The pyrolytic behavior of cellulose in lignocellulosic biomass: a review
Dekui
Shen
ab,
Rui
Xiao
*a,
Sai
Gu
b and
Kaihong
Luo
b
aSchool of Energy and Environment, Southeast University, Nanjing, China 210012. E-mail: 101011398@seu.edu.cn; Fax: +86-025-83795508; Tel: +86-025-83795726
bEnergy Technology Research Group, School of Engineering Sciences, University of Southampton, Highfield, Southampton, SO17 1BJ, UK. E-mail: ds1t07@soton.ac.uk; Fax: +44-023-89593232; Tel: +44-023-80596506
First published on 19th October 2011
Abstract
Pyrolysis is estimated to be one of the most promising methods to convert biomass to diverse products (such as syn-gas, liquid fuel and charcoal), while its application has the potential for alleviating the fossil fuel crisis and environmental deterioration. Cellulose, a linear homopolymer of glucopyranose residues linked by β-1, 4-glycosidic bonds, is the most principal component in biomass (accounting for more than 50% by weight). The research on the pyrolytic behavior of cellulose is particularly beneficial for achieving a better understanding of the pyrolytic behavior of biomass, also promoting its direct applications in terms of fuels, chemicals and bio-materials. The studies on pyrolysis of cellulose are extensively reported in the categories of the following four issues: (1) the physico-chemical properties of cellulose in lignocellulosic biomass; (2) the on-line pyrolysis study of cellulose; (3) the off-line pyrolysis study of cellulose; (4) the interactions with other chemical components under pyrolytic conditions. The information on pyrolysis of cellulose concerning the configuration of cellulose in biomass, the mass loss along with the evolution of volatiles against temperature, the yield of products, the proposed chemical pathways for cellulose decomposition and secondary cracking of the fragments would be vigorously discussed as well as the way-forward in this field, with thanks to the valuable contributions from the leading global researchers and their groups.
 Dekui Shen | Dekui Shen got his bachelor degree in “Thermal Dynamic Engineering” and double degree in “Communication Technology” at Zhejiang University in 2003. He obtained his PhD degree in “Thermo-physics” in State Key Laboratory of Clean Energy Utilization in Zhejiang University in 2008. Then, he started his post-doctoral research work in the Energy Technology Research Group at the University of Southampton. After two years, he got the position as associate professor from Southeast University, concentrating his research on thermo-chemical utilization of biomass and molecular dynamics simulation on the thermo-chemical process. |
 Rui Xiao | Rui Xiao got his bachelor degree in “Thermal Dynamic Engineering” at Xi’An Jiao Tong University in 1994 and his masters degree from the Institute of Thermal Engineering in Southeast University in 1997, then worked at the Southeast University as a lecturer. He achieved his PhD degree at Institute of Thermal Engineering in Southeast University in 2005. In 2006, he got the title of professor in Southeast University. He is an expert in the clean utilization of coal, CO2 capture and bio-fuels. |
1 Introduction
The utilization of lignocellulosic biomass (such as wood, logging residues, crops and agricultural waste), for producing energy and fuels or extracting chemical feedstock, is of globally-growing interest thanks to it being renewable, widely-distributed, indigenous and environmentally friendly compared to fossil fuels.1Pyrolysis technology is well-established as one of the most promising methods to convert biomass into different products (syn-gas, bio-liquid, char and chemicals). Its application may essentially diversify the energy-supply in many situations, leading to a more secure and sustainable global energy-supply structure in the future.2Cellulose, a linear homopolymer of glucopyranose residues linked by β-1,4-glycosidic bonds, is the most principal chemical component in different lignocellulosic biomass (accounting for more than 50% by weight), compared to the other two main components (hemicellulose and lignin). The study on pyrolysis of cellulose would be particularly beneficial for achieving a better understanding of the pyrolytic mechanism of biomass and facilitating its direct application in terms of fuels, chemicals and bio-materials (Fig. 1). Therefore, research concerning the pyrolysis of cellulose has been extensively reported in the literature during the past half-century and can be categorized into four fundamental issues (Fig. 1).
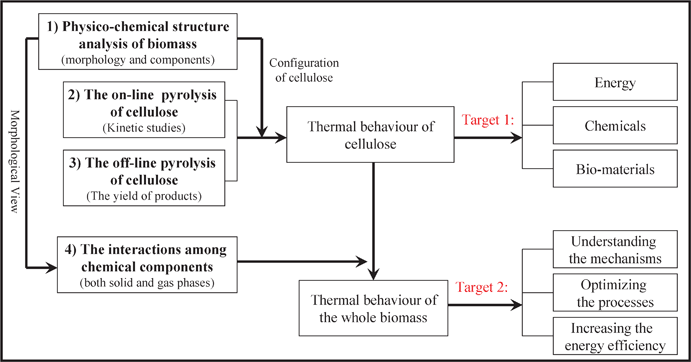 | ||
| Fig. 1 The fundamental issues and targets concerning the pyrolysis of cellulose. | ||
(1) The physico-chemical structural analysis of biomass is concerning the morphological analysis of the biomass cell-wall structure, the distribution and configuration of cellulose, which would facilitate not only the direct utilization of biomass as a bio-material, but also the improvement of the conversion processes of biomass to fuels or chemicals.
(2) The on-line pyrolysis study of cellulose is concentrated on the solid mass loss versus temperature or time (along with the evolution of the volatiles) and kinetic models, mostly employing isothermal and dynamic thermo-gravimetry analysis coupled with or without Fourier transformation infrared spectrometry (FTIR) or mass spectrometry (MS).
(3) The off-line pyrolysis study of cellulose is to examine the yield of the main products (gas, liquid and solid), variation of the compositions in gaseous or liquid products influenced by the intrinsic characteristics and experimental conditions, in order to optimize the pyrolysis process for energy and/or chemicals production.
(4) The interactions among the three main components under pyrolytic conditions is to introduce the possible interaction mechanism of the components in biomass, in terms of the mass loss process, the evolution of the volatiles and the yield of the specific products. This would help to improve the understanding of pyrolysis of the whole biomass system from the pyrolytic behavior of the individual components.
The studies of pyrolysis of cellulose concerning the above four fundamental issues is vigorously discussed in this work (especially for work reported during the past 25 years), where the way-forward for this field is also specified. This would supply a conceptual guide for the improvement of cellulose utilization and optimization of the thermal-conversion process of biomass.
2 The cell-wall structure of biomass and the configuration of cellulose
The morphological structure of lignocellulosic biomass has been studied regarding the distribution and inter-linkages of the chemical components, and their configuration.3,4 This facilitates not only a better understanding of the physico-chemical properties of biomass, but also the improvement of the conversion processes (such as pyrolysis) of biomass to fuels or chemicals.With a growing interest in lignocellulosic biomass as a potential substituent for fossil fuels, the pyrolysis of biomass should be dramatically examined. Consequently, the cell-wall model of lignocellulosic biomass, the distribution of the chemical components (especially cellulose), and the configuration of cellulose is discussed in the following sections, which will help understand the remarkable characteristics of cellulose pyrolysis and its interactions with two other main components (hemicellulose and lignin).
2.1 The cell-wall structure of biomass
The model of the cell-wall of woody biomass, firstly proposed by Fengel and Wegener3 and further developed by Dumitriu5 (Fig. 2), is well-established and involves the cell-wall structure and the distribution of the chemical components in different cell wall layers.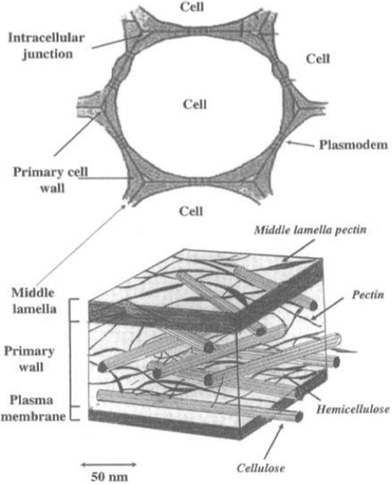 | ||
| Fig. 2 The schematic representation of the proposed cell wall along with the location of the main polysaccharides components by Dumitriu.5 | ||
The cell wall could be morphologically divided into three distinct zones: middle lamella, primary cell wall and secondary wall.5 The middle lamella is shared by two contiguous cells and is composed almost entirely of pectic substances. The primary cell walls are composed of cellulose microfibrils and an interpenetrating matrix of hemicelluloses, pectins, and proteins. Cellulose forms the framework of the cell walls, hemicelluloses cross-link noncellulosic and cellulosic polymers, and pectins provide structural support to the cell wall. The secondary cell walls are derived from the primary walls by thickening and inclusion of lignin into the cell wall matrix and occur inside the primary wall. The transition from primary to secondary cell wall synthesis is marked by the cessation of pectin deposition and a noted increase in the synthesis and deposition of cellulose/hemicellulose and lignin. The cellulose and non-cellulosic polysaccharides of the secondary cell wall are qualitatively distinct from those found in the primary cell walls.
The relevant study6 evidenced that if cellulose is deposited actively between S1 and S3 developmental stages (especially in the middle part of S2 stage), hemicellulose (xylan) deposition occurs in the S1 to early S2 and again in the S3 developmental layers. Successive deposition of hemicellulose (xylan) onto the cell wall increases the microfibril diameter. The large amounts of hemicellulose (xylan) that accumulated on microfibrils appear to be globular but are covered with lignin after they are deposited. The information about the distribution of the main components (hemicellulose, cellulose and lignin) in the cell wall layers of lignocellulosic biomass is quantitatively reported in the literature.7 However, the details concerning the inter-linking/bond relationship (such as the H-bond among the polysaccharide molecules and lignin-carbohydrate coalescence) between the chemical components in the cell walls of wood are not well examined in the literature.
2.2 The configuration of cellulose
As far as the chemical components of biomass were concerned, a distinction should be made between the main macromolecular cell-wall components—cellulose, hemicellulose (polyoses) and lignin.3Cellulose is a uniform component in all lignocellulosic biomass, while the proportions and chemical composition of lignin and hemicellulose differ in different biomass. The configuration of cellulose in lignocellulosic biomass would be discussed, with regard to its content, isolation methods, the characterization of the macromolecules and the inter-linkages among the units.Cellulose is the prominent chemical component in lignocellulosic biomass, accounting for approximately 50% by weight. The methods for isolating and/or determining cellulose from biomass could be summarized as:3
(1) Separation of the main portions of hemicellulose and residual lignin from cellulose;
(2) Direct isolation of cellulose from lignocellulosic biomass, including purification procedures (such as the pulping process);
(3) Determination of the cellulose content by total hydrolysis of biomass, cellulose with subsequent determination of the resulting sugars.
In any isolation method cellulose cannot be obtained in a pure state, thus the purification always plays an important role in the cellulose isolation process. Through the relevant methylation experimental studies,3,5 the primary structure of cellulose is shown to be a linear homopolymer of glucose having the D configuration and connected by β-(1–4) glycosidic linkages (Fig. 3). It is found that the units of the cellulose molecular chain are bound by β-(1–4) glycosidic linkages, showing that the adjacent glucose units are linked by dehydration between their hydroxylic groups at carbon 1 and carbon 4.
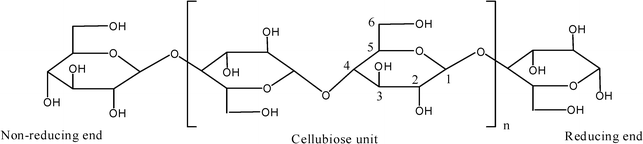 | ||
| Fig. 3 The central part (cellubiose unit) of a cellulose molecular chain with reducing and non-reducing end groups. | ||
The stabilization of the long cellulose molecular chains in ordered systems originates in the presence of functional groups which are able to interact with each other. The functional groups of the cellulose chains are the hydroxyl groups, three of which are linked to each glucopyranose unit. These OH-groups are not only responsible for the supramolecular structure but also for the chemical and physical behavior of the cellulose through the hydrogen bond (H-bond). The OH-groups of cellulose molecules are able to form two types of hydrogen bond depending on their site at the glucose unit.3 The hydrogen bonds between OH-groups of adjacent glucose units in the same cellulose chain are called intramolecular linkages, which give a certain stiffness to the single chain. The hydrogen bonds between OH-groups of adjacent cellulose chains are called intermolecular linkages, which are responsible for the formation of supramolecular structures. The primary structures, consisting of a number of cellulose chains through the hydrogen bonds in a superhelicoidal fashion, are the cellulose microfibrils, which build up the framework of the whole cell walls.5
Two chain ends of the cellulose chain are chemically different (Fig. 3). One end has a D-glucopyranose unit in which an anomeric carbon atom is involved in a glycosidic linkage, whereas the other end has a D-glucopyranose unit in which the anomeric carbon atom is free. This cyclic hemiacetal function is in an equilibrium in which a small proportion is an aldehyde, which gives rise to reducing properties at this end of the chain, so that the cellulose chain has a chemical polarity, while the OH-group at the C4 end of the cellulose chain is an alcoholic hydroxyl and therefore non-reducing. The molecular weight of cellulose varies widely depending on the origin of the sample. As cellulose is a linear polymer with uniform units and bonds the size of the chain molecule is usually defined as degree of polymerization (DP). The degrees of polymerization of the plant-cellulose as well as the technical cellulose products are estimated from 15300 for capsules to 305 for rayon fibers.5
3 The on-line pyrolysis of cellulose
The thermogravimetric (TG) analysis method, either dynamic heating process or isothermal heating process, is well-established for on-line pyrolysis of biomass and its components (cellulose, hemicellulose and lignin). The mass loss of the solid sample could be exactly recorded versus temperature/time. The chemical kinetic models for the biomass and its components are proposed from the analysis of the different mass loss stages and validated through the correlation between the predicted data and the experimental mass loss curve. Since the specific chemical phenomena and the prediction of the volatile yields are rarely referred to in those models, TGA coupled with FTIR, GC, MS or other advanced analytical equipment has been recently employed to investigate the evolution of the volatile along during the pyrolysis process. This facilitates the understanding of the possible chemical reactions for the depolymerization of macromolecules and the secondary cracking of the primary fragments.The on-line pyrolysis of cellulose has been vigorously studied for many years.7–21 When heated either isothermally (where the heating temperature is constant) or dynamically (where the heating temperature is mostly linearly varied) under an inert atmosphere, cellulose initially undergoes depolymerization and dehydration of the macromolecules, and then various fragmentation, elimination and condensation reactions to produce non-condensible volatiles, condensable vapor (liquid tar after cooling) and a carbonaceous char as the solid residue. A large number of studies have been published describing the investigation of the kinetics of pyrolysis of cellulose, and have proposed chemical reaction schemes for both primary and subsequent decomposition steps. The outstanding contribution in the kinetics study of cellulose pyrolysis was given by groups led by Broido and Shafizadeh about 30–40 years ago and have been carefully reviewed by Antal and Varhegyi, and compared with their work in ref. 14,22–24.
Here, we present a systematic overview of the development of the kinetics of cellulose pyrolysis, involving recent studies implemented by other groups led by Piskorz, Di Blasi, Banyaz, Agrawal, Wooten, Hosoya and so on. Several controversial points addressed in previous studies are intensively discussed, which concern the existence of the intermediate anhydrosugars, secondary cracking of volatiles and the formation of char residue.
3.1 Kinetic study without advanced analytic instruments
Historically, it was perhaps Broido's group that firstly called attention to the intriguing phenomena of cellulose pyrolysis and proposed the established kinetic scheme in 1960s.25,26 As described in Scheme 1 (Fig. 4),26,27 the decomposition of cellulose can be represented through two competing reactions: the first step is estimated to be important at low temperatures and slow heating rates, accounting for the slight endothermic formation of anhydrocellulose below 280 °C detected by DTA. At about 280 °C a competitive, more endothermic unzipping reaction is initiated for the remained cellulose, leading to tar formation. The third step presents the exothermic decomposition of anhydrocellulose to char and gas.
Broido's kinetic scheme was re-examined by Argawal,13 revealing that the rates of anhydrocellulose formation are comparable to those of the depolymerization process only in one case for temperatures of ∼270 °C in the isothermal, fixed-bed conditions. Then, the mechanism is approved through the isothermal, fluid-bed experiments in the temperature range 250–300 °C, providing a complete set of kinetic data for the Broido model.13 It is worth noting that the formation of the anhydrocellulose as an intermediate product is undetectable in the experiments, and no kinetic data for the char forming reaction are reported in the above publications. These ambiguities stimulated global research interest in the kinetic studies of cellulose pyrolysis, resulting in a vigorous debate in the following years.
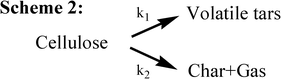
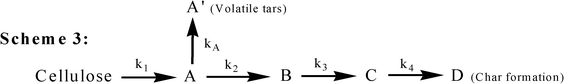
In 1975, Broido and Nelson examined the effect of thermal pretreatments at 230–275 °C on the cellulose char yields varying from 13% (no thermal pretreatment) to over 27%.10 They employed large samples of cellulose (100 mg of shredded cellulose, and 7 cm × 3 cm sheets, individually wrapped several layers deep around a glass rod), which might incur char formation from solid–vapor interactions during the prolonged thermal pretreatment. The previous kinetic model (Scheme 1) is correspondingly improved as described in Scheme 2 (Fig. 5), eliminating the formation of the anhydrocellulose as an intermediate product. One year later, Broido28 reported a kinetic analysis of a 1000 h (about 6 weeks) study of cellulose pyrolysis under vacuum by thermogravimetry (TG) at 226 °C. The analog computer was employed to simulate mass loss according to the model given in Scheme 3 (Fig. 6). The product “A” (later called “active cellulose”) formed through step 1 performs as an important intermediate for the subsequent formation of both volatiles and char. Some data at high temperatures were also analyzed and activation energies for the steps were correspondingly reported. Later with regard to Scheme 3, Varhegyi et al. found a better fit to the thermo-analytical curve when the active cellulose (“A”) was eliminated from the reaction model as described in Scheme 4 (Fig. 7) Nevertheless, the Scheme 3 for cellulose pyrolysis proposed by Broido28 has become the foundation for almost all subsequent research in this field23.
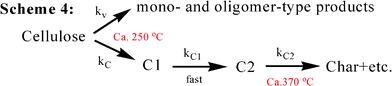
In 1979, Shafizadeh's group undertook a kinetic study of cellulose pyrolysis using pyrolysis apparatus consisting of a horizontal, cylindrical and electrically heated furnace containing a Pyrex tube where another Pyrex tube was placed.29 A 250 mg sample of cellulose was tested in the pyrolysis apparatus at temperatures ranging from 259 to 341 °C, and a chemical reaction model for cellulose, displayed in Scheme 5 (Fig. 8), was proposed to give a good fit to the weight loss data through numerical integration.
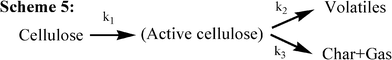
Scheme 5 is slightly different from those proposed by Broido and co-workers but largely confirms the previous findings. This is called the “Broido-Shafizadeh model”.23,30–32 At low temperatures (259–295 °C), the initiation period (characterized by an accelerating rate of weight loss33) has been explained as a formation of “active cellulose” through the depolymerization process (reduction of the DP) with an activation energy of 242.8 kJ mol−1. Then, the “active cellulose” undergoes the two competitive reactions to produce either char and gas (activation energy 153.1 kJ mol−1) or primary volatiles (197.9 kJ mol−1). At high temperatures (above 295 °C), no initial period of accelerating rate of weight loss was observed in Shafizadeh's study.29 Thus a cellulose degradation mechanism was described simply via two competitive first-order reactions, where the formation of “active cellulose” is eliminated from Scheme 5. This mechanism is then confirmed by Antal and Varhegyi's TGA study of cellulose pyrolysis with the heating rate of 40 K min−1, obtaining an activation energy, 238 kJ mol−1, for the formation of volatiles and 148 kJ mol−1 for the formation of char and gas.14
The primary volatiles from cellulose pyrolysis are postulated to be anhydrosugars of which Levoglucosan appears to be the predominant product from cellulose.12 In 1982, Shafizadeh improved the mechanism (Scheme 5) since the volatiles (anhydrosugars) displayed in Scheme 5 act not only as the main primary product from cellulose, but also as the intermediates for the formation of small fragments through secondary cracking reactions.34 Although the role of vapor–solid interactions in the char formation was referred to in Schafizadeh et al.'s study33 (1979), “… pyrolysis of levoglucosan is approved to give some residual char, and it has been suggested that char formation is not a primary step but is a result of repolymerization of volatile material”, this relationship between the char formation and volatiles is not involved in the mechanism. Actually, the char formation through the vapor–solid interactions is also observed and presented in some other publications.12,22,23,32,35–37
Varhegyi proposed a mechanism for cellulose pyrolysis in a sealed crucible, displayed in Scheme 6 (Fig. 9).22 The kinetic Scheme 6 confirms the secondary char formation from the repolymerization of intermediate (volatiles) catalyzed by the water from the dehydration reaction, giving a good fit to nine different DSC experiments completed by Mok et al.35 However, the structure and composition of the char residue from cellulose pyrolysis is ambiguous and the variation of its characteristics is difficult to detect, limiting the understanding of the chemical pathways for the char formation.

The argument between Antal–Varhegyi and Broido–Shafezadeh is remarkable, concerning the existence of “active cellulose” during the pyrolysis of cellulose. Antal and Varhegyi showed that no evidence was found to support the inclusion of the initiation step displayed in Scheme 5 (titled as “Broido-Schafezadeh model”). This step must proceed at an immeasurably high rate at conditions of interest, or it does not exist.23 The kinetic Scheme 7 (Fig. 10) proposed by Banyasz et al.38 fully excluded the notion of “active cellulose” and explained the experimental facts solely by cellulose depolymerization through two reaction channels, confirming the results by Antal and Varhegyi. Moreover, the weight-loss data of cellulose pyrolysis at primarily higher temperatures and heating rates have been more successfully predicted by omitting the active cellulose intermediate.32

In 1995, Antal et al. concluded that the pyrolysis behavior of cellulose samples is well represented by a simple, single-step, irreversible, first-order rate law with a single high activation energy.23 Most of the kinetic models discussed above (mainly derived from TGA studies) are possibly outstanding for simulating the mass loss curve of cellulose, but not indicating any information about the formation of specific products. Besides, the uncertainty on the existence of “active cellulose” (intermediate), the ambiguity of the char formation still persists, which needs to be specified, requiring the employment of the advanced analytical equipment (such as FTIR, GC-MS, HPLC, NMR and so on).
3.2 Kinetic study with advanced analytic instruments
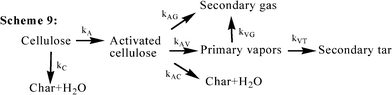
With the development of the advanced analytical equipment, the depolymerization of cellulose and he formation of cellulose with a lower DP (active cellulose) at low temperatures has been evidenced in several reports,32 even under flash pyrolysis conditions.17,39–41 In 2002, Lede et al. directly observed a transient “intermediate liquid compound” in small pellets of cellulose that had been heated by radiant flash pyrolysis in an imaging furnace, which is characterized by HPLC/MS and found to be composed predominantly of anhydro-oligosaccharides (such as levoglucosan, cellobiosan and cellotriosan).41 In the slow heating experiments of cellulose, Wooten32 revealed that intermediate cellulose (IC) is an ephemeral component that appears and then disappears over the course of 60 min of heating at 300 °C, while the rapid disappearance of IC in samples that have been heated at only a slightly higher temperature (i.e., 325 °C) further demonstrates the transient nature of IC. This behavior clearly identifies the compound(s) as a reaction intermediate, and the authors correspondingly associated this intermediate compound with the “active cellulose” in the Broido and Shafezadeh kinetic models (Scheme 3 and Scheme 5).28,29 Thus, some recent research has obtained the formation of “active cellulose” as an intermediate during cellulose pyrolysis, as presented in Scheme 8 (Fig. 11)12,42 and Scheme 9 (Fig. 12).39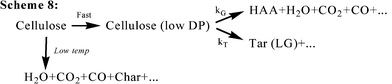
 | ||
| Fig. 12 The kinetic model for cellulose pyrolysis proposed by Diebold (1994)39 and similarly proposed by Wooten et al. (2004).32 | ||
Both Scheme 8 and Scheme 9 establish the formation of water, primary char and “active cellulose” during the low temperature step of cellulose pyrolysis, but the char formed by the repolymerization of the cellulose with low DP (“activated cellulose”) is not considered in Scheme 8.
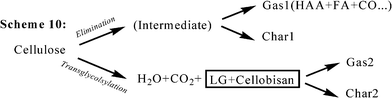
Previously, Bradbury et al.29 and Antal23 suggested that char formation might result from the repolymerization of volatile materials such as levoglucosan. This phenomenon is approved by Hosoya,36 presenting that the secondary char from cellulose is formed from the repolymerization of anhydrosugars (levoglucosan). The experimental data from Wooten et al.'s study32 show that a precursor–product relationship does exist between intermediate cellulose (“active cellulose”) and the aliphatic and aromatic components of the char. The kinetic Scheme 10 (Fig. 13) proposed by Mamleev20 also confirms this finding, elucidating that the intermediate whether levoglucosan or cellobisan are all performed as the precursor for the formation of char.
The comprehensive kinetic model for pyrolysis of cellulose (Scheme 11) (Fig. 14) was proposed by Shen,43 confirming that levoglucosan acts as the primary product and also the precursor for the secondary reactions forming small fragments and char.31 This scheme also combines the results from both Hosoya36 and Wooten.32 However, the kinetic parameters for this model need to be further identified for specific reactions, facilitating its application in industry.
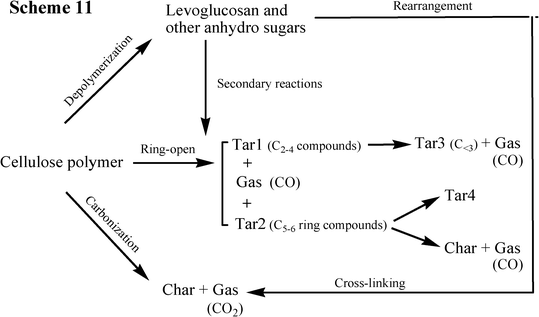 | ||
| Fig. 14 The proposed kinetic scheme for the cellulose pyrolysis by Shen (2010).43 | ||
Nowadays, it might be not difficult to show the existence of “active cellulose” or other important (intermediate) products with the help of the advanced analytical equipment, but the chemical reaction mechanism for cellulose pyrolysis is still ambiguous and controversial. One of the possible routes to improve the understanding of the structure changes of cellulose molecules and formation of the specific products is to study the thermal decomposition of the relevant derivatives, together with molecular dynamics simulations (MDS) which are established for estimating the specific chemical pathways from a microscopic point of view. Moreover, the identification of intermolecular hydrogen bonding between the different molecular chains would be another uncertainty for understanding the pyrolytic behavior of cellulose, especially for the initial stage of cellulose pyrolysis.
4 The off-line pyrolysis of cellulose
Compared to the on-line pyrolysis study of cellulose, the off-line pyrolysis of cellulose is mostly carried out under relatively high temperatures (above 400 °C) or at a high heating rate (more than or around 1000 °C s−1)12,32,36,40,44–48 and sometimes under low temperature heating (below 400 °C),49,50 and is concerned with the following issues: (1) the distributions of the gas, liquid and solid products; (2) the formation of the specific compounds and the pyrolytic chemical pathways. How these two issues may be influenced by the pyrolytic reactors and the variables like temperature, residence time, heating rate, pressure, particle size, catalytic salts and crystallinity is extensively examined in the literature, in order to promote the product specificity, maximize the yield and improve the understanding of the pyrolytic mechanism.In this work, the emphasis is on the effects of the predominant factors such as the reactor type, temperature or heating rate, residence time on the distributions of the products (gas, liquid and solid) from cellulose pyrolysis. Considering the complexity of chemical constituents in gas and liquid products, the attention would be confined to those few compounds which have been established to be reproducible in good yield (such as levoglucosan, hydroxyacetaldehyde, furfural, CO, CO2 and so on), in order to meet the interests of potential industrial applications.
4.1 The distributions of gas, liquid and solid products
Regarding the commercialization of pyrolytic technology for bio-energy conversion, the designed pyrolytic reactor involving the variation of the operating parameters (temperature, residence time, pressure and so on) has remarkable effects on the threshold of the specific product yield and the operating cost of the process.51–54 Most of the reactors for the fast (off-line) pyrolysis of biomass to produce bio-oil or fuel gases is summarized by Bridgewater,55 and are estimated in terms of product yield, feed size, input gas, complexity and so on (Table 1). It has been proven that the fluidized bed reactor is one of the promising technologies for biomass thermal conversion due to the high-efficient heat transfer and ease of scale-up, which has potential for commercial practice.56–60 Microwave pyrolysis, termed as a novel thermo-chemical technology for converting biomass to solid, liquid and gas fuels, is of growing interest thanks to its low requirement on energy input during the process, flexibility of the feedstock size and high quality products (low oxygen content in char and bio-oil). The yield of the products from cellulose through different pyrolysis reactors would be intensively discussed, with regards to the effect of operating conditions such as temperature, residence time and condensing patterns.| Reactor | Liquid yield wt (%) | Feed size | Input gas | Complexity | Scale-up | Statusa |
|---|---|---|---|---|---|---|
| a Demo scale is estimated to be 200–2000 kg h−1, pilot scale is 20–200 kg h−1 and lab scale is <20 kg h−1. | ||||||
| Fluidized bed | 75 | Small | High | Medium | Easy | Demo |
| CFB | 75 | Medium | High | High | Easy | Pilot |
| Entrained gas flow | 65 | Small | High | High | Easy | Lab |
| Vaccum | 60 | Large | Low | High | Hard | Demo |
| Rotating cone | 65 | Very small | Low | High | Hard | Pilot |
| Ablative | 75 | Large | Low | High | Hard | Lab |
| Auger | 65 | Small | Low | Low | Easy | |
Pyrolysis in a fluidized-bed reactor. The outstanding contribution on a study of cellulose pyrolysis in a fluidized bed reactor was made by the research group led by Scott and Piskorz at the University of Waterloo in Canada.12,17,42,46,61–63 A bench scale atmospheric pressure fluidized bed unit using sand as the fluidized solid with a feeding rate of 30 g h−1 of biomass was designed to investigate the yield of liquid product at different temperatures in an inert nitrogen atmosphere with an apparent vapor residence time of approximately 0.5 s.62 Piskorz12 reported the pyrolytic behavior of the two types of cellulose (S&S powdered cellulose with an ash content of 0.22% and Baker TLC microcrystalline cellulose with an ash content of 0.04%) in the fluidized bed reactor, giving a distribution of the gas, liquid and solid products at temperatures from 450 to 550 °C, summarized in Table 2. The yield of organic products in the liquid phase (except water) from the S&S powdered cellulose ranges from 58.58% to 67.81% of the moisture and ash free feed at temperatures from 450 to 550 °C, reaching the maximum at 500 °C. Comparatively, the yield of organic products from the Baker TLC microcrystalline cellulose at 500 °C is determined to be 90.1%. Moreover, the yield of char for S&S powdered cellulose at 500 °C is 3.4%, compared to 1.0% for Baker TLC microcrystalline cellulose.
| Author(s) | Sample | Pyrolysis reactor | Conditions | Yield of products (wt%) | |||
|---|---|---|---|---|---|---|---|
| T/°C | Residence time (s) | Gas | Liquid a (water) | Char | |||
| a The yield of liquid product including water. b The pressure of the reactor is 5 psig of helium pressure. c The operating pressure in the furnace is 5 atm. d Including solid product (char); 5.4% compared to 1.3% for Avicel pH-102 crystalline cellulose. | |||||||
| M.R. Hajaligol, et al. (1982)65 | No. 507 filter paper | Screen-heating Pyrex reactor (fixed bed) | 400∼1000 | 0∼ | 5.25∼46.97 | 16.37∼83.35 | 3.32∼78.37 |
| W.S.L. Mok and M.J. Antal (1983)66 | Whatman filter paper | Two-zone tubular micro reactor (fixed bed)c | 800 | 1∼18 | 62∼71 | — | 15∼23 |
| R.G. Graham, et al. (1984)67 | Avicel pH-102 crystalline cellulose | Downflow entrained bed (fluidized) reactor | 750∼900 | < 0.6 | 74.7∼98.1 | 0.7∼15.8d | — |
| J. Piskorz, et al. (1986)12,42 | S&S powdered cellulose | Fluidized bed reactor | 450∼550 | 0.53∼0.56 | 8.49∼17.89 | 68.75∼75.59 (7.35∼10.17) | 4.2∼8.53 |
| Baker TLC crystalline cellulose | 500 | 0.48 | 5.1 | 94.7 (4.6) | 1.0 | ||
| D. Radlein, et al. (1991)46 | Commercial SS-144 crystalline cellulose | Fluidized bed reactor | 500 | < 0.5 | 7.8 | 83.3 (10.8) | 5.4 |
| Avicel pH-102 crystalline cellulose | 500 | < 0.5 | 3.9 | 89.6 (6.1) | 1.3 | ||
| Y.F. Liao (2003)31 | Filter paper with ash content of 0.01% | Gravitational feeding reactor (Fixed bed) | 300∼1090 | 0.1∼1.4 | 1.5∼60.2 | 6.0∼86.3 | 1.8∼92.5 |
| Aho, et al. (2008)47 | Microcrystalline cellulose powder | Batch-operating fluidized bed reactor | 460 | <1.5 | 32.3 | 47.6 (24.5) | 20.1 |
| T. Hosoya, et al. (2007)36 | Cellulose powder from Toyoroshi Co. | Cylindrical furnace and tube reactor (fixed bed) | 800 | 30 | 12.9 | 77.1 (5.1) | 10 |
| D.K. Shen and S. Gu (2009)21 | Microcrystalline cellulose powder | Batch-operating fluidized bed reactor | 420∼730 | 0.44∼1.32 | 20.1∼42.5 | 30.6∼72.2 | 1.03∼47.4 |
These results confirm that a large amount of the inorganic salts in the ash content promotes the formation of the condensed structure through the catalytic effects, inhibiting the cracking of the macromolecules and enhancing the yield of solid product.21,30,31,34,46,64 Several years later, the pyrolysis of two further types of cellulose (commercial SS-144 crystalline cellulose and Avicel pH-102 crystalline cellulose) were also studied in a fluidized bed by Piskorz's co-worker (Radlein, et al.).46 The yields of the products are shown in Table 2. The temperature, 500 °C, regarded as the optimal condition for producing bio-oil from cellulose in the fluidized bed reactor, gives yields of organic products of 72.5% for commercial SS-144 crystalline cellulose and 83.5% for Avicel pH-102 crystalline cellulose. The difference should also be attributed to the catalytic effect of inorganic salts in the ash, since the yield of char for commercial SS-144 crystalline cellulose is 5.4% compared to 1.3% for Avicel pH-102 crystalline cellulose.
Recently, Aho47 conducted the pyrolysis of softwood carbohydrates under a nitrogen atmosphere in a batch-operating fluidized bed reactor, where quartz sand was used as a bed material and the load of the raw material was approximately 10 g. All sand was kept in the reactor by a net at the upper part of the reactor. The evolved vapors were cooled in the four consecutive coolers with a set point of −20 °C, while between the third and fourth cooler the vapors were passed through a water quench with a pH value of 3 to avoid the absorption of CO2. The furnace temperature was kept at 490 °C until the release of non-condensable gases stopped, while the temperature in the reactor was about 460 °C. The vapor residence time was estimated to be less than 1.5 s based on the height of the reactor and the actual fluidizing gas velocity. The distribution of the products from cellulose (microcrystalline cellulose powder) is shown in Table 2, giving the low yield of organic products of 23.1% and high yield of char as 20.1%. The condensation of the vapors was estimated to be insufficient, while the values for gases and char can be considered reliable. It should be mentioned that the mass balance of the experiment could not be satisfactorily completed, due to its current reactor set-up (especially the vapor-cooling and liquid-precipitating system).
A similar batch-operating fluidized bed reactor was designed by Shen and Gu, in order to study the fast pyrolysis of biomass and its components with the variation of temperature and vapor residence time under inert atmosphere21,68,69. No bed material was applied and the load of the raw material was about 5 g. The solid product was captured by the carbon filter, while the evolved hot vapors were cooled through two U-tubes immersed in ice-water (0 °C) and dry ice-acetone (−30 °C), respectively. The distribution of the products from the pyrolysis of microcrystalline cellulose at temperatures between 420 and 730 °C with a residence time from 0.44 to 1.32 s is given in Table 2. It is estimated that the yield of liquid product reaches its maximum of 72.2% at a temperature of 580 °C with a residence time of 0.44 s. The higher temperature and long residence time promotes the decomposition of the macromolecules and cracking of the volatiles, enhancing the yield of gases and reducing the solid product.21
Pyrolysis in entrained-bed reactor. Graham67 designed a complicated entrained bed reactor to investigate the fast pyrolysis of cellulose, which has a similar or even higher heating rate than that of a fluidized bed. The rapid heat transfer and thorough mixing between the particulate solids and feed are accomplished in two vertical gas–solid contactors: Thermovortactor and Cryovortactor. The biomass or other carbonaceous fuel is rapidly mixed with the hot particulate solids in a Thermovortactor. The suspension passed through a downdraft entrained-bed (fluidized) reactor allowing the individual setting of temperatures, and then was quenched by the cold solids in the Cryovortactor and cooled through a cooling coil submerged in a water tank. The solids were then separated in the mass balance filter and the gas was collected in sampling bags. The feeding rate is less than 1 kg h−1 and the total elapsed time from the Termovortactor inlet to the cryovortactor exit is typically less than 600 ms. The yield of the gas and liquid (heavy fraction including tar and char) products at the temperature from 750 to 900 °C is shown in Table 2. The low yield of liquid product (less than 20%) is mainly due to the high reactor temperature and the inefficient cooling method. Moreover, the mass balance is not convincing, since the heavy fraction of the vapors may condense on the vessels of the Cryovortactor and solid separator.67 It should be noted that the high yield of gases is attributed to the enhanced heat transfer through the pre-mixing between the biomass and solid heat carrier before being fed to the pyrolysis reactor, compared to that of the fluidized bed reactor.
The residence time (both solid and vapor) in the fluidized or entrained bed reactors could be narrowly changed (normally less than 1 s), because of the confinement of the minimum gas velocity for solid fluidization. Therefore, the fixed bed reactors are designed for investigating the effect of not only temperature but also residence time on the yield of products and their specificity.31,36,44,66 Liao31 designed a fixed bed reactor (quartz tube with a sample-holder in the middle), the temperature of which could be changed from 0 to 1100 °C. The filter paper with an area 18 × 50 mm (about 2 g) is fed gravitationally to the reactor from the top, and the carrier gas (nitrogen) brings the evolved volatiles and some char fragments through the carbon filter. The purified volatiles are then cooled through the three traps consecutively: (1) the mixture of water and ice (0 °C); (2) the mixture of acetone and dry ice (−30 °C); and (3) assisting cooling agent (−45 °C). The yield of the products (gas, liquid and char), at temperatures from 300 to 1090 °C with a (vapor) residence time between 0.1 to 1.4 s, was determined by the carrier gas velocity and is extensively discussed by Liao31 (shown in Table 2–3), while the mass balance for all the experiments is convincingly located between 96% and 101.5%. With the same vapor residence time (carrier gas velocity), the yield of the liquid product complies with a Gaussian distribution with temperature, giving a maximum of 86.29% (including 15.72% water) at around 600 °C with a residence time of 0.1 s. It is estimated that the long residence time promotes the yield of gases, due to the sufficient secondary reactions of the volatiles. The yield of gases is increased from 1.5% to 60.2% monotonously with temperature (from 300 to 1090 °C). It needs to be noted that the duration of each experiment, corresponding to the sample heating-up and holding time, is not specified in the work.
| Compound | Yield wt% of feedstock | Method | Authors |
|---|---|---|---|
| a Calculated out from the available experimental data | |||
| Levoglucosan | 20%∼60% | Vacuum pyrolysis | Shafizadeh et al.33 |
| 400 °C∼650 °C | |||
| 2.1% | Fluidized bed reactor | J. Piskorz, et al.12 | |
| S & S powdered cellulose | |||
| 500 | |||
| 25.2% | Fluidized bed reactor | ||
| Baker TLC microcrystalline cellulose | |||
| 500 °C | |||
| 42.03%a | Fixed bed | Y.F. Liao31 | |
| Whatman filter paper | |||
| 580 °C | |||
| 5-Hydroxymethylfurfural | <1% | Fluidized bed reactor | J. Piskorz, et al.12 |
| Baker TLC microcrystalline cellulose | |||
| 500 °C | |||
| <1%a | Fixed bed | Y.F. Liao31 | |
| Whatman filter paper | |||
| 450 °C∼930 °C | |||
| Hydroxyacetaldehyde | 18% | Fluidized bed reactor | J. Piskorz, et al.12 |
| S & S powdered cellulose | |||
| 500 °C | |||
| 8% | Fluidized bed reactor | ||
| Baker TLC microcrystalline cellulose | |||
| 500 °C | |||
| 1.95%∼13.52%a | Fixed bed | Y.F. Liao31 | |
| Whatman filter paper | |||
| 450 °C∼930 °C | |||
| Acetol | 1.1% | Quartz Tube reactor | Hosoya et al.36 |
| 880 °C | |||
| 3.2% | Fluidized bed reactor | J. Piskorz, et al.12 | |
| S & S powdered cellulose | |||
| 500 °C | |||
| 0.7% | Fluidized bed reactor | ||
| Baker TLC microcrystalline cellulose | |||
| 500 °C | |||
| 0.48%∼4.22%a | Fixed bed | Y.F. Liao31 | |
| Whatman filter paper | |||
| 450 °C∼930 °C | |||
| CO | 15% | Screen-heating reactor | Hajaligol et al.44 |
| 750 °C | |||
| 11.1%∼20.2% | Batch-operating fluidized bed reactor | D.K. Shen and S. Gu21 | |
| 420 °C∼730 °C | |||
| 11.44%a | Entrained down-flow reactor | Graham67 | |
| 900 °C | |||
| CO2 | 3% | Screen-heating reactor | Hajaligol et al.44 |
| 750 °C | |||
| 8.6%∼10.1% | Batch-operating fluidized bed reactor | D.K. Shen and S. Gu21 | |
| 420 °C∼730 °C | |||
| H2 | 3.25% a | Entrained down-flow reactor | Garham et al.67 |
| 900 °C | |||
| 0.2%∼0.7% | Batch-operating fluidized bed reactor | D.K. Shen and S. Gu21 | |
| 420 °C∼730 °C |
Pyrolysis in fixed-bed reactor. The pyrolysis of cellulose in a tube (fixed bed) reactor made of Pyrex glass was investigated by Hosoya et al.36 Compared to the study of Liao,31 the cellulose sample is horizontally fed to the furnace and the carrier gas is not employed which means that the vapor residence time could not be set individually. It is estimated that thirty seconds are enough for completing the pyrolysis since no volatile product formation is observed after longer pyrolysis time. The evolved volatiles are retained in the reactor with the solid residue during the whole pyrolysis process. After 30 s pyrolysis, the reactor is pulled out from the furnace and cooled with air flow for 1 min at room temperature. The tar (liquid product) condensed on the reactor vessel is extracted by i-PrOH and water. The amounts of the gaseous, tar and char fractions are determined gravimetrically after pyrolysis and extraction, giving the result at the temperature of 800 °C in Table 2. It should be mentioned that the temperature of the reactor is not evenly distributed during the pyrolysis process, because the bottom of the tube reactor was placed at the center of the cylindrical furnace. Most of the evolved volatiles are condensed in the upper part of the reactor, but do not suffer from the vigorous secondary cracking due to the long (solid) residence time. Thus, the yield of liquid product (77.1% including water) is not visibly different from that of Liao's results at a temperature of 810 °C with a shorter vapor residence time (74.39%).31
Another Pyrex cylindrical tube (fixed bed) reactor was made by Hajaligol et al.,44 where the cellulose sample was held and heated by the porous stainless screen connected to the brass electrodes of the reactor. The system allows independent variation of the following reaction conditions: heating rates (100–100 000 °C s−1), final temperatures (200–1100 °C), sample residence (holding) time at final temperature (0–∝ s). Similar to the experimental set-up of Hosoya,36 the vapor residence time could not be individually changed while the carrier gas is not employed. Part of the evolved vapors is rapidly diluted and quenched in the reactor vessel during the operation, because most of the gas within the reactor remains close to room temperature. The other part of the evolved vapors is purged out of reactor vessel with helium and cooled down through two downstream traps: (1) U-tube packed with glass wool immersed in dry ice/alcohol (−77 °C) and (2) the same trap in liquid nitrogen (−196 °C). The char retained on the screen is determined gravimetrically. The mass balance for each case is around 100%, giving convincing yields for the products at temperatures 400–1000 °C with a sample holding time 0–30 s in Table 2. It is concluded44 that tar yield (liquid product) increases with temperature to a maximum of about 65% at around 700 °C and then decreases with further temperature increases, since the sample residence time is zero. With a long residence time (for example 30 s), the yield of liquid product at 400 °C remarkably increases to 83.35%, due to the sufficient heating-up time for the complete pyrolysis of cellulose. Comparatively, the yield of liquid product at 500 °C with zero holding time is only 16.37% and the yield of char is 83.63% (where the mass balance is 105%), because of the incomplete decomposition of cellulose.
A two-zone tubular micro reactor (fixed bed) was designed by Mok and Antal66 to investigate the effect of vapor residence time on the yield of products from cellulose pyrolysis. Zone A is operated for 15 min for complete solid phase pyrolysis, while Zone B is maintained at 700 °C for vapor phase cracking. The char is determined gravimetrically and the gases are collected by the replacement of water. Unfortunately, the tar collection is not possible with that apparatus. The results of the product distribution at a temperature of 800 °C with a vapor residence time 1–18 s are shown in Table 2. The long vapor residence time and high pressure (5 atm) promote the secondary cracking of volatiles, enhancing the yield of the gas product.
Pyrolysis in a microwave reactor. The microwaves can be firstly used to activate biomass (cellulose as the feedstock) to solid, liquid and gas products, as shown by Allanet al. in 1970s.70 After 2000, two research groups (one is led by J. H. Clark from the University of York in the UK and the other by Y. Fernandez and J. J. Pis from the National Institute of Carbon in Spain) have published a large number of remarkable results on microwave pyrolysis (MWP) of biomass and its components (such as cellulose and lignin).49,50,71–74
The studies of the research group led by Fernandez and Pis are mainly concentrated on high-temperature microwave pyrolysis (more than 400 °C) of biomass.72,74 The feedstock sample (coffee hulls) being rich in cellulose, is made into cylindrical pellets (approximately 3 mm in diameter and 2 cm in length). The pyrolysis of the sample (15 g of pellets) was carried out in an electrical furnace (called CP-conventional pyrolysis) and in a single mode microwave oven at 500, 800, and 1000 °C, observing the variation of the yield of products (char, oil and gases) and their properties (element content and heating value). The electrical furnace was initially heated up to the corresponding pyrolysis temperature, so that the temperature of sample rose quickly. In the case of microwave heating, the sample was placed in an identical quartz reactor, which was then placed in the centre of the microwave guide.75 The evolved volatiles passed through five consecutive condensers placed in an ice bath, the last three of which contained dichloromethane, while the carbonaceous residue was separated from the receptor by sieving. The gas yield was evaluated by weight difference. It is found that the yield of char, oil and gas from pyrolysis of sample under microwave heating is 30.21%, 7.90% and 65.28% by weight of feedstock at 500 °C and changed to 22.70%, 8.58% and 68.72% at 1000 °C. Compared to that of conventional pyrolysis by electrical heating, the formation of the gas products (especially syngas CO + H2) is remarkably enhanced under microwave pyrolysis and the oxygen content in char and oil is significantly reduced increasing their heating value. Most of the above findings on microwave pyrolysis of biomass are also approved by other researchers.48,76
Research led by Clark has made a remarkable contribution on the microwave pyrolysis of biomass under low temperature (less than 350 °C).49,50,73 A Milestone ROTO SYNTH Rotative Solid Phase Microwave Reactor is used for microwave pyrolysis of wheat straw.49 The average sample mass was between 150 and 200 g. The sample was heated at a rate of 17 °C min−1 to a maximum temperature of 180 °C as measured by in situ temperature probes. The condensable fraction produced during the process was collected through a vacuum unit. The yield of solid, liquid and gas products was estimated to be 29%, 57% and 14% by weight of feedstock at 180 °C. Compared to that of conventional pyrolysis under relatively high temperature,77 the oxygen content of the bio-oil obtained from low-temperature microwave pyrolysis is significantly reduced facilitating the following upgrading processes.49 The microwave pyrolysis of cellulose was carried out at temperatures between 100 °C and 300 °C in a CEM Discovery laboratory microwave, regarding the yield of char and its formation mechanism. The high-quality char, where more energy from feedstock is conserved, could be produced with the adjustment of the low pyrolysis temperature. A temperature of 180 °C was estimated to be a key turning point in the microwave degradation of cellulose, favoring the understanding that the production of fuels is allowed at dramatically lower temperatures than those required under conventional pyrolysis (electrical heating). The energy conserved in solid, oil and gas products is evaluated to be balanced for the whole process. In terms of an industrial process, the low-temperature microwave technology can be easily adapted to a variety of biomass to produce a uniform char which can be handled by the end users.
With regards to the above discussion, microwave pyrolysis under both high and low temperatures is estimated to be one of the promising technologies to achieve high-quality solid (low oxygen content), liquid (low oxygen content and water content) and gas (low energy input and high syngas concentration) fuels with a low cost, helping to achieve sustainable development through the utilization of renewable alternatives (biomass) instead of fossil fuels.
4.2 The formation of the specific compounds
The volatiles (both condensable and non-condensable) that evolve from cellulose pyrolysis under moderate or high temperatures are very complicated, and most of which have been identified by employing advanced analytical equipment such as FTIR, GC-MS, HPLC, NMR and so on. A variety of pyran and furan derivatives (C5–6 ring-containing compounds), aliphatic oxygenated C2–4 organic compounds and light species/gases (such as light hydrocarbons, CO and CO2) can be obtained, and the extensive lists together with their spectrometric/chromatograghic patterns and the yields are available in the literature, where the results are remarkably affected by the pyrolytic reactor, operating conditions, condensing method and sample sources. Due to the great potential as feedstocks for fuel and chemicals production, some products established in good yields (such as levoglucosan, furfural, hydroxyacetaldehyde, acetol, CO, CO2 and so on) (Fig. 15) were vigorously investigated regarding the chemical mechanism for their formation and fractionation. The yields of most nominated compounds and gases from the pyrolysis of cellulose are extensively summarized in Table 3.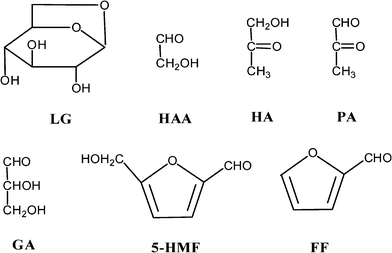 | ||
| Fig. 15 The chemical structures of the typical compounds in bio-oil from cellulose pyrolysis: LG: levoglucosan, HAA: hydroxyacetaldehyde, HA: Hydroxyactone, PA: pyruvic aldehyde, GA: glyceraldehyde, 5-HMF: 5-hydroxymethyl-furfural and FF: furfural. | ||
Inasmuch as the cellulose samples have somewhat different ash contents, the different levoglucan yield may be due to the well-known effect of inorganic cations in reducing tar yields by promoting other fragments or char formation.46 Richards and co-workers established the extraordinary influence of salts and metal ions on the productivity of volatiles (especially levoglucosan and hydroxyacetaldehyde), presenting that the addition of alkali and Ca2+ cations to ash-free cellulose reduced the yield of levoglucosan while other metal ions (particularly Fe3+ and Cu2+) enhanced the yield of levoglucosan.83,84 In accordance with the findings of Richards's laboratory, Piskorz et al. observed very dramatic increases in the yields of levoglucosan (more than 30% by weight) from various celluloses after a mild sulfuric acid-wash pretreatment.42 The profound effects of inorganic substances on the product from carbohydrates were also evidenced by Van der Kaaden through the matrix study on amylase pyrolysis using Curie-point pyrolysis, concluding that carbonyl compounds, acids and lactones are released by alkaline and neutral matrices while furans and anhydrohexoses are favored under neutral and acidic conditions.85
The experimental conditions as well as the purity of cellulose and inorganic additions appear to have an important effect on the yield of levoglucosan. The yield of levoglucosan produced from the S &S powdered cellulose pyrolysis in a fluidized bed increases with the temperature, reaches its maximum at a temperature of 500 °C and then decreases with the elevated temperature.46 This is consistent with the results from Shen's work using fluidized bed reactors, giving the maximum yield of levoglucosan at a temperature of 530 °C.21 A great deal of specific work studying pyrolysis oils produced from Whatman filter paper at temperatures from 400 °C to 930 °C in the fixed bed reactor confirmed that the formation of levoglucosan is mainly located at temperatures between 450 °C and 650 °C, obtaining the maximum yield at 580 °C (about 58.37% by weight of pyrolysis oil).31 Moreover, the yield of levoglucosan decreases with long vapor residence time at a temperature of 600 °C, while most of the small fragments (low molecular weight volatiles) increase notably. These phenomena increase the interest in looking into the chemical mechanism of the levoglucosan formation and its secondary cracking during the cellulose pyrolysis.
An established standpoint presents that the formation of levoglucosan is initiated by the disruption of the cellulose chain, primarily at the 1,4 glucosidic linkage in the macromolecule, followed by intramolecular rearrangement of the cellulosic monomer units.18,21,31,33,46 The actual mechanism of levoglucosan formation remains controversial. Golova favors a free-radical mechanism through the successful validation of the data on the effects of free-radicals.86 Shafizadeh, arguing by analogy with the reactions of model phenyl glucosides, prefers a heterolytic mechanism.33 Essig and Richards83 proposed that the hydroxyl group (–OH) of the free chain ends further depolymerizes the short chain through transglycosylation accompanied with the release of levoglucosan.
Another unsettled issue is whether the depolymerization of macromolecules (disruption of cellulose chain) takes place by a concerted “unzipping” process or by random breaking of the cellulose chain. Briodo et al.87 found that crystalline cellulose and undergoes a large change in DP before weight loss occurs. Similarly, Basch and Lewin88 proposed that if cellulose depolymerized by an unzipping process then the number of free chain ends, as reflected by DP, will influence the initiation rate. Radlein46 reported that one cellulose sample which had been heated to 180 °C for several hours and had a very low DP appeared to give an abnormally high yield of levoglucosan. While the unzipping process may well operate at low temperature, there is evidence that it is inapplicable under fast pyrolysis conditions due to the significant amounts of cellobiosan and higher anhydro-oligomers in cellulose pyrolysates.46 The correlation between the yield of levoglucosan and DP of cellulose sample under fast pyrolysis conditions needs to be specified, attracting the interests for further study.
The possible chemical pathways for the primary decomposition of cellulose monomers (Fig. 16) and secondary cracking of levoglucosan and other primary fragments were comprehensively overviewed and developed by Shen and Gu, revealing the possible chemical information of the typical compound formation from cellulose pyrolysis21 (Fig. 17). The usual view on the mechanism of levoglucosan cracking is that the lower molecular weight products are formed by fragmentation of principal intermediates like levoglucosan and cellobiosan as discussed by Pouwels et al.81 Such a scheme is also indicated by the data of Shafizadeh and Lu who showed that similar low molecular weight products (such as furfural, 5-HMF, glycolaldehyde, hydroxyacetone, acetic acid, formic acid and light species) from cellulose pyrolysis can be formed by direct pyrolysis of levoglucosan,79 which is consistent with the observation by Hosoya et al. through the NMR identification of levoglucosan pyrolysis volatiles.37 Evans et al.89 even concluded that both cellulose and levoglucosan were pyrolyzed at various residence times and give similar cracking patterns and products by using a flash pyrolysis-mass spectrometric technique.
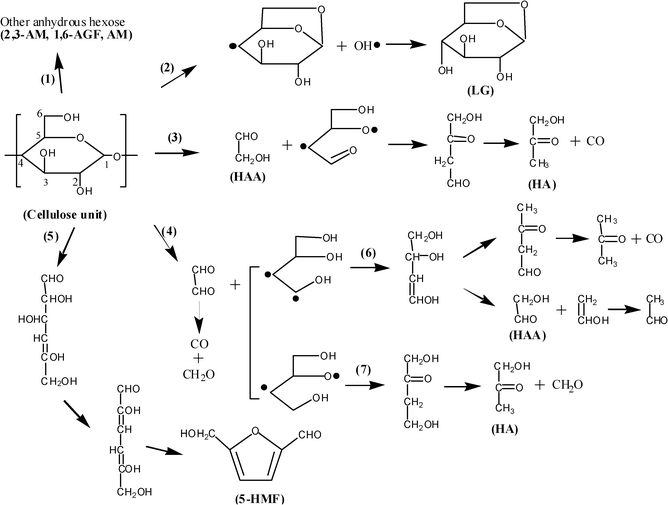 | ||
| Fig. 16 The speculative chemical pathways for the primary decomposition of cellulose monomer.21 | ||
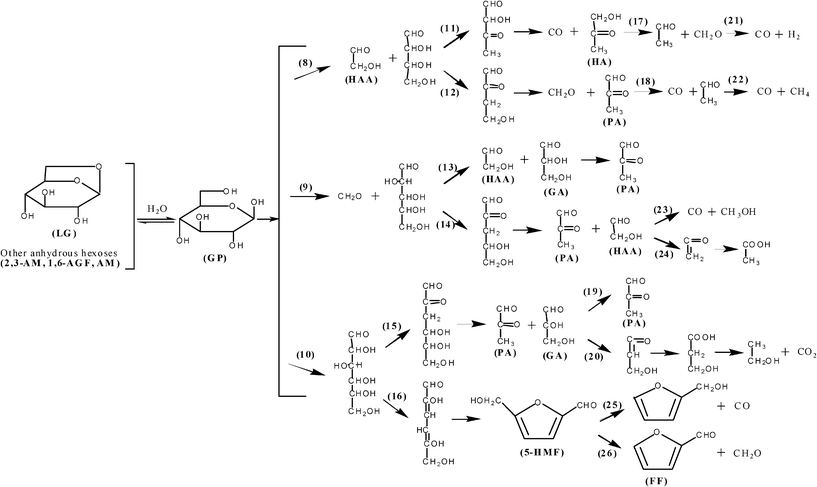 | ||
| Fig. 17 The speculative chemical pathways for the secondary decomposition of the anhydrosugars (especially levoglucosan).21 | ||
However, Richards45 has argued that it is more likely that hydroxyacetaldehyde, known as one of the prominent products from cellulose pyrolysis (chemical pathway (3) in Fig. 16), forms directly from cellulose by a plausible mechanism involving dehydration followed by a retro-Diels–Alder reaction but not from the secondary cracking of levoglucosan. Li et al.18 reported that no detectable hydroxyacetaldehyde is observed by FTIR during levoglucosan pyrolysis in the two-zone pyrolysis reactor, indicating that levoglucosan might not be the major precursor of hydroxyacetaldehyde in cellulose pyrolysis. The two major pathways are then recognized to be active during cellulose pyrolysis: one leading to the formation of levoglucosan as a relatively stable product and the second to yield low molecular products particularly hydroxyacetaldehyde. The experimental studies of cellulose pyrolysis with the addition of inorganic substances show that conditions which result in the selective formation of levoglucosan realize a very low yield of hydroxyacetaldehyder and vice versa, confirming the competitive nature of the above two pathways.4,12,23,83,84,90
Regarding the notable argument on the relationship between levoglucosan and hydroxyacetaldehyde, Liao31 conducted the pyrolysis of both cellulose and levoglucosan under different temperature and vapor residence time in a fixed bed. For cellulose pyrolysis, the yield of levoglucosan is increased and then decreased with the elevated temperature reaching the maximum at a temperature of 580 °C, while the yield of hydroxyacetaldehyde is monotoneously increased with the temperature. Under a fixed temperature (610 °C), the long vapor residence time favors the yield of small fragments (especially hydroxyacetaldehyde) remarkably at the expense of levoglucosan, showing the plausibly “consecutive mechanism” between them. For levoglucosan pyrolysis, no hydroxyacetaldehyde (even some other prevalent volatiles from cellulose pyrolysis) is detected at a temperature of 610 °C with a short residence time 0.1 s, confirming the “competitive mechanism” between levoglucosan and hydroxyacetaldehyde. But under the same temperature with a long residence time 1 s, almost all the volatiles from cellulose are released from levoglucosan pyrolysis, enhancing the “consecutive mechanism” between levoglucosan and hydroxyacetaldehyde. Similar quantitative results are reported by Shen and Gu91 for cellulose pyrolysis in a fluidized bed reactor at different temperatures and vapor residence times. The published data by Piskorz et al.42 reporting the variation of levoglucosan and hydroxyacetaldehyde yields with temperature are compatible with either mechanism.
The experimental results summarized above plainly reveal the hybrid relationship between levoglucosan and the low molecular weight fragments (particularly hydroxyacetaldehyde) during cellulose pyrolysis: both competitive and consecutive (Fig. 16 and Fig. 17). However, the predominance of the nominal mechanism during cellulose pyrolysis is still ambiguous for specifying the hydroxyacetaldehyde (or other low molecular weight volatiles) formation and the extent of levoglucosan secondary decomposition, due to the widely varied experimental conditions and inorganic additions.
Furfural and 5-hydroxymethyl-furfural, categorized as furan derivatives, are another two important C5–6 ring-containing compounds in the product list of cellulose pyrolysis.12 Although the yield of these two compounds is less than 1% by weight of fed cellulose, they are notably identified from the pyrolysis oil (GC-MS) spectrum of cellulose.12,21,31,36,47,78,81 The effect of experimental conditions (temperature and vapor residence time) on yield of furfural and 5-hydroxymethyl-furfural is fully discussed by Liao31, showing that the formation of furfural is notably enhanced by the increased temperature and residence time while the yield of 5-hydroxymethyl-furfural is only increased with the elevated temperature. It is observed that these two compounds could be produced from levoglucosan pyrolysis under a suitable vapor residence time, showing the “consecutive mechanism” between them (Fig. 17). Moreover, furfural is found to be one of the important secondary cracking products from 5-hydroxymethyl-furfural pyrolysis. The commonly accepted standpoint concerning the chemical pathway for furfural and 5-hydroxymethyl-furfural is that levoglucosan or cellulose monomer undergoes a ring-opening reaction to form the C6 aliphatic intermediate, followed by hemiacetal reaction between C-2 and C-5 to form the furan-ring structure after the formation of the acetone-structure on position C–2 through dehydration reactions (chemical pathway (5) in Fig. 16 and chemical pathway (16) in Fig. 17).31,79 The 5-hydroxymethyl-furfural could be decomposed to furfural together with release of formaldehyde through the de-hydroxylmethyl reaction, furan methanol through the de-carbonylation reaction, or 5-methyl-furfural through the de-hydroxyl reaction (chemical pathway (24) and (25) in Fig. 17).21,31,92 It is concluded that furfural and 5-hydroxymethyl-furfural are both competitively and consecutively produced with levoglucosan, while 5-hydroxymethyl-furfural is another source for the formation of furfural.
Moreover, the study of cellulose (Whatman filter paper) pyrolysis in a fixed bed reactor by Liao31 indicates that hydroxyacetaldehyde is an important compound in the condensed liquid product, the yield of which is notably increased from 3% to 19% by weight of liquid product with the elevated temperature (450 to 930 °C). The similar quantitative result is reported by Shen and Gu21 studying the cellulose pyrolysis in a fluidized bed reactor under various temperatures and residence times. But the experimental data published by Piskorz et al.42 shows that the yield of hydroxyacetaldehyde by weight of fed cellulose increases with temperature and starts to decrease at a temperature of 610 °C. Since the yield of liquid product against temperature changes comparably with the yield of hydroxyacetaldehyde,12,21,31,42 the apparent yield of hydroxyacetaldehyde by weight of fed cellulose performs a Gaussian distribution with temperature even though its relevant yield by weight of liquid product is monotonously increased with temperature.
Since no other C2 or C3 product appears in the same yield as hydroxyacetaldehyde, an intermediate or primary products are formed early in the decomposition process through monomer ring cleavage (Fig. 16). The most acceptable standpoint for hydroxyacetaldehyde formation is proposed by Shafizadeh and Lai (chemical pathway (3) in Fig. 16), presenting that hydroxyacetaldehyde, assumed as the precursor for glyoxal, was produced mainly from the C–1 and C–2 position of the glucopyranose.79 This scheme is similar to that proposed by Byrne et al.78
Through the examination of bond energies in the monomer unit by Frankiewicz93 and interatomic distance for β-D-glucose by Sutton,94 it was shown that the length for the C–2 to C–3 bond and for C–1 and O–ring linkage is slightly larger than other similar bonds. This finding is confirmed by Madorsky et al.95 who pointed out that the C–O hemiacetal bond on the ring is thermally less stable than C–C bonds. This information offers support to the hypothesis that the initial ring cleavage of the cellulose monomer tends to occur frequently at these two locations, yielding a two-carbon fragment and a four-carbon fragment, while the two-carbon fragment is rearranged to a relatively stable product, hydroxyacetaldehyde, and the four-carbon fragment can undergo a number of rearrangements of dehydration, scission and decarbonylation to yield a variety of lower molecular weight products.12 This chemical pathway for the formation of hydroxyacetaldehyde is well presented in the study of Liao31 and Shen et al.21 (Fig. 16). They also suggested that almost all of the positions on the pyran-ring could contribute to hydroxyacetaldehyde formation, involving the examples on C–2 to C–3 or C–5 to C–6 positions plausibly through the cracking of the five carbon fragment from initial cleavage of monomer on the bonds of C–1 to C–2 and hemiacetal C–O (chemical pathway (9) in Fig. 17). However, this suggestion should be evidenced through the bond energy examination and atomic label technology on the model compound.
Acetol (1-hydroxy-2-propanganone), regarded as another major product, is perhaps firstly reported by Lipska and Wodley96 in their study of isothermal cellulose pyrolysis at 315 °C. Moreover, some cellulose fast pyrolysis studies have also evidenced acetol as a major component in the products. For instance, Hosoya et al.36 obtained an acetol (in the i-PrOH-soluble fraction) yield of 1.1% by weight of feed sample from the cellulose pyrolysis at a temperature of 800 °C in a sealed tube. Two cellulose samples pyrolysed at a temperature of 500 °C in a fluidized bed reactor by Piskorz et al.12 gave an acetol yield of 3.2% for S & S Powdered cellulose and 0.7% for Baker TLC microstalline cellulose by weight of feed sample, which is possibly due to the well-known effect of inorganic salts. Meanwhile, the authors12 observed that the acetol yield from S & S Powdered cellulose pyrolysis notably increases with temperature. This phenomenon is also evidenced by the work of Liao31 studying cellulose pyrolysis in a fixed bed reactor and fluidized bed reactor respectively, obtaining a range of acetol yields by weight of liquid product from 0.8% to 6% at temperatures from 450 °C to 930 °C.
In 1972, Shafizadeh and Lai79 proposed a possible chemical pathway for acetol formation from levoglucosan decomposition as the rearrangement of the four-carbon fragment from the primary pyran-ring cleavage, while the other two-carbon fragment might be the precursor for hydroxyacetaldehyde. A similar reaction scheme was reported by Byrne et al. in 196678 and proposed again by Piskorz et al.12 in 1986. Meanwhile, pyruvaldehyde was also proposed to be formed through the rearrangement of the four-carbon fragment, competing with the formation of acetol (Fig. 17). It could be found that enol-structure from the dehydration between the conjoint carbon is the intermediate for the acetone-structure, while the dehydration is between C–5 and C–6 for acetol formation and between C–4 and C–5 for pyruvaldehyde formation. According to Benson's rules on energy grounds,97acetol should be favored over the alternative possibility of pyruvaldehyde. This speculation is evidenced by Piskorz,12 Liao31 and Shen and Gu21 studying cellulose fast pyrolysis in a fixed bed reactor or fluidized bed reactor, obtaining higher yields of acetol over pyruvaldehyde (Fig. 16 and Fig. 17). Moreover, other chemical pathways for acetol and pyruvaldehyde formation from the five-carbon fragment or ring-opened six-carbon intermediate are proposed by Liao,31 which are then summarized in levoglucosan secondary cracking pathways by Shen and Gu.21 However, the prevalent one for their formation, which might be affected by experimental conditions, is not specified, while their secondary cracking to CO and aldehyde-compounds could be readily determined.
Among a number of the detectable pyrolysis products from cellulose, some products, such as acetic acid, aldehyde, methanol, formaldehyde and so on, are less frequently discussed in the literature due to their low yields.12,31,44,46,64,66,67,98 In an investigation on the formation of an acidic product, Kang et al.99 proposed a mechanism of hydration of ketene which is formed from the dehydration of alcohol-aldehyde structure (chemical pathway (24) in Fig. 17). This reaction scheme for carboxyl group formation was well-established by the following researchers,12,21,31,36,46,61,68 most of whom did not specify its position on the pyran-ring. The possible chemical pathways for cellulose primary reactions and volatile secondary cracking are systematically summarized by Shen and Gu,21 giving a number of pathways for the formation of these low molecular weight oxygenated compounds.
Mok and Antal66 investigated the effect of residence time on the yield of main gas products from cellulose pyrolysis at a pressure of 5 psig, concluding that CO2 formation was notably enhanced by the longer residence time while CO was inhibited. A different result was reported by Liao31 where CO is remarkably favored by the longer residence time while CO2 is changed slightly, which is further confirmed by Shen and Gu.21 Evans et al.89 proposed that carboxyl groups, formed through the hydration of ketenes, are the precursors for producing CO2, while CO is mainly produced through the decarbonylation reaction of aldehyde-type species. Since the ketene structure, which is related to the formation of acidic compounds (containing carboxyl group), is mainly formed during the low temperature stage, CO2 is the primary product of cellulose pyrolysis, and thus it is not remarkably influenced by reaction temperature. Comparatively, high reaction temperature favors the vigorous secondary tar cracking reactions, especially the carbonyl-group containing fragments, in order to enhance the formation of CO steadily and rapidly. This reaction mechanism is summarized from the results of the researchers,12,18,21,31,37,46,89 however the preference of the carbon on the pyran-ring for CO and CO2 formation is not specified. From the study of thermal decomposition of levoglucosan, Shafizadeh and Lai79 suggested that CO2 was produced primarily from the C–1 and C–2 positions as well as hydroxyacetaldehyde, while the production of CO was less specific, but the information for cellulose pyrolysis was not ruled out.
It needs to be noted that the mole fraction of hydrogen (H2) is also important as well as CO and CO2 and constitutes approximately 21% of the product gas at a reaction temperature of 900 °C in the study of Garham et al.67 Quantitatively, a similar result is reported by Hajaligol et al.,44 also finding that the yield of H2 is noticeably increased at high temperatures (more than 800 °C), while no hydrogen is observed at low reaction temperatures. This implies that high reaction energy is required for the formation of hydrogen through the secondary tar cracking reaction. Li et al.18 proposed that formaldehyde is a precursor for hydrogen formation, together with the evolution of CO through the secondary cracking at around 550 °C. The same chemical scheme is proposed again by Liao31, Hosoya37 and Shen and Gu,21 also giving the possible chemical pathway for hydrocarbons formation through the decarbonylation of aldehyde-type compounds together with the production of CO. It is also observed that both hydrogen and hydrocarbon formation are favored by the elevated temperature, confirming the enhancement of temperature on the secondary tar cracking reactions proposed above together with the evolution of CO. Since hydrogen is the important synthesis gas for methanol and other synthesis, the new methods coupled with thermal technology but with low heating energy input, such as catalytic hydrothermal conversion technology,100–102 are attracting global interest to specify hydrogen formation from cellulose.
The typical compounds from cellulose pyrolysis are extensively discussed in the above studies, regarding the variation of the yield with experimental conditions (residence time and temperature), and the possible chemical pathways for their formation and cracking. It is commonly accepted that levoglucosan is the most prevalent product in the primary volatiles from cellulose pyrolysis, which could further decompose into various low molecular weight compounds (C2–4 compounds or light gases). However, the preference of the various primary reactions and secondary tar (especially levoglucosan) cracking reactions under widely varied experimental conditions with or without the catalysts needs to be further determined in order to identify and promote the specific compound formation. The commonly-accepted chemical pathways need to be essentially estimated through advanced theory and/or technology analysis, such as molecular dynamics simulation (MDS).
5 The interactions among the components in lignocellulosic biomass under pyrolytic conditions
The constituent polymers from lignocellulosic biomass, i.e.polysaccharides (cellulose and hemicellulose) and lignin, are pyrolyzed in different ways.30 The polysaccharides form anhydrosugars, furans, aldehydes, ketones and carboxylic acids as their primary volatile products, while the volatiles from lignin mainly consist of low molecular weight aromatic compounds with guaiacyl-units or phenolic-units. To date, many researchers have extensively studied the pyrolysis of the real biomass and proposed reaction models by assuming that pyrolysis of the main chemical components (cellulose, hemicellulose and lignin) takes place independently without interactions among the three components.103–107 They stated that pyrolysis of biomass can be explained based on a linear superposition of the three components. Yang et al.108 showed that the pyrolysis of the synthesized biomass samples containing two or three of the biomass components indicated negligible interaction among the components. A computational approach was made firstly to predict the weight loss of a synthesized biomass from its composition in cellulose, hemicellulose and lignin, and secondly to predict the proportions of the three components of a biomass. The results calculated for the weight loss of the synthesized biomass are quite consistent with the experimental results. However, results for predicting the composition of the biomass in terms of cellulose, hemicellulose and lignin were not very satisfactory, possibly due to the ignorance of interactions among the components.From the morphological view of the plant cell-wall as discussed in section 2, the main chemical components (cellulose, hemicellulose and lignin) would not perform individually without the intrinsic interactions of the whole biomass system during pyrolysis.3,5,109,110 The interactions among the chemical components of woody biomass under pyrolytic conditions are of growing interest during recent years, in order to gain better understanding of the pyrolytic mechanism of the whole biomass system from the pyrolysis of individual components.109,111–113
Hosoya et al.109 investigated cellulose-hemicellulose and cellulose-lignin interactions during pyrolysis at a gasification temperature of 800 °C for 30 s in a tube reactor, while a cellulose sample mixed with hemicellulose (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, wt/wt) was prepared by grinding a cellulose-hemicellulose mixture in mortar and cellulose sample mixed with MWL (milled wood lignin) (2:1, wt/wt) was prepared by adding cellulose to the 1,4-dioxane solution (0.5 ml) of MWL followed by evaporation of the solvent. In the cellulose-hemicellulose pyrolysis, the experimental and estimated yields were not much different although the tar (total) yield tended to decrease slightly with a small increase in the char yields by mixing. The results indicate that cellulose-hemicellulose interaction is not significant in gas, tar and char yields. In the cellulose-MWL pyrolysis, more significant deviations were observed between the experimental and estimated yields of char and tar fractions; char yield decreased with the increasing yield of the tar total fraction by mixing. Tar composition was also substantially affected by mixing cellulose with MWL, showing that the yield of the i-PrOH-soluble fraction substantially increased from 52.1% to 68% while the yield of water-soluble fractions substantially decreased from 14.5% to 2.8%. These results suggest that nature of the tar fraction is significantly altered from the water-soluble to i-PrOH-soluble products by the mixing of cellulose with MWL.
:1, wt/wt) was prepared by grinding a cellulose-hemicellulose mixture in mortar and cellulose sample mixed with MWL (milled wood lignin) (2:1, wt/wt) was prepared by adding cellulose to the 1,4-dioxane solution (0.5 ml) of MWL followed by evaporation of the solvent. In the cellulose-hemicellulose pyrolysis, the experimental and estimated yields were not much different although the tar (total) yield tended to decrease slightly with a small increase in the char yields by mixing. The results indicate that cellulose-hemicellulose interaction is not significant in gas, tar and char yields. In the cellulose-MWL pyrolysis, more significant deviations were observed between the experimental and estimated yields of char and tar fractions; char yield decreased with the increasing yield of the tar total fraction by mixing. Tar composition was also substantially affected by mixing cellulose with MWL, showing that the yield of the i-PrOH-soluble fraction substantially increased from 52.1% to 68% while the yield of water-soluble fractions substantially decreased from 14.5% to 2.8%. These results suggest that nature of the tar fraction is significantly altered from the water-soluble to i-PrOH-soluble products by the mixing of cellulose with MWL.
Moreover, the interactions among the components for the characteristic secondary char-formation were also investigated, including photographs of the reactors after pyrolysis and tar extraction.109 The wood polysaccharide samples form the secondary char at the upper side of the reactor while vapor phase carbonization of the products from lignin leads to the formation of secondary char from the bottom to upper side continuously. In cellulose-hemicellulose pyrolysis, these char-forming behaviors were explainable as combined behaviors of the individual cellulose and hemicellulose pyrolysis. On the other hand, the cellulose–MWL pyrolysis substantially reduced the vapor phase secondary char formation from MWL.
With the characterization of the compositions in tar by GC-MS, GPC and NMR, it was found that the experimental yield of levoglucosan and 1,6-anhydro-β-D-glucofuranose is similar to the estimated value in the cellulose-hemicellulose pyrolysis. These values are substantially different in the cellulose-MWL pyrolysis, where levoglucosan yield is enhanced while 1,6-anhydro-β-D-glucofuranose production is visibly inhibited. Thus, lignin may affect the anhydrosugar formation differently, because of the different formation mechanisms for anhydrosugars. As for the MWL-derived products, formation of guaiacol, 4-methylguaiacol and 4-vinylguaiacol was enhanced in the presence of cellulose. Effects on other products were very small. Although a detailed mechanism is unknown at present, these results are considered to be related to the inhibition of the vapor phase carbonization of the MWL-derived products.
Later, Hosoya and co-workers114 extended their research on interactions between cellulose and lignin at 600 °C under different pyrolysis times (40–80 s) in two types of reactor. Type A reactor is for investigating the vapour phase interactions, where cellulose and lignin are separately placed at the bottom. Type B reactor is for investigating the solid–liquid phase interactions, where cellulose, mixed with lignin, is placed at the bottom. In 40 s, the lignin and cellulose samples formed solid char in the inner sample holder and the bottom of the reactor, respectively. Secondary char from lignin was also observed around the reactor wall in 80 s. This secondary char formation was effectively inhibited in co-pyrolysis with cellulose. Since the solid–liquid-phase interaction is not effective in the type A reactor, this inhibitory effect arises from the vapor-phase interaction. Similar inhibition of the secondary char formation was observed also in the type B reactor. In 40 s, the tar yield increased from 45.7% (estimated) to 55.1% (experimental) with the suppressing formation of char (32.5–28.3%) and water (16.7–11.6%) in the type B reactor. The influences in the type A reactor were comparatively small. The water yields are usually correlated with the char yields, since dehydration is the main process in char formation. These influences suggest that the solid–liquid-phase interaction in the early stage of pyrolysis (primary pyrolysis stage) enhances the tar formation instead of water and char.
For cellulose pyrolysis, the gas yield increased from 5.0% to 31.1% with the decreased yield of tar from 54% to 29.3% after 40 s and 80 s, although the char yields were not so different (17.7% to 17.0%). Thus, gasification of the cellulose derived tar components proceeded in this period. Contrary to this, gas formation from lignin was comparatively small. Under co-pyrolysis conditions, the experimental gas yields (80 s) (type A: 31.4%, type B: 28.9%) were greater than the estimated one (24.2%), while the estimated and experimental gas yields in 40 s were similar (type A: 4.7%, type B:5.0%, estimated: 5.0%). Accordingly, gas formation in the period 40–80 s (secondary reaction stage) was accelerated in co-pyroysis.
It needs to be noted that the vapor-phase interactions were significant at a longer pyrolysis time of 80 s when the methoxyl groups of the lignin-derived volatiles were cleaved homolytically. The vapor-phase interactions accelerated the gas formation from the cellulose-derived volatiles with suppressing the vapor phase char formation of the lignin-derived volatiles. The yields of methane and catechols from lignin also increased greatly instead of the formation of o-cresols. Most of these influences are explained with a proposed interaction mechanism, in which the cellulose-derived volatiles act as H-donors while the lignin-derived volatiles (radicals) act as H-acceptors. Although the interactions between the cellulose and lignin in both solid–liquid- and vapor-phase are extensively investigated, the results could not interpret the intrinsic mechanism of the interaction among the components in the real wood system because of the neglect of the inter-linkages and morphological relationship between the chemical components.
A time profile of evolution rates of gas and tar in steam gasification of model biomass samples at a temperature of 673 K were examined by Fushimi et al.115 using a continuous cross-flow moving bed type differential reactor to elucidate the interaction among the major biomass components (cellulose, xylan and lignin) during gas and tar evolution. Two types of model biomass samples (sample A: mixture of cellulose (65%) and lignin (35%) with a ball-mill for 5 h; sample B: mixture of cellulose (50%), xylan (23%) and lignin (27%) with a ball-mill for 5 h) were used for the experiment. In the steam gasification of sample A, the evolution of water-soluble tar and gaseous products (CO, H2, CH4 and C2H4) are significantly suppressed by the interaction between cellulose and lignin. The primary (initial) decomposition of lignin is hindered by the interaction with pyrolysate of cellulose, which is different from the result from Hosoya et al.114 The CO2 evolution appreciably enhanced and the evolution of water-soluble tar delays. These results may imply that the volatilization of water soluble tar derived from cellulose is suppressed by lignin and then the decomposition of char derived from polymerized saccharides and lignin takes place, emitting mainly CO2.
From the results using sample B, it was found that the addition of xylan greatly enhances the evolution of gases (CO, H2, CH4 and C2H4) and accelerates the evolution of water-soluble tar and CO2, implying that enhancement of the decomposition of water-soluble tar into gases and/or xylan decomposes into gases without significant interaction with cellulose or lignin. In addition, yields of the major tar components (levoglucosan, furfural and 5-methylfurfural) were measured using HPLC. It was observed that the interaction among cellulose, xylan and lignin suppresses the evolution of levoglucoasan and significantly increases the evolution rate of 5-methylfurfural. There is an insignificant influence of interaction among cellulose, xylan and lignin for furfural evolution.
In order to establish a link of the pyrolysis gas yield from the biomass and its main composition, experimental flash pyrolysis of several biomasses and the model compounds (xylan, cellulose and lignin) at a temperature of 950 °C with a gas residence time of about 2 s was carried out by Couhert et al.113 using an entrained flow reactor (EFR). The synthesized biomass, made by mixing the three components, is described as a simple mix where the products are mixed in equal mass proportion with a spatula in a container, and mixed and then co-ground to thin elements using a laboratory ball mill. During the pyrolysis of simple mixes, the three components devolatilized separately. Interactions are likely to occur outside the particles. During the pyrolysis of intimate mixes, reactions can occur outside the particles in the same way as during the pyrolysis of simple mixes but additional interactions may occur inside the particles. As one component devolatilizes inside the particle, it is submitted to an atmosphere with very high concentrations in gas and condensable vapors; the gases formed are in close contact with the solids of other components. There are also probably interactions inside the particles because the CO2 yield of the intimate mix is higher than the CO2 yield of the simple mix. An attempt was then made to predict gas yields of any biomass according to its composition, but an additivity law does not allow the gas yields of a biomass to be correlated with its fractions of cellulose, hemicellulose and lignin. It is concluded that interactions occur between compounds and that mineral matter influences the pyrolysis process.
It is confirmed that the interactions among the components of wood under pyrolysis conditions are insufficiently investigated in the literature. Some issues concerning the interactions among components need to be further addressed for gaining a better understanding in this field: (1) the component-mixed sample to simulate/represent the original physico-chemical information among the components in the real biomass; (2) the effect of experimental conditions (temperature, residence time, pressure and so on) and reactor type on the interactions among the components during pyrolysis; (3) specificity of the chemical mechanisms of the interactions among the components in vapor-phase, solid–liquid-phase or morphological-phase. This would be beneficial for expressing pyrolysis of biomass through the pyrolysis of individual components in biomass.
6 Conclusions and the way-forward
With regard to the above discussion, the development and challenges of the four fundamental issues for the pyrolysis of cellulose in lignocellulosic biomass can be summarized:The cell-wall model for lignocellulosic biomass, divided into three main zones, is well-established to represent its morphological structure and distribution of the prominent chemical components (hemicellulose, cellulose and lignin) in different zones. This would facilitate the direct utilization of biomass as bio-material and the improvement of the conversion process of biomass to fuels and chemicals. It needs to be noted that the existing cell-wall model is mostly applicable for woody biomass, while that for other lignocellulosic biomass (such as crops, straws and grass) should be further identified.
For on-line pyrolysis of cellulose, the main mass loss stage(s) of the components of woody biomass versus temperature is well presented, together with the corresponding evolution of the volatiles. However, the initial stage of the cellulose pyrolysis, mainly related to the intermolecular hydrogen bonding and that between the different molecular chains, needs to be clarified for gaining better understanding of the whole pyrolytic behavior of cellulose. The kinetic models for cellulose pyrolysis are improved towards tracking the mass loss process of solids along with the formation of typical products with the help of advanced analytic instruments (such as FTIR, GC, NMR and so on).
For off-line pyrolysis of cellulose, the yield of the products is tightly allied to the reactor type, temperature, residence time and condensing method. It is commonly accepted that levoglucosan is the most prevalent product in the primary volatiles from cellulose pyrolysis, which could be further decomposed into various low molecular weight compounds (C2–4 compounds or light gases). However, the preference of the various primary reactions and secondary tar (especially levoglucosan) cracking reactions under widely varied experimental conditions with or without the catalysts needs to be further determined, in order to identify and promote the specific compound formation. The proposed chemical pathways for cellulose pyrolysis need to be essentially estimated through advanced theory and/or technology analysis, such as molecular dynamics simulation (MDS).
The interactions among the main chemical components of lignocellulosic biomass under pyrolytic conditions are remarkably evidenced, regarding the differences between the estimated yield of products and variation of the specific compositions and the experimental data. This proves that the interactions among the components should be significantly considered for gaining a better understanding of the pyrolysis of the biomass system. The component-mixed sample representing the original physico-chemical information between the components in real biomass is required for revealing the intrinsic interaction mechanism between them under pyrolytic conditions, favoring to predict the pyrolytic behavior of biomass from pyrolysis of its individual components.
Acknowledgements
The authors greatly acknowledge the funding support from National Key Basic Research Programs found by MOST of China (2012CB215306 and 2010CB732206) and the projects supported by National Natural Science Foundation of China (51106030 and 51076031).References
- P. McKendry, Bioresour. Technol., 2002, 83, 37–46 CrossRef CAS.
- A. Demirbas, 2008, London: Springer.
- D. Fengel and G. Wegener, 1984, Berlin: Walter de Gruyter.
- D. N. S. Hon and N. Shiraishi, 2001, Marcel Dekker, Inc.
- S. Dumitriu, 2005, New York, Marcel Dekker.
- T. Awano, K. Takabe and M. Fujita, Protoplasma, 2002, 219(106), 22–29 Search PubMed.
- D. Fengel, Wood Sci. Technol., 1969, 3, 203–217 CrossRef CAS.
- A. J. Stamm, Ind. Eng. Chem., 1956, 48, 418–425 CrossRef.
- D. F. Arseneau, Can. J. Chem., 1971, 49, 632–638 CrossRef CAS.
- A. Broido and M. A. Nelson, Combust. Flame, 1975, 24, 263–268 CrossRef CAS.
- F. Shafizadeh and A. G. W. Bradbury, J. Appl. Polym. Sci., 1979, 23, 1431–1442 CrossRef CAS.
- J. Piskorz, D. Radlein and D. S. Scott, J. Anal. Appl. Pyrolysis, 1986, 9, 121–137 CrossRef CAS.
- R. K. Agrawal, Can. J. Chem. Eng., 1988, 66, 413–418 CrossRef CAS.
- G. Varhegyi, E. Jakab and M. J. Antal, Energy Fuels, 1994, 8, 1345–1352 CrossRef CAS.
- I. Milosavljevic and E. M. Suuberg, Ind. Eng. Chem. Res., 1995, 34, 1081–1091 CrossRef CAS.
- R. Bilbao, J. F. Mastral, M. E. Aldea and J. Ceamanos, J. Anal. Appl. Pyrolysis, 1997, 39, 53–64 CrossRef CAS.
- J. Piskorz, P. Majerski, D. Radlein, A. Vlasars-Usas and D. S. Scott, J. Anal. Appl. Pyrolysis, 2000, 56, 145–166 CrossRef CAS.
- S. Li, J. Lyons-Hart, J. Banyasz and K. Shafer, Fuel, 2001, 80, 1809–1817 CrossRef CAS.
- G. Dobele, G. Rossinskaja, T. Dizhbite, G. Telysheva, D. Meier and O. Faix, J. Anal. Appl. Pyrolysis, 2005, 74, 401–405 CrossRef CAS.
- V. Mamleev, S. Bourbigot and J. Yvon, J. Anal. Appl. Pyrolysis, 2007, 80, 141–150 CrossRef CAS.
- D. K. Shen and S. Gu, Bioresour. Technol., 2009, 100, 6496–6504 CrossRef CAS.
- G. Varhegyi, P. Szabo, W. S. L. Mok and M. J. Antal, J. Anal. Appl. Pyrolysis, 1993, 26, 159–174 CrossRef CAS.
- M. J. Antal and G. Varhegyi, Ind. Eng. Chem. Res., 1995, 34, 703–717 CrossRef CAS.
- G. Varhegyi, M. J. Antal, E. Jakab and P. Szabo, J. Anal. Appl. Pyrolysis, 1997, 42, 73–87 CrossRef CAS.
- A. Broido and F. J. Kilzer, Fire Research Abstract and Reviews, 1963, 5, 157 Search PubMed.
- F. J. Kilzer and A. Broido, Pyrodynamics, 1965, 2, 151–163 CAS.
- A. Broido and M. Weinstein,Kinetics of solid-phase cellulose pyrolysis. in The 3rd International Conference on Thermal Analysis, 1971, Wiedemann (Ed.), Birkhauser Verlag: Basel Search PubMed.
- A. Broido, 1976, Academic Press, New York.
- A. G. W. Bradbury, Y. Sakai and F. Shafizadeh, J. Appl. Polym. Sci., 1979, 23, 3271–3280 CrossRef CAS.
- D. B. Colomba, J. Anal. Appl. Pyrolysis, 1998, 47, 43–64 CrossRef.
- Y. F. Liao, 2003, PhD Thesis, ZheJiang University, HangZhou, China Search PubMed.
- J. B. Wooten, J. I. Seeman and M. R. Hajaligol, Energy Fuels, 2004, 18(1), 1–15 CrossRef CAS.
- F. Shafizadeh, R. H. Furneaux, T. G. Cochran, J. P. Scholl and Y. Sakai, J. Appl. Polym. Sci., 1979, 23, 3525–3539 CrossRef CAS.
- F. Shafizadeh, J. Anal. Appl. Pyrolysis, 1982, 3, 283–305 CrossRef CAS.
- W. S.-L. Mok, M.J. Antal, P. Szabo, G. Varhegyi and B. Zelei, Ind. Eng. Chem. Res., 1992, 31, 1162–1166 CrossRef CAS.
- T. Hosoya, H. Kawamoto and S. Saka, J. Anal. Appl. Pyrolysis, 2007, 78, 328–336 CrossRef CAS.
- T. Hosoya, H. Kawamoto and S. Saka, J. Anal. Appl. Pyrolysis, 2008, 83, 64–70 CrossRef CAS.
- J. L. Banyasz, S. Li, J. L. Lyons-Hart and K. H. Shafer, J. Anal. Appl. Pyrolysis, 2001, 57, 223–248 CrossRef CAS.
- J. P. Diebold, Biomass Bioenergy, 1994, 7(1–6), 75–85 CrossRef CAS.
- O. Boutin, M. Ferrer and J. Lede, J. Anal. Appl. Pyrolysis, 1998, 47, 13–31 CrossRef CAS.
- J. Lede, F. Blanchard and O. Boutin, Fuel, 2002, 81, 1269–1279 CrossRef CAS.
- J. Piskorz, D. Radlein, D. S. Scott and S. Czernik, J. Anal. Appl. Pyrolysis, 1989, 16(2), 127–142 CrossRef CAS.
- D. K. Shen and S. Gu, Cellulose Chemistry and Technology, 2010, 44(1-3), 79–87 CAS.
- M. R. Hajaligol, J. B. Howard, J. P. Longwell and W. A. Peters, Industrial and Engineering Chemistry, 1982, 21(3), 457–465 CAS.
- G. N. Richards, J. Anal. Appl. Pyrolysis, 1987, 10(2), 251–255 CrossRef CAS.
- D. Radlein, J. Piskorz and D. S. Scott, J. Anal. Appl. Pyrolysis, 1991, 19, 41–63 CrossRef CAS.
- A. Aho, N. Kumar, K. Eranen, B. Holmbom, M. Hupa, T. Salmi and D. Y. Murzin, Int. J. Mol. Sci., 2008, 9, 1665–1675 CrossRef CAS.
- H. Lei, S. Ren and J. Julson, Energy Fuels, 2009, 23, 3254–3261 CrossRef CAS.
- V. L. Budarin, J. H. Clark, B. A. Lanigan, P. Shuttleworth, S. W. Breeden, A. J. Wilson, D. J. Macquarrie, K. Milkowski, J. Jones, T. Bridgeman and A. Ross, Bioresour. Technol., 2009, 100, 6064–6068 CrossRef CAS.
- V. L. Budarin, J. H. Clark, B. A. Lanigan, P. Shuttleworth and D. J. Macquarrie, Bioresour. Technol., 2010, 101, 3776–3779 CrossRef CAS.
- A. A. C. M. Beenackers, Renewable Energy, 1999, 16, 1180–1186 CrossRef.
- P. Garcia-bacaicoa, J. F. Mastral, J. Ceamanos, C. Berrueco and S. Serrano, Bioresour. Technol., 2008, 99, 5485–5491 CrossRef CAS.
- A. Kumar, K. Eskridge, D. D. Jones and M. A. Hanna, Bioresour. Technol., 2009, 100, 2062–2068 CrossRef CAS.
- A. V. Bridgwater, J. Anal. Appl. Pyrolysis, 1999, 51, 3–22 CrossRef CAS.
- T. Bridgwater, 2008, UK: CPL.
- A. N. Garcia, R. Font and A. Marcilla, Journal of Analytical and Applied Pyrolysis, 1995, 31 Search PubMed.
- M. N. Islam, R. Zailani and F. N. Ani, Renewable Energy, 1999, 17, 73–84 CrossRef CAS.
- D. Mohan, C. U. Pittman and P. H. Steele, Energy Fuels, 2006, 20(3), 848–889 CrossRef CAS.
- H. J. Park, Y. K. Park and J. S. Kim, Fuel Process. Technol., 2008, 89(8), 797–802 CrossRef CAS.
- J. L. Zheng, W. M. Yi and N. N. Wang, Energy Convers. Manage., 2008, 49(6), 1724–1730 CrossRef CAS.
- J. Piskorz, D. S. Scott and D. RadleinPyrolysis oils from biomass: producing, analyzing and upgrading. in Symposium Series, vol. 376. 1988: Washington, DC: American Chemical Society Search PubMed.
- D. S. Scott and J. Piskorz, Can. J. Chem. Eng., 1982, 60(5), 666 CrossRef CAS.
- D. S. Scott and J. Piskorz, Can. J. Chem. Eng., 1984, 62(3), 404–412 CrossRef CAS.
- D. K. Shen, S. Gu, K. H. Luo, A. V. Bridgwater and M. X. Fang, Fuel, 2009, 88, 1024–1030 CrossRef CAS.
- R. H. Mohammed, J. B. Howard and J. P. Longwell, Journal of Industrial and Engineering Chemistry, 1982, 21, 457–465 Search PubMed.
- W. S. L. Mok and M. J. Antal, Thermochim. Acta, 1983, 68, 155–164 CrossRef CAS.
- R. G. Graham, L. K. Mok, M. A. Bergougnou, H. I. D. Lasa and B. A. Freel, J. Anal. Appl. Pyrolysis, 1984, 6, 363–374 CrossRef CAS.
- D. K. Shen, S. Gu and A. V. Bridgwater, J. Anal. Appl. Pyrolysis, 2010, 87(2), 199–206 CrossRef CAS.
- D. K. Shen, S. Gu, K. H. Luo, S. R. Wang and M. X. Fang, Bioresour. Technol., 2010, 101(15), 6136–6146 CrossRef CAS.
- G. G. Allan, B. B. Krieger and D. W. Work, J. Appl. Polym. Sci., 1980, 25, 1839–1859 CrossRef CAS.
- J. A. Menendez, A. Dominguez, M. Inguanzo and J. J. Pis, J. Anal. Appl. Pyrolysis, 2004, 71(2), 657–667 CrossRef CAS.
- A. Dominguez, J. A. Menendez, Y. Fernandez, J. J. Pis, J. M. V. Nabais, P. J. M. Carrott and M. M. L. R. Carrott, J. Anal. Appl. Pyrolysis, 2007, 79(1–2), 128–135 CrossRef CAS.
- V. L. Budarin, Y. Zhao, M. J. Gronnow, P. S. Shuttleworth, S. W. Breeden, D. J. Macquarrie and J. H. Clark, Green Chemistry, 2011, DOI, 10, 1039 Search PubMed.
- Y. Fernandez and J. A. Menendez, J. Anal. Appl. Pyrolysis, 2011, 91(2), 316–322 CrossRef CAS.
- A. Domingez, J. A. Menendez, M. Inguanzo and J. J. Pis, Fuel Process. Technol., 2005, 86, 1007–1020 CrossRef.
- F. Yu, S. Deng, P. Chen, Y. Liu, Y. Wang, A. Olson, D. Kittelson and R. Ruan, Appl. Biochem. Biotechnol., 2007, 137–140, 957–970 CrossRef CAS.
- Nexant. Ltd., The exploitation of pyrolysis oil in the refinery, 2008 Search PubMed.
- G. A. Byrne, D. Gardner and F. H. Holmes, J. Appl. Chem., 1966, 16, 81 CrossRef CAS.
- F. Shafizadeh and Y. Z. Lai, J. Org. Chem., 1972, 37, 278–284 CrossRef CAS.
- H. R. Schulten and W. Gortz, Anal. Chem., 1978, 50, 428 CrossRef CAS.
- A. D. Pouwel, G. B. Eijkel and J. J. Boon, J. Anal. Appl. Pyrolysis, 1989, 14, 237 CrossRef.
- K. Kawamoto, H. Morisaki and S. Saka, J. Anal. Appl. Pyrolysis, 2009, 85, 247 CrossRef.
- M. G. Essig, G. N. Richard and E. M. Schenck, 1989, New York: J. Wiley & Sons.
- G. N. Richards and G. C. Zheng, J. Anal. Appl. Pyrolysis, 1991, 21, 133–146 CrossRef CAS.
- A.V.d. Kaaden, J. Haverkamp, J.J. Boon and J.W.D. Leeuw, J. Anal. Appl. Pyrolysis, 1983, 5, 199 CrossRef.
- O. P. Golova, Russ. Chem. Rev., 1975, 44, 687 CrossRef.
- A. Briodo, A. C. Javier-Son, A. C. Ouano and E. M. Barrall, J. Appl. Polym. Sci., 1973, 17(12), 3627 CrossRef.
- A. Basch and M. Lewin, Journal of Polymer Sciences, 1973, 11, 3071 CAS.
- R. J. Evans, T. A. Milne and M.N. Soltys, in 16th Biomass Thermochemical Conversion Contractors Meeting. 1984. Battelle Pacific Northwest Lab. Search PubMed PNL-SA-12403 .
- W. Pan and G. N. Richards, J. Anal. Appl. Pyrolysis, 1989, 16, 117–126 CrossRef CAS.
- D. K. Shen, S. Gu, K. H. Luo and A. V. Bridgwater, Energy Fuels, 2009, 23, 1081–1088 CrossRef CAS.
- E. J. Shin, M. R. Nimlos and R. J. Evans, Fuel, 2001, 80, 1697–1709 CrossRef CAS.
- T. C. Frankiewicz, Proc. Specialists workshop on fast pyrolysis biomass. 1980, Solar Energy Research Institute: Golden, CO.123-136 Search PubMed.
- L. E. Sutton, 1958, The Chemical Society, London Spec. Pub.
- S. L. Madorsky, V. E. Hart and S. Straus, Journal of Research of the National Bureau of Standards, 1958, 60, 343 CAS.
- A. E. Lipska and F. A. Wodley, J. Appl. Polym. Sci., 1969, 13, 851 CrossRef CAS.
- S. W. Benson, 1976, New York: Wiley, 2nd edn.
- T. Funazukuri, Fast pyrolysis of cellulose in a micro fluidized bed, in Department of Chemcial Engineering 1983, University of Waterloo, Waterloo Search PubMed.
- J. C. Kang, P. H. Chen and W. R. Johnson, 1976, New York, Academic Press.
- M. Watanabe, H. Inomata and K. Arai, Biomass Bioenergy, 2002, 22, 405–410 CrossRef CAS.
- M. Osada, T. Sato and M. Watanabe, Energy Fuels, 2004, 8(2), 327–333 CrossRef.
- X. H. Hao, L. J. Guo, X. M. Zhang and Y. Guan, Chem. Eng. J., 2005, 110, 56–65 CrossRef.
- R. Alen, E. Kuoppala and P. Oesch, J. Anal. Appl. Pyrolysis, 1996, 36, 137 CrossRef CAS.
- R. S. Miller and J. A. Bellan, Combust. Sci. Technol., 1997, 126, 97 CrossRef CAS.
- J. Teng and Y.C. Wei, industrial and Engineering Chemistry Research, 1998, 37(3806) Search PubMed.
- J. J. M. Orfao, Fuel, 1999, 78, 349–358 CrossRef CAS.
- J. J. Manya, E. Velo and L. Puigjanaer, Ind. Eng. Chem. Res., 2004, 42, 434 CrossRef.
- H. P. Yang, R. Yan, H. P. Chen, C. G. Zheng, D. H. Lee and D. T. Liang, Energy Fuels, 2006, 20, 388–393 CrossRef CAS.
- T. Hosoya, H. Kawamoto and S. Saka, J. Anal. Appl. Pyrolysis, 2007, 80, 118–125 CrossRef CAS.
- S. Dammstrom, L. Salmen and P. Gatenholm, BioResources, 2009, 4(1), 3–14 Search PubMed.
- N. Worasuwannarak, T. Sonobe and W. Tanthapanichakoon, J. Anal. Appl. Pyrolysis, 2007, 78, 265–271 CrossRef CAS.
- G. Wang, W. Li and B. Li, et al. , Fuel, 2008, 87(4–5), 552–558 CrossRef CAS.
- C. Couhert, J. M. Commandre and S. Salvador, Fuel, 2009, 88, 408–417 CrossRef CAS.
- T. Hosoya, H. Kawamoto and S. Saka, J. Anal. Appl. Pyrolysis, 2009, 85(1–2), 237–246 CrossRef CAS.
- C. Fushimi, S. Katayama and A. Tsutsumi, J. Anal. Appl. Pyrolysis, 2009, 86, 82–89 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |