1), Vmicro: micropore volume (mL g−1), wmacro: width of macropores (μm), wmeso: averaged width of mesopores (nm), wmicro: averaged width of micropores (nm) b FA: furfuryl alcohol, PF: phenol/formaldehyde resin, RF: resorcinol/formaldehyde resin c I: impregnation in the solution, CVI: chemical vapor infiltration | |||||||
| MCM-48 | Cubic, Ia3d uniform channels, interpenetrating & non-interpenetrating channel system | CMK-1 | Sucrose (Ic) | S BET a = 1380, bimodal wmesoa = 3.0, Vmesoa = 1, and Vmicroa = 0.3 | 108 | ||
| Al-implanted MCM-48 | (SNC-1) | PFb (I) | (2θ = 1.6 & 2.7°) | S BET = 1257, wmeso = 2.3 | 109 | ||
| Different alkyl chains Cn (n = 12–20) SBET = 1140, wmeso = 0.96–1.14 | CMK-1 | Sucrose, FAb (I) | a a = 8.0–9.4 | S BET = 1700, wmeso = 3.4, Vmeso = 1.0 | 111 | ||
| Silylated MCM-48 | Divinylbenzene (I) | Cubic, I4132 | S BET = 1200, wmeso = 2.4 | 116 | |||
| CMK-1 | Sucrose (I) | (2θ = 1.6 & 2.7°) | S BET = 1500, Vta = 0.92, SBET = 970, Vt = 0.63 | 122 | |||
| Propylene (CVIc) | (2θ = 1.6 & 2.7°) | ||||||
| Calcined MCM-48 | CMK-1 | Sucrose (I) | I41/a | 120 | |||
| Al-implanted MCM-48 | CMK-4 | Acetylene (CVI) | Ia3d | ||||
| SBA-1 | Cubic, Pm3a | CMK-2 | Sucrose (I) | Cubic | 132 | ||
| SBA-15 | Hexagonal, P6mm uniform channels interconnected by micropores | Channels with a diameter of 9.2 nm | CMK-3 | Sucrose (I) | 2D-Hexagonal | S BET = 1520, Vt = 1.3 mL g−1, wmeso = 4.5 nm | 112 |
| CMK-3 | FA (I) | 2D-Hexagonal | Cylinders with inner dia. of 5.9 and the spaces between them (4.2) | 115 | |||
| 823 K treated, SBET = 850, Vt = 1.03 | CMK-3 | Sucrose (I) | 2D-Hexagonal | S BET = 1160, Vtotal = 1.04 | 114 | ||
| 1153 K treated, SBET = 350, Vt = 0.49 | 2D-Hexagonal | S BET = 1160, Vtotal = 1.08 | |||||
| 1243 K treated, SBET = 220, Vt = 0.23 | Random | S BET = 750, Vtotal = 0.53 | |||||
| HTAB/C16EO8 = 3/0–0/3 | CMK-3 | Sucrose (I) | Hexagonal | w meso = 2.2–3.3 | 118 | ||
| Rod-like morphology, 1–2 μm long, under hydrothermal condition | Sucrose (I) | 2D-Hexagonal | S BET = 1823, Vt = 2.23, wmeso = 5.8 | 121 | |||
| + NaCl with ca. 0.2 μm dia. | CMK-3 | FA (I) | Mesopores (wmeso = 3.9 nm) & macropores (wmacroa = 550 nm) | 130 | |||
| MCM-41 | Hexagonal, P6mm uniform channels without interconnecting pores | (C-41) | Sucrose, FA (I) | Nanochannels separated randomly | Microporous, SBET = 1100 | 111 | |
| Al-implanted | (M41-C) | PF (I) | Random | Microporous, wmicro = 0.6 | 110 | ||
| Removed amphiphiles completely leaf-like morphology and Al-implanted | (C1) | Sucrose | Periodical arrays random | Nanowire arrays, wmeso = 2.0–2.4 | 124 | ||
| (C2) | FA (I) | Broad distribution of wmeso | |||||
| MSU-H | Hexagonal wormholes, ordered | w meso = 7.6–11.9 nm | Sucrose (I) | 2D-Hexagonal | S BET = 1228, wmeso = 3.9, Vt = 1.26 | 117 | |
| w meso = 2.4–27 nm | — | FA (I) | Wormholes | w meso = 2–10 nm, unimodal (2.9 nm and bimodal (2.9 & 14 nm) | 128 | ||
| Ordered mesopores | Al-implanted HMS | (SNU-2) | PF (I) | Ordered, 2θ = 2.2° | S BET = 1056, bimodal wmeso = 2.0, wmicro = 0.6 | 110 | |
| FA, PF (I) | Ordered, 2θ = 1.8° | Unimodal and bimodal | 127 | ||||
| Colloidal silica | Colloidal particles | Monodispersed spheres, sintered | Phenol resin (mixing) | Interconnected pores | 3D periodical alignment of pores | 135 | |
| Stabilized by cetyltrimethyl-ammonium bromide | RFb (mixing) | S BET = 1512, Vmeso = 3.6 , wmeso = 12 | 136 | ||||
| Tetraethoxy silane | FA (mixing) | S BET = 1170, Vt = 1.27, wmeso = 4 | 137 | ||||
| Monolith, interconnected meso- and macropores | FA (I) | Interconnected meso- & macropores | w meso = 4.3 nm, wmacro = 0.5–30 | 141 | |||
| Tetramethyl orthopsilicate + cyclodextrin | Cyclodextrin (mixing) | Microporous, random | S BET = 1970, Vt = 1.0, wmicro = 1.6 nm | 142 | |||
The pores (channels) in the template silicas are replicated in the carbons by either impregnation or CVI of a carbon precursor, followed by carbonization and removal of the templates. The pore symmetry in these silica-templated carbons was measured by powder XRD pattern at a very low diffraction angle, as shown on the silica templates and the carbons synthesized by using each template in Fig. 10. When MCM-48 was used as a template, the resultant carbons cannot replicate the symmetry of the template exactly, having a symmetry as I4132, I4/a or lower, probably due to them collapsing after the removal of the template.108–110,116
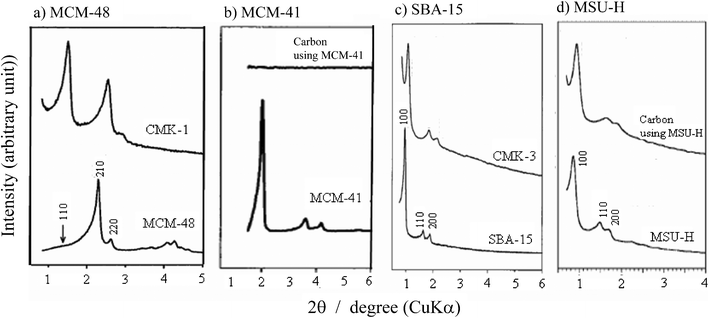 | ||
| Fig. 10 XRD patterns at low diffraction angle for the template silicas and the resultant carbon. | ||
Into a silica MCM-48, sucrose was impregnated with sulfuric acid through their aqueous solution. After the carbonization at a temperature of 800–1100 °C, the template silica was dissolved in an aqueous solution containing NaOH and ethanol to isolate the porous carbon.108 Other sugars, glucose and xylose, were successfully used as carbon precursors. The resultant carbon, denoted as CMK-1, had an SBET of 1380 m2 g−1 and ordered mesopores with a width of 3.0 nm, together with micropores 0.5–0.8 nm in width; a Vmeso of 1.1 cm3 g−1 and Vmicro of 0.3 cm3 g−1. An ordered pore structure was confirmed by the diffraction peaks at 1.6 and 2.7° in 2θ (Cu-Kα), which is different from the symmetry of the template MCM-48 (Ia3d) (Fig. 10a). In order to use the phenol/formaldehyde resin as the carbon precursor, aluminum was implanted onto the wall of mesopores of template silicas, such as MCM-48 and MCM-41, to generate strong acid catalytic sites on the channel wall for the polymerization of the resin.109,110Silylation of the pore surface of MCM-48 was effective to get highly ordered mesopores in the resultant carbon.116 The pore structure symmetry, Ia3d, in the template MCM-48 was successfully replicated in the carbon by the repetition of the impregnation/drying step.119 By applying CVI of acetylene at 800 °C to fill the pores in the template MCM-48, the symmetry of the template could be preserved in the resultant carbon even after carbonization at 900 °C and removing the template (designated as CMK-4).120
Mesoporous MCM-48 type silicas with different pore sizes were synthesized by using alkyltrimethylammonium/co-surfactant mixtures and used as templates for the preparation of CMK-1 type ordered mesoporous carbons by impregnation of sucrose from its sulfuric acid solution, followed by carbonization at 900 °C.111 The mesopore volume and size in the resultant silicas could be changed by using alkyltrimethylammonium with different alkyl chain lengths consisting of different ncarbon atoms (n = 12, 14, 16, 18 and 20), but the carbons prepared from these silica templates had almost the same mesopore volume and size, 1 cm3 g−1 and 3.4 nm respectively. In order to synthesize ordered mesoporous carbon, the repeated impregnation/carbonization of sucrose to fill the pores in the template completely, the addition of the optimum amount of sulfuric acid and the carbonization at a temperature above 600 °C were essential.115
The carbon prepared by using MCM-41 as a template contained disordered micropores, probably due to the collapse of the template framework upon its removal, and consisted of carbon nanowires randomly separated (designated as CMK-41),111 as shown in the XRD in Fig. 10b. However, MCM-41 after complete removal of the organic amphiphiles by microwave digestion could give self-supported carbon nanowire arrays, probably because adjacent nanowires are irregularly connected by thin rods.124 By using Al-implanted MCM-41 with a leaf-like morphology and furfuryl alcohol, carbon nanowires with diameters of 4–5 nm were packed side-by-side without any regularity.
Carbons with ordered mesopores in the same symmetry as the template SBA-15 were obtained by impregnation of sucrose aqueous solution with sulfuric acid, followed by carbonization at 900 °C, which was designated as CMK-3,112 as shown in Fig. 10c. The TEM images of CMK-3 are shown in Fig. 11. The resultant carbon had the SBET of 1520 m2 g−1 and Vtotal of 1.3 cm3 g−1, being due to the channels with diameters of 4.5 nm. The carbon obtained by using furfuryl alcohol consisted of two kinds of mesopores, channels with the inner diameter of 5.9 nm and mesopores formed between the adjacent channels.115 To obtain ordered mesoporous carbons, the calcination temperature of the sucrose-impregnated template SBA-15 had to be below 880 °C, because SBA-15 calcined at 970 °C gave a disordered pore arrangement.114 The size of the mesopore in the carbon increased from 2.2 to 3.3 nm upon increasing the thickness of the SBA-15 template wall from 1.4 to 2.2 nm, which was controlled by changing the ratio of hexadecyltrimethylammonium bromide/polyoxyethylene hexadecyl ether-type surfactants.118 By using SBA-15 particles with a rod-like morphology, carbon CMK-3 rods were obtained, having an SBET of 1823 m2 g−1, Vtotal of 2.23 cm3 g−1 and mesopore width of 5.8 nm.121 From a powder mixture of the SBA-15/PFA composite with NaCl of about 0.2 μm particles, followed by the compression under a pressure of 1000 kg m−3, a CMK-3 monolith was obtained, which consisted of mesopores 3.7 nm in width and macropores 550 nm in width.130 By CVI of acetonitrile into the mesopores of SBA-15 at 950–1100 °C, ordered mesoporous N-doped carbons were prepared.129
 | ||
| Fig. 11 Lattice fringe image and diffraction pattern (inserted) for the carbon (CMK-3) prepared by using a silica SBA-15 as a template.112 Reproduced with the permission of the American Chemical Society. | ||
The mesoporous silica MSU-H, which had a porous framework similar to SBA-15, was also used as a template for the preparation of mesoporous carbons.117 The pore structure symmetry in the template MSU-H was preserved in the resultant carbon with a small shrinkage, smaller than the case of SBA-15 (Fig. 10d). The carbon obtained from sucrose at 900 °C had a sharp peak in the pore size distribution at 3.9 nm.117 The carbons obtained from furfuryl alcohol with paratoluene sulfonic acid at 800 °C by using MSU-H templates with different mesopore sizes from 2.4 nm to 27 nm had the mesopore sizes from 2 nm to 10 nm.128 By controlling the amount of carbon precursor impregnated, mesoporous carbons with unimodal (2.9 nm width) and bi-modal (2.9 and 14 nm widths) pore structures could be prepared from furfuryl alcohol127,128 and also from phenol resin.127 The size of the mesopores in the carbon were controlled from 3.8 to 10.5 nm, resulting in the decreases of SBET and Vtotal from 1337 to 848 m2 g−1 and from 1.60 to 1.25 cm3 g−1, respectively, by infiltration of the mixture of sucrose with different amounts of boric acid (0–25 mol%).131Boric acid added with sucrose may change to boron oxide and borosilicate during carbonization, and may result in an increase in mesopore size in the carbon.
Mesophase pitch could be used as a carbon precursor for the ordered mesoporous silica templates, but its impregnation and carbonization above 750 °C had to be done in an autoclave.123 After carbonization at 900 °C, the diffraction lines indexed as 002 and unsymmetrical 10 for carbon were clearly detected, indicating turbostratic stacking of the carbon layers. A petroleum pitch with a low softening point between 114 and 122 °C could be impregnated into the templates MCM-48 and SBA-15 at 302 °C under atmospheric pressure, resulting in mesoporous carbons.125
Colloidal silicas were also used as templates to synthesize porous carbons.134–143 A porous carbon with a 3D periodical alignment was prepared from a phenolic resin and a silica template, which was prepared by sintering SiO2 spheres having uniform diameter in a range of 150–300 nm.135 A mixture of silica colloid stabilized by cetyltrimethylammonium bromide with resorcinol/formaldehyde resulted in mesoporous carbon with SBET of 1512 m2 g−1 and Vmeso of 3.6 cm3 g−1 due to mesopores having a width of approximately 12 nm.136 Through a sol–gel process using tetraethoxy silane in the presence of furfuryl alcohol, followed by carbonization at 800 °C, a carbon having an SBET of 1170 m2 g−1 and Vtotal of 1.27 cm3 g−1 composing of 67% mesopores centered at 4 nm width was prepared.137 The mixtures of a mesophase pitch with colloidal silica in ethanol was kept at 260 °C, slightly higher than the softening point of the pitch, for a short time (30 min) to penetrate colloidal particles into the pitch and then carbonized at 900 °C to result in mesoporous carbon.138 The Vtotal of the resultant carbons was 1.0–1.6 cm3 g−1 mainly due to mesopores with a width of approximately 13–24 nm. Colloidal spherical silicas 8 nm in width and of elongated particles (5–20 nm in diameter and 40–300 nm in length) were used as the template by mixing with resorcinol/formaldehyde in aqueous solution to obtain mesoporous carbons with an SBET of 600–900 m2 g−1 and Vtotal of 1.0–2.0 cm3 g−1.140 By using a silica template, which was synthesized through a sol–gel route, carbons with fully interconnected mesopores approximately 4.3 nm in width and macropores approximately 0.5–30 μm in width were prepared in a monolith.141 A microporous carbon monolith was prepared by carbonization of the mixture of cyclodextrin and tetramethyl orthosilicate with sulfuric acid catalyst, which had an SBET of 1970 m2 g−1, Vtotal of 1.0 cm3 g−1 and micropore width centered at 1.6 nm.142Silica-templated carbon films with a size larger than 15 × 25 mm2 were prepared by spin-coating of an acidic aqueous solution of sucrose with tetraethyl orthosilicate onto silicon wafers, followed by carbonization at 400 °C for 4 h and dissolution of the silica template.139 The resultant carbon films had an SBET of 2600 m2 g−1 and Vtotal of 1.4 cm3 g−1, consisting mainly of mesopores with the width centered at about 2.4 nm. Carbon films were prepared by using a colloidal silica with a large size (20–80 nm), which consisted of uniform spherical pores and had a large pore volume (5–9 cm3 g−1).143 By using silica templates, of which the pore surface was modified by dimethylchlorosilane, mesoporous carbons were prepared from acrylonitrile, which had a large pore volume (1.5–1.8 cm3 g−1) with primary contribution of the mesopores and relatively low microporosity.144 From the mixtures of polystyrene (PS) latex and colloidal silica, macroporous carbons with mesopores were prepared, which gave relatively good EDLC performance.145 Macropores were controllable by PS latex and mesopores by silica colloid.
Onion-like carbon-silica composite vesicles were prepared through aqueous emulsion of a low-molecular-weight resol-type phenol resin as a carbon precursor, tetraethylorthosilicate as a silica source, commercially available surfactant F127 as a template, and 1,3,5-trimethylbenzene as an organic co-solvent.146 These vesicles could be converted into onion-like mesoporous carbon vesicles having bi-modal pores of 4–23 and 66–82 nm widths.
Mesocellular carbon foams with pore sizes larger than 20 nm were prepared by selecting mesocellular aluminosilicates with various cell and window sizes as the templates and phenol/formaldehyde as the carbon precursor.147
Silica-templated mesoporous carbons were fostered by prospects of their applications as adsorbents for large molecules,148–151hydrogen152 and CO2,153catalyst supports,113,154 template for inorganic nanostructures130,155–159 and also for electrodes of electric double-layer capacitors.109,110,140,160–172
High electrical conductivity was required for some applications of nanoporous carbons, such as catalyst supports, the heat treatments at high temperatures in an inert atmosphere were carried out on silica-templated carbons.173–177 On most of the nanoporous carbons, however, their surface area and pore volume decreased markedly, though electrical conductivity increased, by high temperature treatment. On an ordered mesoporous carbon prepared from a sucrose by using mesoporous silica MCM-48 as a template, a gradual decreases in SBET, Vtotal and active surface area (ASA) were reported; from 2000 to 38 m2 g−1 in SBET, from 1.10 to 0.05 cm3 g−1 in Vtotal and from 62 to 1 m2 g−1 in SAS with a temperature increase from 900 to 2500 °C, even though interlayer spacing d002 decreased from 0.382 to 0.337 nm.176 When a poly(vinyl chloride), which was known to give a graphitizing carbon under conventional carbonization, was used as a carbon precursor for templating carbonization using mesoporous silica SBA-15, the SBET and Vtotal of the resultant mesoporous carbons decreased from 930 to 260 m2 g−1 and from 1.09 to 0.34 cm3 g−1, respectively, although electrical conductivity increased from 0.3 to 4.2 S cm−1 by heat treatment at 2300 °C.174
3.3 MgO-templated carbons
Nanoporous carbons were prepared by using MgO particles as an inorganic template178–186 and the results were reviewed in two papers; focusing on the procedure of pore control187 and on applications in relation to their pore structure.188 A mixture of MgO precursor, which gives nano-sized MgO particles after its pyrolysis, with carbon precursor was heat-treated at 900 °C for 1 h in an inert atmosphere. From the carbon-coated MgO particles thus obtained, template MgO was dissolved out using a diluted acid at room temperature to isolate the carbon formed. MgO was selected as a template mainly because of its chemical and thermal stability, no structural and compositional changes, no reaction with carbon up to the carbonization temperature of the carbon precursors, and ease of dissolvability into a diluted acidic solution.Different MgO precursors were used, MgO itself, magnesium acetate Mg(CH3COO)2, citrate Mg3(C6H5O7)2, gluconate Mg(C11H22O14), and hydroxy-carbonate 3MgCO3·Mg(OH)2. Poly(vinyl alcohol) PVA was used as the carbon precursor in most of the work. A coal tar pitch, hydroxyl propyl cellulose HPC, poly(ethylene terephtharate) PET, poly(amic acid) consisting of pyromellitic dianhydride and 4,4′-oxydianiline PMDA/ODA, poly(vinyl pyrrolidone) PVP, poly(acrylamide) PAA, and trimethylolmelamine TMM were also used. Mixing of the two precursors was performed in different ratios either in powder (powder mixing) or in solution (solution mixing). After carbonization at 900 °C, the products prepared from most of the mixtures with MgO/carbon precursor ratios larger than 5/5 were obtained as a powder, with no marked aggregation of particles and no white particles found even under a high magnification, revealing that all MgO particles are coated by carbon.
The SBET, total surface area Stotal, microporous surface area Smicro and external surface area Sext determined by αs plot analysis of N2 adsorption isotherms at 77 K are listed for the carbons prepared from the different combinations of MgO and carbon precursors in Table 2. In most of the systems, Sext is predominant and depends strongly on the mixing ratio MgO/carbon precursor, mixing method, either powder or solution mixing, and also the MgO precursor.
| Mixing method | Precursors | Mixing ratio in MgO/precursor | S BET a | α s plot analysis | ||||
|---|---|---|---|---|---|---|---|---|
| S total a | S micro a | S ext a | ||||||
| a S BET: BET surface area, Stotal: total surface area, Smicro: microporous surface area, Sext: external surface area, and Smeso: mesoporous surface area | ||||||||
| Powder mixing | MgO/PVA | 7/3 | 920 | 959 | 822 | 137 | ||
| 5/5 | 789 | 803 | 647 | 156 | ||||
| 3/7 | 546 | 567 | 457 | 110 | ||||
| MgO/HPC | 7/3 | 741 | 723 | 102 | 621 | |||
| 5/5 | 382 | 396 | 60 | 336 | ||||
| 3/7 | 249 | 264 | 40 | 224 | ||||
| MgO/PET | 7/3 | 794 | 810 | 164 | 646 | |||
| 5/5 | 701 | 724 | 167 | 557 | ||||
| 3/7 | 645 | 667 | 159 | 508 | ||||
| Powder mixing | Mg acetate/PVA | 7/3 | 1080 | 961 | 451 | 510 | ||
| 5/5 | 886 | 878 | 466 | 412 | ||||
| 3/7 | 579 | 602 | 283 | 319 | ||||
| Solution mixing | 7/3 | 1800 | 1788 | 87 | 1701 | |||
| 5/5 | 980 | 966 | 65 | 901 | ||||
| 2/8 | 289 | 312 | 26 | 286 | ||||
| Powder mixing | Mg citrate/PVA | 7/3 | 1545 | 1459 | 121 | 1338 | ||
| 5/5 | 1423 | 1346 | 7 | 1339 | ||||
| 3/7 | 1154 | 1102 | 53 | 1049 | ||||
| Solution mixing | 7/3 | — | Difficult to apply αs analysis | |||||
| 5/5 | 1351 | 1253 | 2 | 1251 | ||||
| 3/7 | 1085 | 1055 | 97 | 958 | ||||
| S BET | BJH analysis | |||||||
| S total | S micro | S meso a | ||||||
| Powder mixing | Mg gluconate/PVA | 10/0 | 1200 | 1338 | 897 | 441 | ||
| 7/3 | 813 | 1367 | 677 | 690 | ||||
| 5/5 | 570 | 907 | 235 | 672 | ||||
| 3/7 | 230 | 729 | 130 | 599 | ||||
When commercially available MgO powder with a particle size of approximately 100 nm was used with the carbon precursor in the ratio of 7/3, microporous carbon with an Smicro of about 800 m2 g−1 was obtained.179 From the same MgO/PVA ratio of 7/3, however, mesoporous carbons were obtained in the systems using Mg acetate, citrate and gluconate, particularly by solution mixing in Mg acetate/PVA system. Mesopores are formed by replicating MgO particles, and micropores are formed in the wall of the mesopores. When MgO with a particle size of approximately 100 nm was used, which gave macropores but not mesopores, the surface area of the MgO particles is much smaller than those due to small MgO particles formed from the MgO precursors, and so the thickness of the pore wall is supposed to be much thicker, which seems to lead to the formation of micropores in the wall.
In Fig. 12a, the SBET is plotted for the carbons obtained from the mixtures of Mg acetate with PVA prepared through powder and solution mixing.180 The solution mixing resulted in very high SBET, as high as 1800 m2 g−1, much higher than powder mixing. The principal part of this high surface area was due to a high Sext, as listed in Table 2. As shown in Fig. 12b, the pore size distribution in the carbon prepared through solution mixing shows a sharp pore size distribution at about 13 nm, although a broad distribution is obtained through powder mixing. For Mg citrate/PVA and Mg gluconate/PVA systems, however, no marked differences in the surface area and pore size distributions were observed between powder and solution mixing.
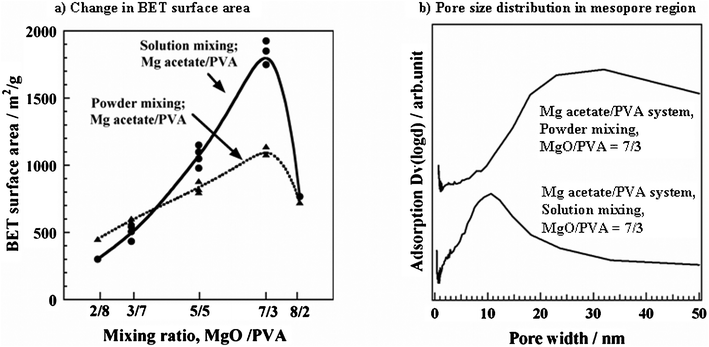 | ||
| Fig. 12 Effect of mixing process of Mg acetate and PVA on BET surface area (a) and pore size distribution (b) of the resultant carbon.181 | ||
For Mg citrate/PVA system, the carbons obtained are mesopore rich, as listed in Table 2. The pore size distribution of these carbons showed a sharp maximum at about 5 nm and the Smeso and Vmeso determined by the BJH method reached about 1600 m2 g−1 and about 1.7 cm3 g−1, respectively. For Mg gluconate/PVA system, the dependencies of the pore structure parameters on the mixing ratio were a little different from those in the Mg citrate/PVA system.182 In Fig. 13a, the Stotal, Smeso and Smicro determined by the BJH method are plotted against the MgO/PVA ratio in the Mg gluconate/PVA system. From the Mg gluconate itself (MgO/PVA = 10/0), Stotal reaches 1300 m2 g−1 and consists mainly of micropores, Smicro of about 900 m2 g−1 and Vmicro is about 0.6 cm3 g−1, though the Smeso and Vmeso are small in comparison with other carbons prepared in this system. In the carbon prepared from MgO/PVA of 7/3 in this system, Smicro is comparable to Smeso. Upon decreasing the MgO/PVA ratio from 7/3, mesopores become predominant. Pore size distribution in the mesopore region is shown for these carbons in Fig. 13b. The mixtures of Mg gluconate with PVA gave a sharp size distribution at 2–4 nm, the pore volume of this size range decreased with decreasing MgO/PVA ratio.
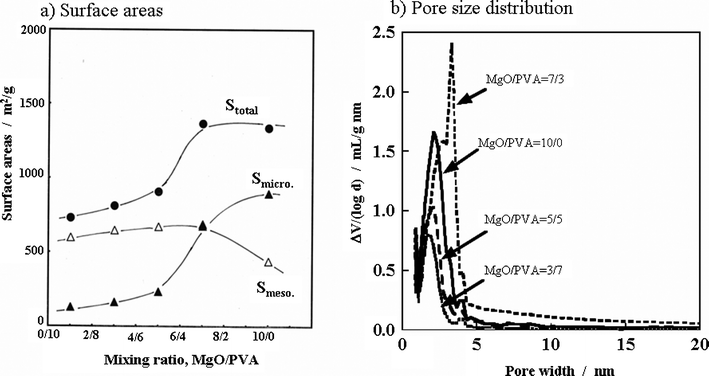 | ||
| Fig. 13 Changes in surface areas, Stotal, Smicro and Smeso (a) and in pore size distribution (b) with mixing ratio MgO/PVA for the carbons prepared in the Mg gluconate/PVA system.182 | ||
A coal tar pitch with a softening point of 85.2 °C was used as a carbon precursor to give mesoporous carbons.184 The Mg acetate/pitch system gave carbons with mesopores centered at around 13 nm and the Mg citrate/pitch system resulted in mesopores centered at around 5 nm, even though the two precursors are mixed in powder. High wettability of the pitches to the MgO surface seems to be the main reason to have a sharp pore size distribution. When a polyimide and PET were used as carbon precursors, however, dispersion of MgO precursor particles in the carbon precursor through the dispersion either in organic solution187 or repeated fusion and crushing185 were needed to get high surface area in the resultant carbons.
The template MgO was experimentally demonstrated to be recycled. Acetic and citric acids were selected to dissolve out MgO from carbon-coated MgO. The recovered Mg acetate and citrate aqueous solutions were mixed with PVA again and subjected to the present procedure. In Table 3, the SBET and carbon yield are listed for the carbons obtained in each cycle. During 5 cycles, almost the same SBET and carbon yield are obtained, revealing that the MgO can be recycled in 100%, by supplying carbon precursor in every cycle.
| Cycle no. | Using acetic acid | Using citric acid | ||||
|---|---|---|---|---|---|---|
| Mg acetate/PVA system MgO/PVA=5/5 | Mg citrate/PVA system MgO/PVA=5/5 | Mg citrate itself | ||||
| S BET (m2 g−1) | Carbon yield (mass%) | S BET (m2 g−1) | Carbon yield (mass%) | S BET (m2 g−1) | Carbon yield (mass%) | |
| 1 | 1210 | 9.8 | 1402 | 26.8 | 1680 | 9.7 |
| 2 | 1185 | 9.6 | 1468 | 25.2 | 1590 | 11.0 |
| 3 | 1262 | 9.3 | 1423 | 25.4 | 1621 | 10.1 |
| 4 | 1203 | 10.1 | 1415 | 26.1 | 1689 | 9.6 |
| 5 | 1249 | 9.1 | 1481 | 25.4 | 1575 | 10.5 |
Porous carbon nanofibers were prepared by electrospinning of the DMF solution of PAN and MgCl2 in different ratios, followed by stabilization at 250 °C, carbonization at 1050 °C and dissolution of MgO with 1 mol L−1H2SO4.189MgCl2 changed to MgOHCl during the stabilization process and to MgO during carbonization. The nanofibers prepared are microporous, have an SBET of 800 m2 g−1 and Vmicro of 3.32 cm3 g−1.
MgO-templated carbon containing nanoparticles of Sn metal in its pores was successfully applied to the anode of lithium ion rechargeable batteries.190 The space neighboring to the Sn particles is supposed to absorb a marked expansion due to its alloying with Li and carbon shell surrounded Sn nanoparticles to disturb their movement during alloying/de-alloying due to charge/discharge cycles of the batteries. MgO-templated mesoporous carbons were used as electrode materials of electric double-layer capacitors (EDLCs).181,186,191–196 Using nitrogen-containing organic compounds as carbon precursors in this procedure, nitrogen-doped nanoporous carbon was prepared, which gave a better EDLC performance.186 By using these mesoporous carbons as one of electrodes of asymmetric EDLCs, the roles of micropores and mesopores, and the predominant contribution of the negative electrode to their capacitor performance were demonstrated.192–194 The procedure for the preparation of carbon-coated MgO was successfully applied on various ceramic particles,197,198 particularly on photocatalyst TiO2 to enhance its performance.199–201
A similar process using an Ni(OH)2 template with phenol in an ethanol solution was proposed to prepare mesoporous carbons and the obtained carbon was reported to give high energy and power densities in both aqueous and organic electrolytes to EDLC.202 Also, nanoparticles of TiO2 were used as a template to have carbon hollow spheres.203 In these cases, HCl and HF had to be used to dissolve the templates (NiO and TiO2). Barium citrate gave porous carbons containing both micropores and mesopores, but much smaller SBET than the carbon prepared from Mg citrate.195,204
3.4 Soft-templated carbons
Mesoporous carbons with an ordered pore structure were successfully prepared by using surfactants as organic templates,205–221 called soft templates here in contrast to inorganic templates (hard templates) as zeolites, silicas and MgO. The key of this process was the direct use of the self-assembly of the surfactant block copolymers as templates, although the same surfactants were used for the preparation of mesoporous silicas (see section 3.2). In most cases of soft-template carbonization, the solvent evaporation-induced self-assembly (EISA) method was applied on the mixture of a carbon precursor with a surfactant.Carbon films with hexagonal arrays of channels (cylindrical mesopores) perpendicular to the film surface were synthesized by using a diblock copolymer, poly(stylene)-block-poly(4-vinylpridine) (PS-P4VP), as a template and resolcinol/formaldehyde as a carbon precursor.205 A solution of PS-P4VP and resorcinol was cast onto a silica substrate to form a film, in which most of the resorcinol molecules were located in the P4VP domain due to the hydrogen-bond association between the basic P4VP blocks and the acidic resorcinol monomers. By the controlled evaporation of the solvent N,N′-dimethylformamide (DMF) in DMF/benzene vapor at 80 °C, a highly-ordered nanostructure of the film was obtained, where the PS domain became the cylinder directed perpendicular to the film surface. This film was exposed to formaldehyde vapor to form a highly cross-linked phenol resin in the P4VP domain and then carbonized to 800 °C. The channels were formed perpendicularly in the carbon film, of which the diameter was 33.7 nm and the wall thickness was 9.0 nm. A crack-free carbon film with thicknesses up to 1 μm and sizes up to 6 cm2 was prepared. By using the same diblock copolymer and a phenol resin with hexamethylenetetramine, the products of pyrolysis up to 600 °C had disordered pores.206
A mixture of a commercially available triblock copolymer, poly(ethylene oxide)-b-poly(propylene oxide)-b-poly(ethylene oxide) (Pluronic F127, PEO106-PPO70-PEO106), resorcinol, triethyl orthoacetate and formaldehyde in a water/ethanol/HCl mixed solution was spin-coated on a silicon substrate and then carbonized up to 800 °C to get mesoporous carbon films.206Resorcinol/formaldehyde (RF) and triethyl orthoacetate (EOA) were the carbon precursors. The periodic structure of RF/EOA and F127 was established in the original organic films due to EISA with a spacing of 9.2 nm, and it was retained even after carbonization at 800 °C although the spacing was shortened to 7.0 nm. The surfactant F127 was decomposed up to the temperature of 400 °C to form mesopores. The carbon films prepared at 800 °C had an SBET of 1354 m2 g−1, Vtotal of 0.743 cm3 g−1 and mesopore width of 5.9 nm. By changing the concentration of the surfactant F127 in the starting solution, a carbon with hexagonal arrays of channels (phenol/F127 = 1/0.012) and that with cubic arrays of mesopores (phenol/F127 = 1/0.005–0.006) could be prepared.209 The former consisting of channels with the diameter of 2.9 nm, SBET of 968 m2 g−1 and Vtotal of 0.56 cm3 g−1, and the latter of the mesopores with the size of about 3.7 nm, SBET of 778 m2 g−1 and Vtotal of 0.44 cm3 g−1. As shown in Fig. 14, N2 adsorption/desorption isotherms for two mesoporous materials, polymers and carbons, are type IV with a pronounced hysteresis and have a sharp pore size distribution. By using phloroglucinol/formaldehyde with F127, mesoporous carbons were prepared in monolith, fiber and film morphologies, much faster and under much milder conditions than by using phenol/ and resorcinol/formaldehydes.210 Through a dual phase separation process, macroporous carbon with mesoporous walls was prepared.211
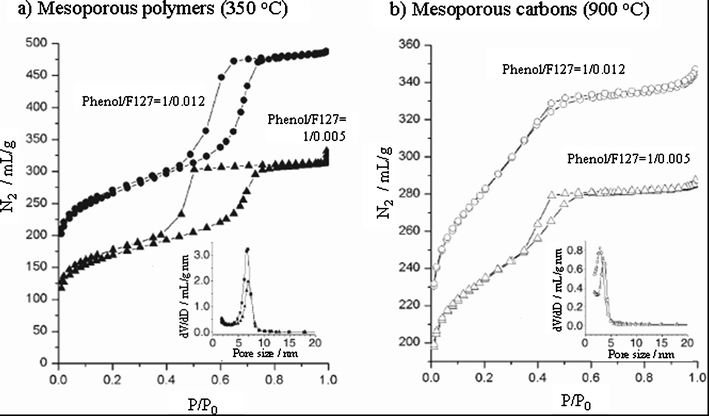 | ||
| Fig. 14 N2 adsorption/desorption isotherms for the mesoporous polymers and the resultant mesoporous carbons and their pore size distribution (inserted).208 Reproduced with the permission of WILEY-VCH Verlag GmbH & Co. | ||
By using another triblock copolymer Pluronic P123 (PEO20-PPO70-PEO20) with resols in aqueous solution, carbon with continuous cubic mesopores was obtained.212 A resin of resorcinol/phloroglucinol/formaldehyde was successfully used for a carbon precursor for the process using P123 template. Other kinds of triblock copolymers of acrylonitrile (AN) and η-butyl acrylate (BA), (AN)45-(BA)530-(AN)45, was carbonized on a cleaved mica or silicon wafer after stabilization at 200–230 °C, but no information on pore structure was presented.213Triblock copolymer poly-(propylene glycol)-block-poly(ethylene glycol)-block-poly(propylene glycol) (PPO)15-(PEO)22-(PPO)15 was also used as a template.214
Different pore structures in the carbon materials were obtained by simply adjusting the ratio of phenol to template surfactant; two-dimensional hexagonal, three-dimensional bicontinuous, body-centered cubic and lamellar.215,216 The compositions in a range of phenol/F127 ratio (in mol) of 1/0.010–0.015 and phenol/P123 of 1/0.007–0.016 gave carbons having channels with the diameter of about 2.8 nm in hexagonal arrangement even after carbonization at 1200 °C, as shown its TEM images of two perpendicular faces in Fig. 15a and 15b.215 From the compositions in a range of phenol/F127 of 1/0.003–0.008 and phenol/F108 (PEO132-PPO50-PEO132) of 1/0.005–0.010, carbons with mesopores with a mean diameter of 3.8 nm arranged in a cubic symmetry was synthesized, as shown in Fig. 15c and 15d. Its N2 adsorption/desorption isotherm was type-IV with clear hysteresis at P/P0 of around 0.5. The carbon, in which bicontinuous mesopores with a mean size of 2.3 nm arranged in a cubic symmetry, was obtained from the compositions in a very narrow range of phenol/P123 of 1/0.018–0.019. By increasing the phenol/P123 ratio to 1/0.022–0.027, a lamellar mesostructure was formed.
 | ||
| Fig. 15 TEM images for the resultant ordered mesoporous carbons with hexagonal symmetry (a and b) and cubic symmetry (c and d).215 | ||
Ordered mesoporous carbons were prepared through a basic aqueous solution of phenol/formaldehyde and triblock copolymer templates.216 A similar effect of mixing ratio of carbon precursor/F127 was reported, together with the result on the F127/P123 mixing.217
When a water–ethanol solution of 1,5-dihydroxynaphthalene, formaldehyde and F127 with the formaldehyde/F127 ratio of 1/0.003 was used, the carbon having the channels, which were arranged perpendicular to the film surface in a hexagonal symmetry and had a diameter of 6.5 nm, was obtained.219 From the composite prepared by deposition of benzyl alcohol at 300 °C into a film of F127 on a silicon substrate, a ultrathin carbon film with a thickness of about 15 nm was prepared, in which the channels with diameters of 9.4 nm were running along the film surface and parallel with each other.220
The stability of the hexagonal symmetry arrays of the channels in the film of phenol/formaldehyde and triblock copolymer composites and that after removal of the template at 350 °C was discussed by taking the constraint due to wetting of the thin film with the substrate, and the contraction stress during the template removal into account.221 The removal of the template at 350 °C from the composite film templated by F127 changed the pore structure from two-dimensional hexagonal symmetry to a disordered structure, but the films templated by P123, which had shorter PEO segment than F127, could give well-ordered channels after the removal of the template. The composition of phenol/P123 to give ordered channels was markedly narrowed for thin films in comparison to bulk powders, the range of 1/0.010–0.013 for the film but 1/0.008–0.016 for the powder.
Carbons having ordered macropores, of which walls contained ordered mesopores, were synthesized via a dual-templating technique using poly(methyl methacrylate) (PMMA) and triblock copolymer as templates.222 A solution of phenol/formaldehyde and F127 was added into sedimented colloidal crystal pieces of PMMA, followed by evaporation of the solvent, cross-linking and heating at 450 °C to have phenol resin monolith. Carbon monoliths were obtained by heating up to 900 °C. The size of the macropores formed by the template of PMMA spheres and that of the mesopores by F127 template was 342–404 and around 3 nm, respectively. Mesopores were formed in either face-centered cubic or two-dimensional hexagonal symmetry.
The frameworks in the mesoporous carbons obtained through this template method was shown to be relatively rigid, even though SBET and Vtotal decreased gradually with increasing heat treatment temperature; 607 m2 g−1 and 0.58 cm3 g−1 after 850 °C treatment and 230 m2 g−1 and 0.30 cm3 g−1 after 2600 °C.218 Mesoporous carbon thus obtained could be activated by using KOH to create micropores with only a slight sacrifice of mesopores.223,224 Impregnation of KOH through its aqueous solution into the carbon and heat-treated at 700 °C for 45 min resulted in an increases in the SBET from 500 to 900 m2 g−1 and Vtotal from 0.70 to 0.87 cm3 g−1, where Vmicro increased from 0.04 to 0.22 cm3 g−1 and Vmeso decreased from 0.66 to 0.64 cm3 g−1.223KOH activation of the mesoporous carbons at 800 °C increased the surface area and pore volume of both the micropores and mesopores.224 The mesoporous carbons were applied to the electrodes of the electric double-layer capacitor with both aqueous and non-aqueous electrolytes and gave a higher capacitance than commercially available activated carbon, much higher after KOH activation.224 Mesoporous carbon with hexagonally-shaped pores prepared from resolcinol-formaldehyde and F127, which was characterized by the measurement of freezing and melting behavior of confined Kr225 and the grand caronical Monte Carlo simulation of adsorption isotherms of Ar at 77.4 and 87.3 K.226
Ordered mesoporous TiC/C composites were prepared by the same procedure by using resol-type phenol, titanium citrate and surfactant F127 in ethanol/water mixture, followed by carbothermal reduction up to 1000 °C.227 Nano-sized crystalline TiC particles with the size of 4–7 nm were confined in the carbon pore walls and could enhance the oxidation resistance of carbon open frameworks. Starting from phenol, tetraethyl orthosilicate (TEOS) and colloidal silica with F127, porous carbons consisting of macropores of 20 or 50 nm caused by dissolution of colloidal silica, mesopores of ∼12 nm by soft-templating process of F127 and micropores of ∼2 nm by dissolution of silica formed from TEOS were prepared.228Activation with CO2 at 850 °C resulted in SBET of 2800 m2 g−1 and Vtotal of 6.0 cm3 g−1. A ethanol/water solution of phenol resin with a surfactant F127 was coated into a porous α-Al2O3 tubular support (average porosity: 40%) by dipping and then carbonized at 600 °C in N2.229Carbon membrane formed on the Al2O3 walls had an SBET of 670 m2 g−1, Vtotal of 0.58 cm3 g−1 and mesopores with sizes of about 4.2 nm. The Al2O3/C composites thus prepared exhibited good permeation properties, high hydrothermal stability and high alkaline resistance.
Carbon foams were prepared using either poly(urethane) or melamine foam as template with impregnation of poly(amide acid), followed by imidization and carbonization.230,231 The templates used, poly(urethane) (PU) and melamine, gave only small amounts of carbon residues after carbonization. Because of the presence of macropores, the activation by using air was effectively performed to increase the micropores, which resulted in the acceleration of water vapor adsorption.230 By using fluorinated polyimide as carbon precursor, microporous carbon foams were prepared by a single carbonization process, having an SBET of 1540 m2 g−1 and Vmicro of 0.63 cm3 g−1.231 Furfuryl alcohol232 and petroleum pitch233 were used as carbon precursor to impregnate into PU-based foams to fabricate carbon foams. Pore structure in template PU foam was controlled by adding small particles of clay, although some decomposition residues from the clay mineral remained in the carbon foams.232 For pitch impregnation to PU foam, a water slurry dispersed small particles of the pitch with a surfactant was used.233
3.5 Other templates
In 1987, carbon foams consisting of glass-like carbon were prepared by using sintered NaCl as a template.234 Commercially available NaCl with the average particle size of 17 μm was molded into a bar having a size of 20.0 × 2.5 × 1.0 cm3 and about 35% porosity, and then sintered at 710 °C. Phenol polymer was impregnated into the sintered NaCl bar through its THF solution and then carbonized at 700 °C. Template NaCl was removed by careful washing with water and HNO3, and then the carbon bar was freeze-dried. The carbon foam thus prepared contained macropores with the size of about 8 μm and had the bulk density of 0.035 to 0.075 g cm−3.Li-form of taeniolite was intercalated with either hydroxylaluminum Al2(OH)5Cl or hydroxylaluminum-zirconium Al1.2Zr0.3Cl and then saturated with an 80% furfuryl alcohol (FA) solution of benzene, followed by polymerization of FA and carbonization at 700 °C.235 The resultant carbons were microporous, depending strongly on the water content of the inorganic matrix, and showed molecular sieving properties. Porous clay heterostructures prepared from natural montmorillonite by intercalation of different surfactants, octyl-, decyl- and dodecyl-amines, and tetraethylorthosilicate, were also used as a template to prepare nanoporous carbons.236Carbon precursor FA was impregnated at room temperature, polymerized at 95 °C, carbonized at 500–800 °C and then the template was removed with 10% HF at room temperature. The pore structure of the carbons obtained consisted of pores with widths of about 2 nm (domain I) and larger mesopores (domain II), the former supposedly being due to the inner structure of the template clays and the latter to aggregation of the clay particles. The carbon obtained by using decyl-amine and carbonizing at 700 °C gave the highest SBET of 1469 m2 g−1 and pore volume of the domain I of 0.68 cm3 g−1. The second impregnation/polymerization of FA made the pore parameter smaller. Similar porous carbons were obtained by using porous clay heterostructures loaded by different metals and sucrose.237 Rice husk gave mesoporous carbon after carbonization and removal of mineral residues (mainly SiO2), which worked as template to give mesopores.238
4 Concluding remarks
4.1 Prospects for templating processes
From the view point of the production of graphite flakes, template carbonization using clays is too complicated and costly, and heat treatment at a high temperature above 2800 °C is necessary. It has to be pointed out that we have still enough resources of natural graphite. However, this process might be worthwhile to consider as one of the routes to prepare graphene,239 even though many experimental studies on both template and carbon precursor are needed. In the present procedure, the carbonization temperature is limited up to 700 °C because the layer structure of MONT is stable only up to 700 °C. As a consequence, the resultant carbon still contains a large amount of hydrogen, nitrogen and/or oxygen, which might remain as lattice defects after further carbonization up to about 1500 °C,21 and the annealing at a high temperature was essential to get highly crystalline flakes. Even after treatment at 1400 °C, lattice fringe images of TEM indicated the stacking of many hexagonal carbon layers, although the formation of a mono-atomic intercalate layer was confirmed on MONT–PAN complexes.21 In order to synthesize graphene through this route, therefore, a new template which is stable up to high temperatures has to be developed in order to keep intercalate layer in between lamellae until the completion of its carbonization. Also, carbon precursors which have high carbon yield after carbonization are desired.Preparation of tubular carbons by using AAO films as templates is considered as one of routes to synthesize carbon nanotubes.10 Although a uniform diameter and length of well-aligned carbon tubules can be obtained through this process, the process is also complicated and costly and high temperature treatment is essential to convert tubular carbons to carbon nanotubes comparable to those obtained by other routes. Filling of metals and metal oxides can be done into tubular carbons formed in the AAO template easily by keeping their outer surfaces clean, more easily than the cases of carbon nanotubes prepared by other techniques, such as ark discharging and catalytic CVD. Holding some drugs inside the tubular carbons in the AAO template can be done easily together with magnetic metal particles, and the resultant tubules can be applied to magnetic drug delivery system after separating from the template.54 This technique, coupled with a high temperature treatment, is also interesting for the preparation of multi-walled carbon nanotubes (MWCNTs) with high homogeneity in tube diameter and length, which can be controlled by selecting the AAO film. The drawbacks of this technique for practical application seem to be a low yield of carbon in an AAO templating process and the necessity to use either a high-concentration HF or NaOH solution in an autoclave for removing the template. These drawbacks may be compensated by excellent functionality of the resultant carbon tubules. Even so, more work is strongly required on the development of more appropriate template materials and carbon precursors.
Templating processes for the control of pore structure in carbons have various advantages and disadvantages in comparison with the conventional activation process. In the activation process, pores are created by the gasification of matrix carbon to CO and/or CO2 through oxidation, in other words, by losing carbon, and the formation of mesopores can be done by sacrificing smaller micropores which are formed either during carbonization of the precursor or through activation, resulting in lowering the final yield of porous carbon and coexisting mesopores with micropores. Ordered micropores and mesopores are very difficult to realize in the carbon through the activation process. The templating processes, on the other hand, can create pores with uniform size and morphology, even in the ordered state, as explained above. However, it has to be pointed out again that most of the templating processes are complicated and costly, more than the production of activated carbons. Phenol resins, PAN, PVA, pitches have been employed as carbon precursors in templating processes, but biomasses have been rarely used, although they are important carbon precursors for the mass production of activated carbons.
4.2 Comparison among templating processes for pore structure control
Different template carbonization processes for pore structure control are compared by listing carbon precursors, resultant nanoporous carbons and template performance in Table 4.| Templates | Carbon precursors | Carbon prepared | Template performance | ||||
|---|---|---|---|---|---|---|---|
| Pore structure | Pore volume & surface area | Removal | Recyclability | ||||
| a CVI: chemical vapor infiltration, FA: furfuryl alcohol, PF: phenol formaldehyde, MP: mesophase pitch, RF: resolcinol-formaldehyde, AN: acrylonitrile, PVA: poly(vinyl alcohol), PET: poly(ethylene terefuthalate), PIs: polyimides, EOA: triethyl orthoacetate, PGF: phloroglucinol-formaldehyde | |||||||
| Inorganic | Zeolites | Zeolite Y, β and L, ZSM-5 | Propylene CVI a + FAa impregnation | Micropores ordered | 3700 m2 g−1, 1.8 cm3 g−185 | 46% HF or NaOH | No |
| 4000 m2 g−1, 1.8 cm3 g−189 | |||||||
| Mesoporous silicas | MCM-48, MCM-41, SBA-1 and SBA-15 | Impregnation of sucrose, FA, PFa and MP.aCVI of acetylene | Mesopores ordered or disordered | 1130 m2 g−1, ∼1.0 cm3 g−1111 | HF or NaOH | No | |
| 1520 m2 g−1, 1.3 cm3 g−1112 | |||||||
| Colloidal silicas | Organic silicates and inorganic silica | Mixing with RF,aANa | Micropores & mesopores disordered | 1512 m2 g−1, 3.6 cm3 g−1136 | HF | No | |
| 2600 m2 g−1, 1.4 cm3 g−1139 | |||||||
| MgO | MgO, Mg acetate, citrate, Mg(OH)2 | Mixing with PVA,a coal tar pitches, PET,a PIsa | Mesopores disordered | 1800 m2 g−1179 | Acetic, citric acids | Easily recycle | |
| 1600 m2 g−1, 1.7 cm3 g−1181 | |||||||
| Organic | Surfactants | Diblock & triblock copolymers | Mixing with RF, EOA,aPGF,aAN | Mesopores ordered or disordered | 1354 m2 g−1, 0.74 cm3 g−1207 | Not needed | No |
| 968 m2 g−1, 0.56 cm3 g−1208 | |||||||
| Organic foams | Urethane & melamine foams | Impregnation of PIs | Macropores & micropores | 1540 m2 g−1, 0.63 cm3 g−1226 | Not needed | No | |
The advantage of zeolite template carbonization is the formation of ordered micropores in the resultant carbons by the replication of zeolite framework. For the exact inheritance from zeolite template, complete filling of pores in the template by carbon and sufficient carbonization in the zeolite channels was required.82,84 Two-step filling by liquid impregnation and CVI and the heat treatment up to 900 °C in the channels of zeolite Y gave very high SBET as 3600 m2 g−1 and ordered micropores of a volume as 2.0 cm3 g−1. The preparation procedure was tried to be simplified by changing the first step (impregnation) to CVI.90 However, it is still complicated; two steps for complete filling of the zeolite pores, carbonization in the zeolite channels and separation of the resultant carbon from zeolite by washing with HF are essential. The zeolite template has to be sacrificed to synthesize microporous carbons. It will be necessary to develop an application, in which the function of zeolite-templated microporous carbon has to be high enough to compensate the cost of this multi-stepped process.
Template carbonization using ordered mesoporous silicas can give ordered mesopores in carbons by inheriting the mesoporous structure in the templates. In order to replicate the regularity in pore structure, complete filling of the mesopores in the template is required, which has been done either by repeated impregnation or by combining the impregnation and CVI processes. When the filling of carbon precursor was not enough, ordered mesopore structure in the carbon can not be retained after removal of the silica template. In some cases, micropores are created in the walls of mesopores, being kept high mesoporosity. The advantage of this templating process using mesoporous silicas is the formation of ordered mesopores in the resultant carbons. However, it has to be pointed out that ordered mesoporous carbons can get under the sacrifice of the template silicas and also that, for the removal of the template silicas, either corrosive HF or NaOH in the autoclave has to be used, which is not desirable for a large-scale production of the carbon.
The soft template method, most of them using either surfactant diblock or triblock copolymers, has an advantage to give ordered mesopores in different symmetries, channels with mesopore-sized diameter in hexagonal symmetry, mesopores in a body-centered cubic symmetry, cubic bicontinuous mesopores and mesopores in random arrays. This was developed to simplify the silica-templating process for the preparation of mesoporous carbons by applying the same process for the preparation of mesoporous silicas to mesoporous carbon precursors. However, it has to be pointed out that the carbon precursors are limited to thermosetting resins and the procedure is still complicated, even though the surfactant template has to be sacrificed for the synthesis of mesoporous carbons. The carbon precursors used are only thermosetting resins, either phenol/, resorcinol/ or phloroglucinol/formaldehyde resins and poly(acrylonitrile). The process for the preparation of phenol/triblock copolymer composite has to be performed under strictly controlled conditions in order to realize the ordered mesopores in carbons, the pH of the phenol/copolymer mixture, solvent evaporation for the self-assembly of copolymer, etc. It may be key for further development of this process into a large scale production to expand the possible carbon precursors and also to create a certain amount of micropores in addition to mesopores. Secondly, KOH activation was reported to be effective.223,224 However, it might be worthwhile to try activation in an air flow, because carbon foams prepared using a PU-foam as a soft-template were possible to be activated in a simple and quick procedure.230
In comparison with other template carbonization processes discussed above, MgO template carbonization has the following advantages: 1) the MgO template is easily removed by a diluted non-corrosive acid, 2) MgO can be recycled, and 3) size and volume of the mesopores are tunable by selecting MgO precursor.187Pore size formed in the MgO-templated carbons is very homogeneous, being comparable with other processes. Therefore, the MgO template carbonization process has a high possibility to apply to the production of mesoporous carbons in a large scale. Now, it is announced that a few hundreds of grams of nanoporous carbon in a batch can be prepared by the MgO-template method.188 It is also one of advantages of this process to be able to use thermoplastic precursors, such as poly(vinyl alcohol) and pitches, without any stabilization process. In many applications, such as most cases of adsorbents and electrodes of energy storage devices, it may be an advantage to get the final nanoporous carbons as a powder without any pulverization processes. A disadvantage of this method is that mesopores can not be obtained in an ordered state, although other methods can give ordered pores. However, it may be that the pores in the carbon do not necessarily need to be ordered in many of the applications of nanoporous carbons.
Pore structures in the carbons prepared from furfuryl alcohol by using various templates, a clay (bentonite), β-zeolite and Al implanted MCM-48, were discussed by focusing on the application for gas separation and storage.240 The carbons from acrylonitrile, furfuryl alcohol, pyrene and vinyl acetate by using various zeolite and montmorillonite were compared on electrochemical performances for energy storage.241
4.3 High temperature behavior of templated carbons
Structural changes with high temperature treatment is known to be governed by the nanotexture of carbon materials, which is classified on the basis of the scheme and degree of preferred orientation of small carbon layers to random, planar, axial and point orientation.6,9,242 The nanotexture in carbon materials is determined by their carbonization process, depending strongly on their precursor and carbonization conditions. The carbonization behavior of a carbon precursor in a nano-sized space of the template (interlayer gallery of clays, nanochannels of AAO films, micropores of zeolites and mesopores of silicas) or with a limited thickness on the template (channel surface of AAO films and the surface of MgO) is supposed to be quite different from that under conventional conditions (without templates). PAN-derived carbon prepared through conventional carbonization is non-graphitizing, but the carbon prepared in the gallery of MONT is easily converted to graphite at a high temperature of 2800 °C,1,21 suggesting that the pyrolysis of PAN is performed in the monolayer of carbon atoms between the MONT layers. In the nanochannel of the AAO film, the nanotexture of the resultant nanotubules and nanofibers depends strongly on the precursor and carbonization conditions: glassy carbon nanopillars were formed from furfuryl alcohol by step-wise carbonization, although a simple carbonization resulted in tubules containing many bubbles,65 and carbon nanofibers with platelet nanotexture (carbon layers preferentially oriented perpendicular to the fiber axis) were prepared from mesophase pitches, PVA and PVC.66,67,69 When hexa(4-dodecylphenyl)-peri-hexabenzocoronene was used with step-wise carbonization, the tube walls had a well aligned platelet nanotexture, although one-step carbonization could not give a well aligned platelet nanotexture and also hexa(4-dodecylphenyl)benzene could not.68 During the pyrolysis of a thermoplastic precursor through MgO-template carbonization to form mesoporous carbons,178 a constraint from the MgO template surface due to the wettability of the pyrolyzed carbonaceous residues is so strong to disturb its flow during the pyrolysis and, as a consequence, the orientation of the nano-sized carbon layers after carbonization can not extend in a large area, which gives a certain influence on the high temperature behavior, e.g., graphitizability, of the resultant mesoporous carbons. The crystallinity of the carbon was experimentally shown to depend on the MgO/PVA ratio, with a lower ratio giving better crystallinity of the resultant carbon and a smaller pore volume.243 In the composites of phenolic precursor and triblock copolymer, symmetrically oriented structure in the template was shown to collapse during the pyrolysis of the template at 350 °C when the thickness of the pore wall was not thick enough.221 The stress from contraction during the pyrolysis of the template and wetting between the pyrolyzed composite film and the substrate were considered to be the dominant factors for collapsing of the oriented pore structure. Such a constraint surely affects the nanotexture in the carbon walls of the resultant mesoporous carbon, even though its oriented pore structure remains. It has been pointed out that an interfacial interaction between the precursor and the matrix (for examples, the Al2O3 wall of AAO films, MgO surface) has a strong influence on the nanotexture of the resultant carbon.242,244 More detailed studies are strongly desired on the structure and nanotexture of the carbon materials prepared through the template carbonization process.These experimental results suggest that the carbon materials prepared through template carbonization are expected to have quite different behaviors at high temperatures, different from our knowledge on graphitization under conventional conditions. Since high electrical conductivity is required for many applications of nanoporous carbons, including catalyst supports and energy storage, high temperature treatment of templated carbons was demanded in order to get high conductivity without loss of functions (for example, high surface area). Heat treatment at high temperatures was carried out on silica-templated carbons, as explained before. For most of the silica-templated carbons, however, their surface area and pore volume decreased markedly by high temperature treatment, though their electrical conductivity increased.172 Also, the graphitization degree remained very low, as their interlayer spacing d002 did not become lower than 0.344 nm even using poly(vinyl chloride) as carbon precursor, which was known to give graphitizing carbon under conventional conditions.174 This low graphitization degree on the silica-templated carbons is probably because thermosetting resins, such as phenol resin, poly(acrylonitrile), etc., were used as carbon precursor. When the mesophase pitch was used as carbon precursor, interlayer spacing decreased noticeably, but the SBET became 115 m2 g−1 after 2500 °C and no ordering of the mesopores was observed.174Catalytic graphitization of a mesoporous carbon prepared by the impregnation of metal (Fe, Ni or Mn) was tried.177 However, it has to be pointed out that the mesoporous structure established during templating is expected to be completely destroyed during catalytic graphitization, even though the heat treatment temperature applied is 900 °C, which is much lower than the temperature that the metal catalyst accelerates graphitization. By using ethanol solution of phenol resin with nickel chloride, polystyrene as macropore-forming agent and triblock copolymer F127 as mesopore-forming agent, porous carbon was prepared through calcination at 600 °C.245 The carbon heat-treated at 1000 °C was reported to be a mixture of graphite with amorphous carbon, in addition of metallic Ni. Although metallic Ni is known to act as catalyst for graphite formation even at 1000 °C,246 it has to be pointed out that the graphite is formed as a separated phase from the pristine carbon. More detailed analysis of the structure is strongly desired.
Since the carbonization behavior of the precursors in a constrained space of the template is quite different, as discussed above, it is difficult to predict the graphitization behavior of various templated carbons at a high temperature above 2500 °C. Fundamental and detailed studies on nanotexture formation during template carbonization and on their structural evolution with heat treatment temperature are strongly desired. Also, it might be necessary to optimize the selections on carbon precursor and heat treatment temperature in order to fulfil the requirements in graphitization degree (in most cases, electrical conductivity) and pore structure.
Acknowledgements
We express our sincere thanks to Prof. T. Kyotani of Tohoku University for his kind cooperation and discussion on this review.References
- T. Kyotani, N. Sonobe and A. Tomita, Nature, 1988, 331, 331–333 CrossRef.
- J. W. Patrick, Porosity in carbons, ed. Edward Arnold, London, 1995 Search PubMed.
- M. Inagaki, New carbons, Control of structure and functions, Elsevier, Amsterdam, 2000, pp. 124–145 Search PubMed.
- H. Marsh, M. A. D. Diaz-Estebanez, Sciences of carbon materials, ed. H. Marsh and F. Rodriguez-Reinoso, Universidad de Alicante, Alicante, 2000353–377 Search PubMed.
- T. Kyotani, Carbon alloys, ed. Yasuda Eet al., Elsevier, Amsterdam, 2003, 109–127 Search PubMed.
- M. Inagaki, F. Kang, Carbon Materials Science and Engineering, Tsinghua Univ. Press, Beijing, 2006, 184–225 Search PubMed.
- E. J. Bottani, Adsorption by Carbons, ed. J. M. D. Tascon, Elsevier, Amsterdam, 2008 Search PubMed.
- M. Inagaki, K. Kaneko and T. Nishizawa, Carbon, 2004, 42, 1401–1417 CrossRef CAS.
- M. Inagaki, New Carbon Mater., 2009, 24, 193–222 CrossRef CAS.
- T. Kyotani, B. K. Pradhan and A. Tomita, Bull. Chem. Soc. Jpn., 1999, 72, 1957–1970 CrossRef CAS.
- B. Sakintunas and Y. Yueruem, Ind. Eng. Chem. Res., 2005, 44, 2893–2902 CrossRef.
- A.-H. Lu and F. Schueth, Adv. Mater., 2006, 18, 1793–1805 CrossRef CAS.
- J. Lee, J. Kim and T. Hyeon, Adv. Mater., 2006, 18, 2073–2094 CrossRef CAS.
- T. Kyotani, Bull. Chem. Soc. Jpn., 2006, 79, 1322–1337 CrossRef CAS.
- F. Su, Z. Zhou, W. Guo, J. Liu, X. N. Tian and X. S. Zhao, Chemistry and Physic of Carbon, , ed. Radovic L 2008, 30, pp. 64–128 Search PubMed.
- T. Kyotani, Carbon, 2000, 38, 269–286 CrossRef CAS.
- A. G. Pandolfo and A. H. Hollenkamp, J. Power Sources, 2006, 157, 11–27 CrossRef CAS.
- A. Stein, Z. Wang and M. A. Fierke, Adv. Mater., 2009, 21, 265–293 CrossRef CAS.
- M. Inagaki, H. Konno and O. Tanaike, J. Power Sources, 2010, 195, 7880–7903 CrossRef CAS.
- N. Sonobe, T. Kyotani and A. Tomita, Carbon, 1988, 26, 573–578 CrossRef CAS.
- N. Sonobe, T. Kyotani, Y. Hishiyama, M. Shiraishi and A. Tomita, J. Phys. Chem., 1988, 92, 7029–7034 CrossRef CAS.
- N. Sonobe, T. Kyotani and A. Tomita, Carbon, 1990, 28, 483–488 CrossRef CAS.
- N. Sonobe, T. Kyotani and A. Tomita, Carbon, 1991, 29, 61–67 CrossRef CAS.
- T. Kyotani, N. Sonobe and A. Tomita, Tanso, 1992, 1992(No.155), 301–306 Search PubMed.
- T. Kyotani, H. Yamada, N. Sonobe and A. Tomita, Carbon, 1994, 32, 627–635 CrossRef CAS.
- T. Kyotani, T. Mori and A. Tomita, Chem. Mater., 1994, 6, 2138–2142 CrossRef CAS.
- N. Sonobe, T. Kyotani, A. Tomita and Y. Hishiyama, Tanso, 1990, 1990(No.141), 38–44 Search PubMed (in Japanese).
- T. Kyotani, K. Suzuki, N. Sonobe, A. Tomita, Y. Chida and R. Hara, Carbon, 1993, 31, 149–153 CrossRef CAS.
- T. Kyotani, L. Tsai and A. Tomita, Chem. Mater., 1995, 7, 1427–1428 CrossRef CAS.
- J. Li, M. Moskovits and T. L. Haslett, Chem. Mater., 1998, 10, 1963–1967 CrossRef CAS.
- G. Che, B. B. Lakshmi, C. R. Marth and E. R. Fisher, Chem. Mater., 1998, 10, 260–267 CrossRef CAS.
- G. Che, B. B. Lakshmi, E. R. Fisher and C. R. Martin, Nature, 1998, 393, 346–349 CrossRef CAS.
- J. S. Suh and J. S. Lee, Appl. Phys. Lett., 1999, 75, 2047–2049 CrossRef CAS.
- J. Li, C. Papadopoulos, J. M. Xu and M. Moskovits, Appl. Phys. Lett., 1999, 75, 367–369 CrossRef CAS.
- J. Li, C. Papadopoulos and J. Xu, Nature, 1999, 402, 253–254 CAS.
- J. S. Lee, G. H. Gu, H. Kim, K. S. Jeong, J. Bae and J. S. Suh, Chem. Mater., 2001, 13, 2387–2391 CrossRef CAS.
- J. Matsui, M. Iko, N. InokumaN, H. Orikasa, M. Mitsuishi, T. Kyotani and T. Miyashita, Chem. Lett., 2006, 35, 42–43 CrossRef CAS.
- J. Matsui, K. Yamamoto, N. Inokuma, H. Orikasa, T. Kyotani and T. Miyashita, J. Mater. Chem., 2007, 17, 3806–3811 RSC.
- T. Kyotani, L. Tsai and A. Tomita, Chem. Commun., 1997, 701–702 RSC.
- G. Che, B. B. Lakshml, C. R. Martin and E. R. Fisher, Langmuir, 1999, 15, 750–758 CrossRef CAS.
- B. K. Pradhan, T. Kyotani and A. Tomita, Chem. Commun., 1999, 1317–1318 RSC.
- B. K. Pradhan, T. Toba, T. Kyotani and A. Tomita, Chem. Mater., 1998, 10, 2510–2515 CrossRef CAS.
- K. Matsui, B. K. Pradhan, T. Kyotani and A. Tomita, J. Phys. Chem. B, 2001, 105, 5682–5688 CrossRef CAS.
- K. Matsui, T. Kyotani and A. Tomita, Adv. Mater., 2002, 14, 1216–1219 CrossRef CAS.
- H. Orikasa, J. Karoji, K. Matsui and T. Kyotani, Dalton Trans., 2007,(34), 3757–3762 RSC.
- K.-H. Kim, H. Orikasa, T. Kyotani and M. Yamaguchi, IEEE Trans. Magn., 2005, 41, 4075–4077 CrossRef.
- X-H. Wang, H. Orikasa, N. Inokuma, Q. -H. Yang, P.-X. Hou, H. Oshima, K. Itoh and T. Kyotani, J. Mater. Chem., 2007, 17, 986–991 RSC.
- K.-H. Kim, M. Yamaguchi, H. Orikasa and T. Kyotani, Solid State Commun., 2006, 140, 491–494 CrossRef CAS.
- Y. Hattori, Y. Watanabe, S. Kawasaki, F. Okino, B. K. Pradhan, T. Kyotani and A. Tomita, Carbon, 1999, 37, 1033–1038 CrossRef CAS.
- H. Touhara and F. Okino, Carbon, 2000, 38, 241–267 CrossRef CAS.
- H. Touhara, J. Inahara, T. Mizuno, Y. Yokoyama, S. Okanao, K. Yanagiuch, I. Mukopadhyay, S. Kawasaki, F. Okinoa, H. Shirai, W.-H. Xu, T. Kyotani and A. Tomita, J. Fluorine Chem., 2002, 114, 181–188 CrossRef CAS.
- T. Kyotani, S. Nakazaki, W. H. Xu and A. Tomita, Carbon, 2001, 39, 782–785 CrossRef CAS.
- H. Orikasa, N. Inokuma, S. Okubo, O. Kitakami and T. Kyotani, Chem. Mater., 2006, 18, 1036–1040 CrossRef CAS.
- H. Orikasa, N. Inokuma, S. Ittisanronnachai, X.-H. Wang, O. Kitakami and T. Kyotani, Chem. Commun., 2008, 2215–2217 RSC.
- S. Ittisanronnachai, H. Orikasa, N. Inokuma, Y. Uozu and T. Kyotani, Carbon, 2008, 46, 1361–1363 CrossRef CAS.
- M. Vijayaraj, R. Gadiou, K. Anselme, J. Dentzer, C. Vix-Guterl, H. Orikasa, T. Kyotani and S. Ittisanronnachai, Adv. Funct. Mater., 2010, 20, 2489–2499 CrossRef CAS.
- W.-H. Xu, T. Kyotani, B.K. Pradhan, T. Nakajima and A. Tomita, Adv. Mater., 2003, 15, 1087–1090 CrossRef CAS.
- Q.-H. Yang, W.-H. Xu, A. Tomita and T. Kyotani, Chem. Mater., 2005, 17, 2940–2945 CrossRef CAS.
- Q. Yang, W. Xu, A. Tomita and T. Kyotani, J. Am. Chem. Soc., 2005, 127, 8956–8956 CrossRef CAS.
- Q.-H. Yang, P.-X. Hou, M. Unno, S. Yamauchi, R. Saito and T. Kyotani, Nano Lett., 2005, 5, 2465–2469 CrossRef CAS.
- T. Kyotani, W.-H. Xu, Y. Yokoyama, J. Inahara, H. Touhara and A. Tomita, J. Membr. Sci., 2002, 196, 231–239 CrossRef CAS.
- J. P. Tu, L. P. Zhu, K. Hou and S. Y. Guo, Carbon, 2003, 41, 1257–1263 CrossRef CAS.
- A. Huczko, Appl. Phys. A: Mater. Sci. Process., 2002, 74, 617–638 CrossRef CAS.
- T. Kyotani, L. Tsai and A. Tomita, Chem. Mater., 1996, 8, 2109–2113 CrossRef CAS.
- S. Rahman and H. Yang, Nano Lett., 2003, 3, 439–442 CrossRef CAS.
- K. Jian, H. S. Shim, A. Schwartzman, G. P. Crawford and R. H. Hurt, Adv. Mater., 2003, 15, 164–167 CrossRef CAS.
- C. Chan, G. Crawford, Y. Gao, R. Hurt, K. Jian, H. Li, B. Sheldon, M. Sousa and N. Yang, Carbon, 2005, 43, 2431–2440 CrossRef CAS.
- L. Zhi, J. Wu, J. Li, U. Kolb and K. Muellen, Angew. Chem., Int. Ed., 2005, 44, 2120–2123 CrossRef CAS.
- H. Konno, S. Sato, H. Habazaki and M. Inagaki, Carbon, 2004, 42, 2756–2759 CrossRef CAS.
- H. Orikasa, T. Akahane, M. Okada, Y. Tong, J. Ozaki and T. Kyotani, J. Mater. Chem., 2009, 19, 4615–4621 RSC.
- B. J. Holliday and C. A. Mirkin, Angew. Chem., Int. Ed., 2001, 40, 2022–2043 CrossRef CAS.
- J. Jang and J. Bae, Angew. Chem., Int. Ed., 2004, 43, 3803–3806 CrossRef CAS.
- N. Nishiyama, T. Zheng, Y. Yamane, Y. Egashira and K. Ueyama, Carbon, 2005, 43, 269–274 CrossRef CAS.
- D. Fujikawa, M. Uota, T. Yoshimura, G. Sakai and T. Kijima, Chem. Lett., 2006, 35, 432–433 CrossRef CAS.
- D. Fujikawa, M. Uota, G. Sakai and T. Kijima, Carbon, 2007, 45, 1289–1295 CrossRef CAS.
- T. Kyotani and A. Tomita, J. Jpn. Petrol. Inst., 2002, 45, 261–270 CAS.
- T. Kyotani, T. Nagai, S. Inoue and A. Tomita, Chem. Mater., 1997, 9, 609–615 CrossRef CAS.
- P. Enzel and T. Bein, Chem. Mater., 1992, 4, 819–824 CrossRef CAS.
- S. A. Johnson, E. S. Brigham, P. J. Ollivier and T. E. Mallouk, Chem. Mater., 1997, 9, 2448–2458 CrossRef CAS.
- T. Cordero, P. A. Thrower and L. R. Radivic, Carbon, 1992, 30, 365–374 CrossRef CAS.
- J. Rodriguez-Mirasol, T. Cordero, L. R. Radovic and J. J. Rodriguez, Chem. Mater., 1998, 10, 550–558 CrossRef CAS.
- Z. X. Ma, T. Kyotani and A. Tomita, Chem. Commun., 2000, 2365–2366 RSC.
- Z. X. Ma, T. Kyotani, Z. Liu, O. Terasaki and A. Tomita, Chem. Mater., 2001, 13, 4413–4415 CrossRef CAS.
- Z. Ma, T. Kyotani and A. Tomita, Carbon, 2002, 40, 2367–2374 CrossRef CAS.
- K. Matsuoka, Y. Yamagishi, T. Yamazaki, N. Setoyama, A. Tomita and T. Kyotani, Carbon, 2005, 43, 876–879 CrossRef CAS.
- T. Kyotani, Z. Ma and A. Tomita, Carbon, 2003, 41, 1451–1459 CrossRef CAS.
- F. Su, X. S. Zhao, L. Lv and Z. Zhou, Carbon, 2004, 42, 2821–2831 CrossRef CAS.
- A. Garsuch, O. Klepel, R. R. Sattler, C. Berger, R. Glaeser and J. Weitkamp, Carbon, 2006, 44, 593–596 CrossRef CAS.
- F. O. M. Gaslain, J. Parmentier, V. P. Valtchev and J. Patarin, Chem. Commun., 2006, 991–993 RSC.
- P.-X. Hou, T. Yamazaki, H. Orikasa and T. Kyotani, Carbon, 2005, 43, 2624–2627 CrossRef CAS.
- P.-X. Hou, H. Orikasa, H. Itoi, H. Nishihara and T. Kyotani, Carbon, 2007, 45, 2011–2018 CrossRef CAS.
- H. Nishiyama, Q.-H. Yang, P.-X. Hou, M. Unno, S. Yamauchi, R. Saito, J. I. Paredes, A. Marinez-Alonso, J. M. D. Tascon, Y. Sato, M. Terauchi and T. Kyotani, Carbon, 2009, 47, 1220–1230 CrossRef.
- K. Takai, T. Suzuki, T. Enoki, H. Nishihara and T. Kyotani, J. Phys. Chem. Solids, 2010, 71, 565–568 CrossRef CAS.
- Y. Kopelevich, R. R. da Silva, J. H. S. Torres, A. Penicaud and T. Kyotani, Phys. Rev. B: Condens. Matter, 2003, 68, 092408 CrossRef.
- P.-X. Hou, H. Orikasa, T. Yamazaki, K. Matsuoka, A. Tomita, N. Setoyama, Y. Fukushima and T. Kyotani, Chem. Mater., 2005, 17, 5187–5193 CrossRef CAS.
- Z. Yang, Y. Xia, X. Sun and R. Mokaya, J. Phys. Chem. B, 2006, 110, 18424–18431 CrossRef CAS.
- Z. Yang, Y. Xia and R. Mokaya, J. Am. Chem. Soc., 2007, 129, 1673–1679 CrossRef CAS.
- M. Kayanuma, U. Nagashima, H. Nishihara, T. Kyotani and H. Ogawa, Chem. Phys. Lett., 2010, 495, 251–255 CrossRef CAS.
- K. Takai, T. Suzuki, T. Enoki, H. Nishihara and T. Kyotani, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 205420 CrossRef.
- Y. Nakashima, T. Matsushita, M. Hieda, N. Wada, H. Nishihara and T. Kyotani, J. Low Temp. Phys., 2011, 162, 565–572 CrossRef CAS.
- F. Su, J. Zeng, Y. Yu, L. Lv, J. Y. Lee and X. S. Zhao, Carbon, 2005, 43, 2366–2373 CrossRef CAS.
- E. N. Coker, W. A. Steen, J. T. Miller, J. Kropf and J. E. Miller, J. Mater. Chem., 2007, 17, 3330–3340 RSC.
- C. O. Ania, V. Khomenko, E. Raymundo-Pinero, J. B. Parra and F. Beguin, Adv. Funct. Mater., 2007, 17, 1828–1836 CrossRef CAS.
- H. Nishihara, H. Itoi, T. Kogure, P. Hou, H. Touhara, F. Okino and T. Kyotani, Chem.–Eur. J., 2009, 15, 5355–5363 CrossRef CAS.
- C. Portet, Z. Yang, K. Y. Gogotsi, R. Mokaya and G. Yushin, J. Electrochem. Soc., 2009, 156, A1–A6 CrossRef CAS.
- T. Kwon, H. Nishihara, H. Itoi, Q.-H. Yang and T. Kyotani, Langmuir, 2009, 25, 11961–11966 CrossRef CAS.
- H. Itoi, H. Nishihara, T. Kogure and T. Kyotani, J. Am. Chem. Soc., 2011, 133, 1165–1167 CrossRef CAS.
- R. Ryoo, S. H. Joo and S. Jun, J. Phys. Chem. B, 1999, 103, 7743–7746 CrossRef CAS.
- J. Lee, S. Yoon, T. Hyeon, S. M. Oh and K. B. Kim, Chem. Commun., 1999, 2177–2178 RSC.
- J. Lee, S. Yoon, S. M. Oh, C.-H. Shin and T. Hyeon, Adv. Mater., 2000, 12, 359–362 CrossRef CAS.
- M. Kruk, M. Jaroniec, R. Ryoo and S. H. Joo, J. Phys. Chem. B, 2000, 104, 7960–7968 CrossRef CAS.
- S. Jun, S. H. Joo, R. Ryoo, M. Kruk, M. Jaroniec, Z. Liu, T. Ohsuna and O. Terasaki, J. Am. Chem. Soc., 2000, 122, 10712–10713 CrossRef CAS.
- S. H. Joo, S. J. Choi, I. Oh, J. Kwak, Z. Kiu, O. Terasaki and R. Ryoo, Nature, 2001, 412, 169–172 CrossRef CAS.
- H. J. Shin, R. Ryoo, M. Kruk and M. Jaroniec, Chem. Commun., 2001, 349–350 RSC.
- S. H. Joo, S. Jun and R. Ryoo, Microporous Mesoporous Mater., 2001, 44–45, 153–158 CrossRef CAS.
- S. B. Yoon, J. Y. Kim and J.-S. Yu, Chem. Commun., 2001, 559–560 RSC.
- S.-S. Kim and T. J. Pinnavaia, Chem. Commun., 2001, 2418–2419 RSC.
- J. S. Lee, S. H. Joo and R. Ryoo, J. Am. Chem. Soc., 2002, 124, 1156 CrossRef CAS.
- H. Yang, Q. Shi, X. Liu, S. Xie, D. Jiang, F. Zhang, C. Yu, B. Tu and D. Zhao, Chem. Commun., 2002, 2842–2843 RSC.
- M. Kaneda, T. Tsubakiyama, A. Carisson, Y. Sakamoto, T. Ohsuna, O. Terasaki, S. H. Joo and R. Ryoo, J. Phys. Chem. B, 2002, 106, 1256–1266 CrossRef CAS.
- C. Yu, J. Fan, B. Tian, D. Zhao and G. D. Stucky, Adv. Mater., 2002, 14, 1742–1745 CrossRef CAS.
- C. Vix-Guterl, S. Boulard, J. Parmentier, J. Weckmann and J. Patarin, Chem. Lett., 2002, 1062–1063 CrossRef CAS.
- T. W. Kim, I. S. Park and R. Ryoo, Angew. Chem., Int. Ed., 2003, 42, 4375–4379 CrossRef CAS.
- B. Tian, S. Che, Z. Liu, X. Liu, W. Fan, T. Tatsumi, O. Terasaki and D. Zhao, Chem. Commun., 2003, 2726–2727 RSC.
- C. Vix-Guterl, S. Saadallah, L. Vidal, M. Reda, J. Parmentier and J. Patarin, J. Mater. Chem., 2003, 13, 2535–2539 RSC.
- J. Parmentier, C. Vix-Guterl, P. Gibot, M. Reda, M. Ilescu, J. Werckmann and J. Patarin, Microporous Mesoporous Mater., 2003, 62, 87–96 CrossRef CAS.
- A. B. Fuertes and D. M. Nevskaia, Microporous Mesoporous Mater., 2003, 62, 177–190 CrossRef CAS.
- S. Alvarez and A. B. Fuertes, Carbon, 2004, 42, 433–436 CrossRef CAS.
- Y. Xia and R. Mokaya, Adv. Mater., 2004, 16, 1553–1558 CrossRef CAS.
- A.-H. Lu, W.-C. Li, W. Schmidt and F. Schueth, Microporous Mesoporous Mater., 2006, 95, 187–192 CrossRef CAS.
- H. I. Lee, J. H. Kim, D. J. You, J. E. Lee, J. M. Lim, W.-S. Ahn, C. Pak, S.H. Joo, H. Chang and D. Seung, Adv. Mater., 2008, 20, 757–762 CrossRef CAS.
- R. Ryoo, S. H. Joo, M. Kruk and M. Jaroniec, Adv. Mater., 2001, 13, 677–681 CrossRef CAS.
- J. Lee, S. Han and T. Hyeon, J. Mater. Chem., 2004, 14, 478–486 RSC.
- M. T. Gilbert, J. H. Knox and B. Kaur, Chromatographia, 1982, 16, 138–146 CAS.
- A. A. Zakhidov, R. H. Baughman, Z. Iqbal, C. Cui, I. Khayrullin, S. O. Dantes, J. Marti and G. Ralchenko, Science, 1998, 282, 897–901 CrossRef CAS.
- S. Han and T. Hyeon, Chem. Commun., 1999, 1955–1956 RSC.
- D. Kawashima, T.. Aihara, Y. Kobayashi, T. Kyotani and A. Tomita, Chem. Mater., 2000, 12, 3397–3401 CrossRef CAS.
- Z. Li and M. Jaroniec, J. Am. Chem. Soc., 2001, 123, 9208–9209 CrossRef CAS.
- J. Pang, X. Li, D. Wang, Z. Wu, V. T. John, Z. Yang and Y. Lu, Adv. Mater., 2004, 16, 884–886 CrossRef CAS.
- S. Han, K. T. Lee, M. Oh and T. Hyeon, Carbon, 2003, 41, 1049–1056 CrossRef CAS.
- A. Taguchi, J. H. Small and M. Linden, Adv. Mater., 2003, 15, 1209–1211 CrossRef CAS.
- B.-H. Han, W. Zhou and A. Sayant, J. Am. Chem. Soc., 2003, 125, 3444–3445 CrossRef CAS.
- K. P. Gierszal, M. Jaroniec, C. Liang and S. Dai, Carbon, 2007, 45, 2171–2177 CrossRef CAS.
- M. Kruk, B. Dufour, E. B. Celer, T. Kowalewski, M. Jaroniec and K. Matyjaszewski, J. Phys. Chem. B, 2005, 109, 9216–9225 CrossRef CAS.
- S-W. Woo, K. Dokko, H. Nakano and K. Kanamura, J. Mater. Chem., 2008, 18, 1674–1680 RSC.
- D. Gu, H. Bongard, Y. Deng, D. Feng, Z. Wu, Y. Feng, J. Mao, B. Tu, F. Schueth and D. Zhao, Adv. Mater., 2010, 22, 833–837 CrossRef CAS.
- J. Lee, K. Sohn and T. Hyeon, J. Am. Chem. Soc., 2001, 123, 5146–5147 CrossRef CAS.
- S. Han, K. Sohn and T. Hyeon, Chem. Mater., 2000, 12, 3337–3341 CrossRef CAS.
- A. Vinu, C. Streb, V. Murugesan and M. Hartmann, J. Phys. Chem. B, 2003, 107, 8297–8299 CrossRef CAS.
- M. Choi and R. Ryoo, Nat. Mater., 2003, 2, 473–476 CrossRef CAS.
- D.-W. Wang, F. Li, G.Q. Lu and H.-M. Cheng, Carbon, 2008, 46, 1593–1599 CrossRef CAS.
- K. Xia, Q. Gao, C. Wu, S. Song and M. Ruan, Carbon, 2007, 45, 1989–1996 CrossRef CAS.
- C. Pevida, T. C. Drage and C. E. Snape, Carbon, 2008, 46, 1464–1474 CrossRef CAS.
- A.-H. Lu, W.-C. Li, Z. Hou and F. Schueth, Chem. Commun., 2007, 1038–1040 RSC.
- A.-H. Lu, W. Schmidt, A. Taguchi, B. Spliethoff, B. Tesche and F. Schueth, Angew. Chem., Int. Ed., 2002, 41, 3489–3492 CrossRef CAS.
- M. Kang, S. H. Yi, H. I. Lee, J. E. Yie and J. M. Kim, Chem. Commun., 2002, 1944–1945 RSC.
- J. Parmentier, C. Vix-Guterl, S. Saadallah, M. Reda, M. Ilescu, J. Werckmann and J. Patarin, Chem. Lett., 2003, 32, 262–263 CrossRef CAS.
- S. S. Kim, L. Shath and T. J. Pinnavaia, Chem. Mater., 2003, 15, 1664–1668 CrossRef CAS.
- A. Sakthivel, S.-J. Huang, W.-H. Chen, Z.-H. Lan, K.-H. Chen, T.-W. Kim, R. Ryoo, A. S. T. Chiang and S.-B. Liu, Chem. Mater., 2004, 16, 3168–3175 CrossRef CAS.
- S. Yoon, J. Lee, T. Hyeon and S. M. Oh, J. Electrochem. Soc., 2000, 147, 2507–2512 CrossRef CAS.
- H. Zhou, S. Zhu, M. Hibino and I. Honma, J. Power Sources, 2003, 122, 219–223 CrossRef CAS.
- K. Jurewicz, C. Vix-Guterl, E. Frackowiak, S. Saadallah, M. Reda, J. Parmentier, J. Patarin and F. Beguin, J. Phys. Chem. Solids, 2004, 65, 287–293 CrossRef CAS.
- C. Vix-Guterl, S. Saadallah, K. Jurewicz, E. Frackowiak, M. Reda, J. Parmentier, J. Patarin and F. Beguin, Mater. Sci. Eng., B, 2004, 108, 148–155 CrossRef.
- A. B. Fuertes, F. Pico and J. M. Rojo, J. Power Sources, 2004, 133, 329–336 CrossRef CAS.
- A. B. Fuertes, G. Lota, T. A. Centeno and E. Frackowiak, Electrochim. Acta, 2005, 50, 2799–2805 CrossRef CAS.
- T. A. Centeno, M. Sevilla, A. B. Fuertes and F. Stoeckli, Carbon, 2005, 43, 3012–3015 CrossRef CAS.
- L. Li, H. Song and X. Chen, Electrochim. Acta, 2006, 51, 5715–5720 CrossRef CAS.
- S. Alvarez, M. C. Blanco-Lopez, A. J. Miranda-Ordieres, A. B. Fuertes and T. A. Centeno, Carbon, 2005, 43, 866–870 CrossRef CAS.
- W. Xing, S. Z. Qiao, R. G. Ding, F. Li, G. Q. Lu, Z. F. Yan and H. M. Cheng, Carbon, 2006, 44, 216–224 CrossRef CAS.
- M. Sevilla, S. Alvarez, T. A. Centeno, A. B. Fuertes and F. Stoeckli, Electrochim. Acta, 2007, 52, 3207–3215 CrossRef CAS.
- K. Xia, Q. Gao, J. Jiang and J. Hu, Carbon, 2008, 46, 1718–1726 CrossRef CAS.
- D. Banham, F. Feng, J. Burt, E. Alsrayheen and V. Birss, Carbon, 2010, 48, 1056–1063 CrossRef CAS.
- Z. Li, M. Jaroniec, Y.-J. Lee and R. Radovic, Chem. Commun., 2002, 1346–1347 RSC.
- A. B. Fuertes and S. Alvarez, Carbon, 2004, 42, 3049–3055 CrossRef CAS.
- S. B. Yoon, G. S. Chai, S. K. Kang, J.-S. Yu, K.P. Gierszai and M. Jaroniec, J. Am. Chem. Soc., 2005, 127, 4148–4149 CrossRef.
- R. Gadiou, A. Didion, S.-E. Saadallah, M. Couzi, J.-N. Rouzaud, P. Delhaes and C. Vix-guterl, Carbon, 2006, 44, 3348–3352 CrossRef CAS.
- M. Sevilla and A. B. Fuertes, Carbon, 2006, 44, 468–474 CrossRef CAS.
- M. Inagaki, S. Kobayashi, F. Kojin, N. Tanaka, T. Morishita and B. Tryba, Carbon, 2004, 42, 3153–3158 CrossRef CAS.
- T. Morishita, T. Suzuki, T. Nishikara, T. Tsumura and M. Inagaki, Tanso, 2005, 226–231 CAS [in Japanese]..
- T. Morishita, T. Suzuki, T. Nishikawa, T. Tsumura and M. Inagaki, Tanso, 2006, 220–226 CAS [in Japanese]..
- T. Morishita, Y. Soneda, T. Tsumura and M. Inagaki, Carbon, 2006, 44, 2360–2367 CrossRef CAS.
- T. Morishita, K. Ishihara, M. Kato, T. Tsumura and M. Inagaki, Carbon, 2007, 45, 209–211 CrossRef CAS.
- T. Morishita, K. Ishihara, M. Kato and M. Inagaki, Tanso, 2007, 19–24 CAS [in Japanese].
- M. Inagaki, M. Kato, T. Morishita, K. Morita and K. Mizuuchi, Carbon, 2007, 45, 1121–1124 CrossRef CAS.
- J. Przepiórski, J. Karolczyk, K. Takeda, T. Tsumura, M. Toyoda and A. W. Morawski, Ind. Eng. Chem. Res., 2009, 48, 7110–7116 CrossRef.
- H. Konno, H. Onishi, N. Yoshizawa and K. Azumi, J. Power Sources, 2010, 195, 667–673 CrossRef CAS.
- T. Morishita, T. Tsumura, M. Toyoda, J. Przepiórski, A. W. Morawski, H. Konno and M. Inagaki, Carbon, 2010, 48, 2690–2707 CrossRef CAS.
- T. Morishita, L. Wang, T. Tsumura, M. Toyoda and H. Konno, Tanso, 2010,(No. 242), 60–68 CAS [in Japanese].
- M.-J. Jung, J. S. Im, E. Jeong, H. Jin and Y.-S. Lee, Carbon Lett., 2009, 10, 217–220 Search PubMed.
- T. Morishita, M. Hirabayashi, Y. Nishioka, T. Okuni, N. Ota and M. Inagaki, J. Power Sources, 2006, 160, 638–644 CrossRef CAS.
- J. A. Fernandez, T. Morishita, M. Toyoda, M. Inagaki, F. Stoeckli and T. A. Centeno, J. Power Sources, 2008, 175, 675–679 CrossRef CAS.
- L. Wang, T. Morishita, M. Toyoda and M. Inagaki, Electrochim. Acta, 2007, 53, 882–886 CrossRef CAS.
- L. Wang, M. Toyoda and M. Inagaki, Adsorpt. Sci. Technol., 2008, 26, 491–495 CrossRef CAS.
- L. Wang, M. Inagaki and M. Toyoda, Tanso, 2009,(No. 240), 230–238 Search PubMed [in Japanese].
- J. Zhou, X. Yuan, W. Xing, W. Si and S. Zhuo, New Carbon Mater., 2010, 25, 370–375 CrossRef CAS.
- Y. Wang and C. Wang, New Carbon Mater., 2010, 25, 376–381 CrossRef CAS.
- M. Inagaki, H. Miura and H. Konno, J. Eur. Ceram. Soc., 1998, 18, 1011–1015 CrossRef CAS.
- M. Inagaki, Y. Okada, H. Miura and H. Konno, Carbon, 1999, 37, 329–334 CrossRef CAS.
- T. Tsumura, N. Kojitani, I. Izumi, N. Iwashita, M. Toyoda and M. Inagaki, J. Mater. Chem., 2002, 12, 1391–1396 RSC.
- B. Tryba, T. Tsumura, M. Junas, A. W. Morawski and M. Inagaki, Appl. Catal., B, 2004, 50, 177–183 CrossRef CAS.
- B. Tryba, A. W. Morawski, T. Tsumura, M. Toyoda and M. Inagaki, J. Photochem. Photobiol., A, 2004, 167, 127–135 CrossRef CAS.
- D. W. Wang, F. Li, M. Liu, G. Q. Lu and H. M. Cheng, Angew. Chem., Int. Ed., 2008, 47, 373–376 CrossRef CAS.
- C. A. Filho and A. J. G. Zarbin, Carbon, 2006, 44, 2869–2876 CrossRef.
- J. Zhou, X. Yuan, W. Xing, W. Si and S. Zhuo, Carbon, 2010, 48, 2765–2772 CrossRef CAS.
- C. Liang, K. Hong, G. A. Guiochon, J. W. Mays and S. Dai, Angew. Chem., Int. Ed., 2004, 43, 5785–5789 CrossRef CAS.
- H. Kosonen, S. Valkama, A. Nykaenen, M. Toivanen, G. T. Brinke, J. Ruokolainen and O. Ikkala, Adv. Mater., 2006, 18, 201–205 CrossRef CAS.
- S. Tanaka, N. Nishiyama, Y. Egashira and K. Ueyama, Chem. Commun., 2005, 2125–2127 RSC.
- Y. Meng, D. Gu, F. Zhang, Y. Shi, H. Yang, Z. Li, C. Yu, B. Tu and D. Zhao, Angew. Chem., Int. Ed., 2005, 44, 7053–7059 CrossRef CAS.
- F. Q. Zhang, Y. Meng, D. Gu, Y. Yan, C. Z. Yu, B. Tu and D. Y. Zhao, J. Am. Chem. Soc., 2005, 127, 13508–13509 CrossRef CAS.
- S. Tanaka, Y. Katayama, M. P. Tate, H. W. Hillhouse and Y. Miyake, J. Mater. Chem., 2007, 17, 3639–3645 RSC.
- C. Liang and S. Dai, Chem. Mater., 2009, 21, 2115–2124 CrossRef CAS.
- C. D. Liang and S. Dai, J. Am. Chem. Soc., 2006, 128, 5316–5317 CrossRef CAS.
- T. Kowalewski, N. V. Tsarevsky and K. Matyjaszewski, J. Am. Chem. Soc., 2002, 124, 10632–10633 CrossRef CAS.
- D. Carriazo, F. Pico, M. C. Gutierrez, F. Rubio, J. M. Rojo and F. del Monte, J. Mater. Chem., 2010, 20, 773–780 RSC.
- Y. Meng, D. Gu, F. Zhang. Y. Shi, L. Cheng, D. Feng, Z. Wu, Z. Chen, Y. Wan, A. Stein and D. Zhao, Chem. Mater., 2006, 18, 4447–4464 CrossRef CAS.
- F. Zhang, Y. Meng, D. Gu, Y. Yan, Z. Chen, B. Tu and D. Zhao, Chem. Mater., 2006, 18, 5279–5288 CrossRef CAS.
- J. Jin, N. Nishiyama, Y. Egashira and K. Ueyama, Microporous Mesoporous Mater., 2009, 118, 218–223 CrossRef CAS.
- X. Wang, C. Liang and S. Dai, Langmuir, 2008, 24, 7500–7505 CrossRef CAS.
- F. H. Simanjuntak, J. Jin, N. Nishiyama, Y. Egashira and K. Ueyama, Carbon, 2009, 47, 2531–2533 CrossRef CAS.
- J. Jin, N. Nishiyama, Y. Egashira and K. Ueyama, Chem. Commun., 2009, 1371–1373 RSC.
- L. Song, D. Feng, N. J. Fredin, K. G. Yager, R. L. Jones, Q. Wu, D. Zhao and B. D. Vogt, ACS Nano, 2010, 4, 189198 Search PubMed.
- Z. Wang, E. R. Kiesel and A. Stein, J. Mater. Chem., 2008, 18, 2194–2200 RSC.
- J. Gorka, A. Zawislak, J. Choma and M. Jaroniec, Carbon, 2008, 46, 1159–1161 CrossRef CAS.
- J. Jin, S. Tanaka, Y. Egashira and N. Nishiyama, Carbon, 2010, 48, 1985–1989 CrossRef CAS.
- K. Morishige, J. Phys. Chem. C, 2011, 115, 2720–2726 CAS.
- Y. Wang, P. T. M. Nguyen, N. Sakao, T. Horikawa, D. D. Do, K. Morishige and D. Nicholson, J. Phys. Chem. C, 2011, 115, 13361–13372 CAS.
- T. Yu, Y. Deng, L. Wang, R. Liu, L. Zhang, B. Tu and D. Zhao, Adv. Mater., 2007, 19, 2301–2306 CrossRef CAS.
- J. Gorka and M. Jaroniec, Carbon, 2011, 49, 154–160 CrossRef CAS.
- S. Tanaka, N. Nishitani, A. Doi and Y. Miyake, Carbon, 2011, 49, 3184–3189 CrossRef CAS.
- M. Inagaki, T. Morishita, A. Kuno, T. Kito, M. Hirano, T. Suwa and K. Kusakawa, Carbon, 2004, 42, 497–502 CrossRef CAS.
- N. Ohta, Y. Nishi, T. Morishita, Y. Ieko, A. Ito and M. Inagaki, New Carbon Mater., 2008, 23, 216–220 CrossRef CAS.
- G. Harikrishnan, T. U. Patro and D. V. Khakhar, Carbon, 2007, 45, 531–535 CrossRef CAS.
- Y. Chen, B. Chen, X. Shi, H. Xu, Y. Hu and Y. Yuan, Carbon, 2007, 45, 2132–2134 CrossRef CAS.
- R. Pekala and R. W. Hopper, J. Mater. Sci., 1987, 22, 1840–1844 CrossRef CAS.
- T. J. Bandoz, J. Jagiello, K. Putyera and J. A. Schwarz, Chem. Mater., 1996, 8, 2023–2029 CrossRef.
- C. Santos, M. Andrade, A. L. Vieira, A. Martins, J. Pires, C. Freire and A. P. Carvalho, Carbon, 2010, 48, 4049–4056 CrossRef CAS.
- D. Nguyen-Thanh and T. J. Bandosz, Microporous Mesoporous Mater., 2006, 92, 47 CrossRef CAS.
- P. M. Yeletsky, V. A. Yakovlev, M. S. Mel'gunov and V. N. Parmon, Microporous Mesoporous Mater., 2009, 121, 34–40 CrossRef CAS.
- M. Inagaki, Y. A. Kim and M. Endo, J. Mater. Chem., 2011, 21, 3280–3294 RSC.
- P. M. Barara-Rodrigues, T. J. Mays and G. D. Moggridge, Carbon, 2003, 41, 2231–2246 CrossRef.
- C. Meyers, S. D. Shah, S. C. Patel, R. M. Sneeringer, C. A. Bessel, N. R. Dollahon, R. A. Leising and E. S. Takeuchi, J. Phys. Chem. B, 2001, 105, 2143–2152 CrossRef CAS.
- M. Inagaki, Carbon, 1997, 35, 711–713 CrossRef CAS.
- K. Shen, Z.-H. Huang, L. Gan and F. Kang, Chem. Lett., 2009, 38, 90–91 CrossRef CAS.
- R. Hurt, G. Krammer, G. Crawford, K. Jian and C. Rulison, Chem. Mater., 2002, 14, 4558–4565 CrossRef CAS.
- C. Huang, R. Doong, D. Gu and D. Zhao, Carbon., 2011, 49, 3055–3064 CrossRef CAS.
- M. Inagaki, K. Fujita, Y. Takeuchi, K. Oshida, H. Iwata and H. Konno, Carbon, 2001, 39, 921–929 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |