Fast repair of DNA radicals in the earliest stage of carcinogenesis suppresses hallmarks of cancer
Rongliang
Zheng
*a,
Zhongjian
Jia
b,
Ji
Li
a,
Shuangsheng
Huang
a,
Ping
Mu
a,
Fangxin
Zhang
b,
Chunming
Wang
a and
Chengshan
Yuan
b
aInstitute of Biophysics, School of Life Sciences, Lanzhou University, Lanzhou, 730000, China. E-mail: zhengrl@lzu.edu.cn; Fax: +86-931-8912561; Tel: +86-931-8911136
bState Key Laboratory of Applied Organic Chemistry, School of Chemistry and Chemical Engineering, Lanzhou University, Lanzhou, 730000, China
First published on 18th October 2011
Abstract
Genomic instability is a characteristic of most cancers and could be directly caused by DNA damage. DNA repair maintains genomic integrity, reducing the onset of cancer. A distinct DNA fast repair process, synonymous fast repair, is initiated and finished in a microsecond time scale, enormously faster by approximately 9 orders than enzymatic repair, and thus eradicates the headstream of DNA damage, DNA radicals exclusively, in the earliest stage of carcinogenesis. The known enzymatic repair cannot repair DNA radicals and mutation, but fast repair can. Therefore, the fast repair mechanism is a defence in the front line in fighting carcinogenesis. If there was no fast repair, certainly much more DNA steady damage would accumulate and would lead to mutation and cancer with high risk. Almost all the tested phytophenols with fast repair activity, except one, are able to suppress the hallmarks of cancer, such as morphological normalization, redifferentiation, decrease of cancer growth and transplanting rate, inhibition of telomerase, metastasis and angiogenesisin vitro and in vivo, reducing cancer incidence and mortality rate in animals.
 Rongliang Zheng | Rongliang Zheng, professor at the Institute of Biophysics, School of Life Sciences, Lanzhou University, China, won the 2009 National Prize for Natural Sciences of China and was selected for the National Outstanding Scientist for life honor in 2010. He graduated from the Graduate School of Peking University in 1956, and was a postdoctoral fellow and visiting professor at Johns Hopkins University, USA, and Brunel University, UK, and a co-supervisor of PhD students at 7 Paris University. His research focuses on the antitumor, antioxidant and fast repair of DNA radical damage activities of natural products from herbs, as well as the role of oxygen radicals in cell carcinogenesis. |
 Zhongjian Jia | Zhongjian Jia is a professor at the College of Chemistry and Chemical Engineering, Lanzhou University, and graduated from the Department of Chemistry, Graduate School of Peking University in 1958. She won the 2009 National Prize for Natural Sciences, and the Gold Cattle Prize awarded jointly by the National Development Program Commission, Ministry of Education and Ministry of Sciences and Techniques of China. Her research field is natural organic chemistry, especially, the extraction, isolation, elucidation and bioactivities of natural products from folk herbs grown in the Northwest Plateau of China. |
 Ji Li | Ji Li received his PhD degree in Prof. Rongliang Zheng's laboratory in 1998. He completed his molecular signaling postdoctoral training at Sichuan University and the National Institute on Aging/NIH during 1998–2002. He was appointed as a research faculty member at Yale University School of Medicine from 2003 to 2007. He then set up his laboratory in the University of Wyoming in 2007. He is currently assistant professor at the State University of New York (SUNY) at Buffalo; his group focuses on investigating the signaling mechanisms of aging-associated cardiovascular diseases. |
 Shuangsheng Huang | Shuangsheng Huang received his PhD degree in cell biology in 2005 under the supervision of Prof. Rongliang Zheng. During his PhD studies, he studied the biphasic regulating effects of reactive oxygen species on angiogenesis. He is currently working at the Northwest University for Nationalities as an associate professor. His interests focus mainly on reactive oxygen species and tumor angiogenesis. |
 Ping Mu | Ping Mu received her PhD degree from the Institute of Biophysics, School of Life Sciences, Lanzhou University in China in 2007, before working as a postdoctoral fellow in Washington State University, USA. During her PhD studies, she focused on the biphasic regulating effects of reactive oxygen species on angiogenesis as well as the anti-angiogenesis and anti-tumorigenesis of natural compounds. |
 Fangxin Zhang | Fangxin Zhang is a chief professor of the Department of Gastroenterology in Lanzhou General Hospital, Gansu, China. He obtained his medical doctor's degree from Xijing Hospital, Fourth Military Medical University, Xi'an, China, in 1997. His research is in the field of digestive tract tumors, especially in early cancer diagnosis and treatment, and in the prevention of plateau gastrointestinal diseases. He won the prize of outstanding researcher in the medical and health sector of Gansu Province. |
1. Introduction
DNA damage or mutagenesis is now documented as a fundamental molecular basis of all forms of cancer.1 The DNA damage is a biomarker of carcinogenesis.2 Accumulation of DNA damage could lead to increased genomic instability that in turn could lead to a more malignant phenotype of tumor. Thus, DNA damage drives cancer development. To prevent DNA damage, organisms have evolved DNA repair machinery. Francis Crick, who won the Nobel prize for the discovery of DNA structure, said in 1974: “I later came to realize that DNA is so precious that probably many distinct repair mechanisms would exist. Nowadays one could hardly discuss mutation without considering repairs at the same time.” Various types of cancer involve defects in DNA repair.3Mismatch repair or replication errors of the genetic code in the form of mutation or chromosomal aberrations increase the DNA damage and risk of cancer. Mice lacking a precise DNA repair activity show hypersensitivity to DNA-damaging agents, genetic instability, and elevated cancer rates.4 Individuals with an inherited impairment in DNA repair capability are often at increased risk of cancer.5 Failure of DNA repair can lead to hereditary nonpolyposis colon cancer,6 the human hereditary Xeroderma pigmentosum,7 which is characterized by about a 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000-fold increased risk of skin cancer, and some forms of breast cancer. Experimental animals with genetic deficiencies in DNA repair often show increased cancer incidence.8 All of this evidence reflects the critical importance of DNA repair in protecting against mutagenesis and carcinogenesis.
000-fold increased risk of skin cancer, and some forms of breast cancer. Experimental animals with genetic deficiencies in DNA repair often show increased cancer incidence.8 All of this evidence reflects the critical importance of DNA repair in protecting against mutagenesis and carcinogenesis.
Not only does DNA damage trigger the development of cancer, but may trigger cancer cell-cycle arrest and death.9 Correspondingly, there are two strategies for cancer prevention relating to DNA repair. One strategy is to inhibit cancer cells’ DNA repair activity, leading to accumulation of extensive and massive DNA steady damage and finally resulting in selective cancer cell death.10 Another potential strategy is oppositely to enhance the repair mechanism, especially the fast repair mechanism, leading to elimination or abolition of DNA damage and finally resulting in cancer prevention. Both strategies illustrate the essentiality of DNA repair mechanisms in cancer prevention and therapy. The results of some recent studies of the latter potential strategy that may shed light on this important area are summarized in this review.
2. Fast repair of DNA radicals
2.1 The significance of fast repair
DNA is very sensitive to carcinogens including radiation, ultraviolet light and genotoxins. Almost all the carcinogens can be metabolized into free radical form. When DNA is attacked by free radicals, the primary products are DNA radicals, synonymous to DNA transient damage.11 A number of these DNA radicals have been identified in living cells, tissues, and in whole animals as well.12 As all other radicals, DNA radicals are chemically extreme active. They may undergo intramolecular rearrangement or react with proteins and lipids to form DNA–protein and DNA–lipid cross-links, and in this case, finally become DNA steady damage.Although DNA steady damage can be repaired by the enzymatic repair system, this takes place over a time scale of hours,13 while DNA transient damage, DNA radicals, are known to possess half-lives of only seconds,14 so the enzymatic repair mechanism cannot repair the transient damage.
Obviously, how to protect against DNA radicals is a serious problem in living things. Fortunately, there is another repair mechanism named “fast repair”, “non-enzymatic repair” or “chemical repair”, which operates by a quite different mechanism and has been recognized only in recent years.15 The fast repair process is initiated and finished within a microsecond time scale,16 and is enormously faster, by approximately 9 orders, than enzymatic repair, hence the term fast repair. Therefore, fast repair is able to eradicate the headstream of DNA damage, DNA radicals, exclusively.
Secondly, although DNA damage can be recognized by enzymes, and, thus, can be repaired by enzymatic repair, a mutation cannot be recognized by enzymes, and, thus, cannot be repaired by enzymes. However, both DNA transient damage and mutation can be prevented in their extremely early stages by fast repair.
2.2 Three disadvantages of enzymatic repair
Although, the major repair system of organisms is the known enzymatic repair, involving a series of enzymes, it is not perfect and has 3 disadvantages:1) DNA repair enzymes themselves can be easily damaged by various stresses and lose their function.17
2) During the processes of aging and disease, or metabolism of xenobiotics, most enzymatic repair activities decrease.18
3) Some repairing enzymes are error prone during DNA replication, and may lead to genomic instability, even tumorigenesis.10
2.3 Mechanism of fast repair
Fast repair works through reacting directly with DNA radicals via one electron transfer processes without any enzymes,15,19 which has been demonstrated in both experimental and theoretical studies.Based on the experimental data, a mechanism involving electron transfer between phytophenols and damaged DNA has been proposed.15 For example, in the repair of the hydroxyl adducts of dGMP by verbascoside (VER), one kind of phytophenol. The primary transient absorption spectrum (λmax = 390 nm) of dGMP–OH˙ declined in a few microseconds, concurrently with the transient absorption spectrum (λmax = 310 nm) of the VER phenoxyl radical (VER–PhO˙) rising, indicating that the dGMO–OH˙ was repaired by VER, and that VER finally became VER–PhO˙. This also means that the fast repair was initiated as soon as the DNA radical was formed; that is the reason why it was called fast repair. The proposed mechanism shows dGMP being attacked by OH˙, becoming dGMP–OH˙, the latter accepting one electron from the phenolic group of verbascoside and then repaired to dGMP. The reactions are as follows:
| dGMP |
| –OH˙ + VER–PhOH → VER–PhO˙ + dGMP–OH |
| − |
| + H |
| + |
| (dGMP–OH) |
| − |
| → dGMP or hydrated dGMP |
 | ||
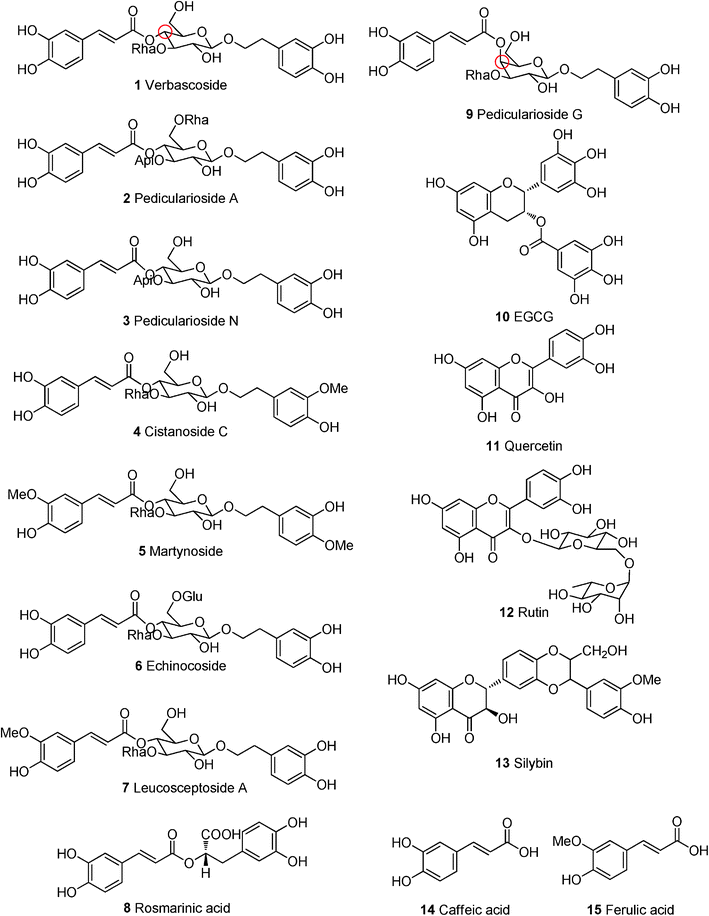
| Fig. 1 Cistanoside C (left side in red) docks into the minor groove of DNA with telomere sequence (TTAGGG)3 (right side). | ||
2.4 Many natural compounds have fast repairing activity
Willson et al.22 first discovered that cysteamine and promethazine can rapidly repair DNA radicals with a reaction rate constant of 2.8 × 109 M−1.s−1. They proposed that this is a new model of DNA repair. Later O'Neill et al.23 further found that a series of endogenous reductants can fast repair DNA radicals. Five catechins of green tea, dopamine and uric acid,24 phytophenols19b including caffeic acid25 and hydroxycinnamic acid derivatives26 can fast repair DNA radicals. Two phenylpropanoid glycosides (PPGs), verbascoside (i.e.acteoside) and angoroside C, extracted from the roots of Scrophularia ningpoensis (traditional Chinese medicine name: Xuanshen) were able to repair the dAMP– and dGMP–OH˙ adducts effectively.27Silybin, a natural flavonoid, isolated from the seeds of Silybum marianum, a traditional Chinese herb, repaired the dGMP–OH˙adduct within tens of microseconds. Other flavones (hesperetin, naringin and naringenin) also had repair activities but which were lower than that of silybin.28Some phenylpropanoid glycosides, one kind of phytophenol, with different structures isolated from Tibetan folk herbs by us,29 and some other phytophenols (Fig. 2) are able to repair all 3 types of DNA radicals: DNA anions, DNA cations and DNA–OH˙ adducts (Table 1). Repair rate constants as high as 109 M−1s−1 were measured using the pulse radiolysis technique and transient absorption spectral method.15
 | ||
| Fig. 2 Structures of some phytophenols. | ||
| Phytophenol | Type of DNA | Damaged DNA | Reference |
|---|---|---|---|
| Verbascoside | Base | T–OH˙ | 16a |
| Base | T˙ − | 36a | |
| Nucleotide | dAMP(–H)˙ | 34a | |
| Nucleotide | dAMP˙ − | 36c | |
| Nucleotide | dCMP(–H)˙ | 34a | |
| Nucleotide | dGMP–OH˙ | 16b | |
| Nucleotide | dGMP–OH˙ | 32c | |
| Nucleotide | TMP˙ − | 36b | |
| Poly-nucleotide | polyG–OH˙ | 32a | |
| Pedicularioside A | Base | T–OH˙ | 32b |
| Base | T˙ − | 36a | |
| Nucleotide | dAMP–OH˙ | 16b | |
| Nucleotide | dGMP–OH˙ | 16b | |
| Nucleotide | TMP˙ − | 36b | |
| Nucleotide | dAMP˙ − | 36c | |
| Nucleotide | dAMP(–H)˙ | 34a | |
| Nucleotide | dCMP(–H)˙ | 34a | |
| Poly-nucleotide | polyC˙+ | 34b | |
| Pedicularioside N | Base | T˙ − | 36a |
| EGCG | Nucleotide | dGMP–OH˙ | 26 |
| Cistanoside C | Nucleotide | dAMP–OH˙ | 16b,34a |
| Nucleotide | dAMP(–H)˙ | 36c | |
| Nucleotide | dAMP˙ − | ||
| Nucleotide | dCMP(–H)˙ | 34a | |
| Nucleotide | dGMP–OH˙ | 16b | |
| Nucleotide | dGMP(–H)˙ | 34a | |
| nucleotide | polyG–OH˙ | 32a | |
| Nucleotide | TMP˙ − | 36b | |
| Poly-nucleotide | polyC˙+ | 34b | |
| Strand of DNA | dsDNA˙+ | 34b | |
| Martynoside | Base | T˙ − | 36a |
| Echinocoside | Base | T˙ − | 48b |
| Leucosceptoside A | Base | T˙ − | 36a |
| Quercetin | Nucleoside | dT˙ − | 36d |
| Nucleotide | dAMP(–H)˙ | 34d | |
| Nucleotide | dCMP(–H)˙ | 34c | |
| Nucleotide | dGMP–OH˙ | 32d | |
| Nucleotide | dGMP(–H)˙ | 34d | |
| Poly-nucleotide | polyC+ | 34d | |
| Rutin | Nucleoside | dT− | 36d |
| Nucleotide | dCMP(–H) | 34d | |
| Nucleotide | dCMP(–H) | 34c | |
| Nucleotide | dGMP–OH˙ | 32d | |
| Nucleotide | dGMP(–H) | 34d | |
| Poly-nucleotide | polyC˙+ | 34d | |
| Silybin | Nucleotide | dGMP–OH˙ | 28 |
| Caffeic acid | Base | T˙ − | 25 |
| Nucleoside | dT˙ − | 25 | |
| Ferulic acid | Nucleotide | dGMP–OH˙ | 26 |
2.5 Fast repair in cells
All experiments mentioned above were carried out in chemical systems. In tissues and cells, the reactions should be more complex. O'Neill and Chapman23b inferred that fast repair activity should exist in cells for a series of endogenous reductants, such as ascorbate, thiols and phenols, etc. However, due to the difficulty of capturing the fast repair process and detecting its products, there have been no studies following up on their speculation until recently. We demonstrated that the fast repair mechanism by rosmarinic acid and verbascoside,30 or by quercetin and rutin31 could also occur in living cells. Thus, the fast repair mechanism of phytophenols may be universal both in vitro and in cells. Obviously, the experiments in cells have more significance in the clinic. Further studies are still required before any firm conclusions are reached.3. DNA radicals disrupt genomic integrity
3.1 DNA–OH˙ adducts
Among thousands of known carcinogens, the hydroxyl radical HO˙ is the simplest and strongest, is extremely short-lived and reacts rapidly with almost every cellular biomolecule including DNA. The hydroxyl radical attacks DNA resulting in the formation of a DNA–OH˙ adduct. We found that attack of DNA by OH˙ radical results in the geometry of DNA being largely perturbed, such as increasing the distance between a base pair, finally leading to the hydrogen bond of the base pair being broken (Fig. 3).21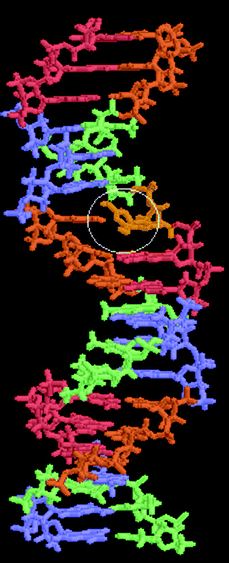 | ||
| Fig. 3 The hydrogen bonds of a base pair are broken in a DNA–OH˙ adduct. | ||
The OH˙ adduction reaction is too fast to prevent, therefore, strategies for elimination of DNA damage induced by OH˙ should focus on the elimination of the secondary DNA radicals by reductant via fast repair. Different structures of phytophenols were proven to fast repair 7 types of DNA–OH˙ adducts, including T–OH˙, dT–OH˙, dAMP–OH˙, dGMP–OH˙, polyG–OH˙, ssDNA–OH˙ and dsDNA–OH˙.16,32
3.2 DNA cations
Many kinds of chemical carcinogens can be activated by metabolic pathways to form oxidizing intermediates, which then oxidize cellular DNA to generate DNA base radical cations.33 The direct energy deposition of radiation in DNA can also generate base radical cations. The OH˙ adducts of purine bases may undergo dehydration to generate radical cation type radicals, dAMP(–H)˙ and dGMP(–H)˙. Different phytophenols are able to fast repair 6 types of DNA cations,34 including dAMP(–H)˙, dCMP(–H)˙, dGMP(–H)˙, polyC˙+, dsDNA˙+ and dsDNA˙+.3.3 DNA anions
DNA bases have a very high intrinsic reactivity with hydrated electrons35 and generate base electron adducts, the synonym, base radical anions, which are responsible for inducing strand breaks and forming stable base lesions. Phytophenols are able to fast repair 4 types of DNA radical anions, including T˙−, dT˙−, TMP˙− and dAMP˙−.36Moreover, phytophenols can fast repair not only damage to bases, deoxynucleosides and deoxynucleotides but also damage to integral DNA, such as poly C, poly G and dsDNA radicals, with the latter being closer to cellular conditions.16b,32a,34
All above 5 different structural levels of DNA radicals, such as base–, nucleosid–, nucleotid–, polynucleotid– or strand DNA–radicals are able to induce DNA strand breaks and form stable damage.37 This demonstrates that the fast repair machinery acts as a rescuer of the genome to maintain genomic integrity.
4. Phytophenols with fast repair activity also suppress hallmarks of cancer
The concept of six fundamental and common hallmarks and two new emerging hallmarks of all cancers was summarized with rather wide acceptance.38 The two relevant papers have been referenced over 10000 times by other research papers. Genomic instability is clearly an enabling characteristic that is causally associated with the hallmarks of cancer. Thus the importance of repairing DNA damage in the course of tumor progression was emphasized by the two papers. In fact, the fast repair machinery maintains genomic integrity and results in decreased cancer incidence. Therefore fast repair of DNA damage will logically be able to prevent incipient cancers from developing and will cure preexisting cancers (Fig. 4).
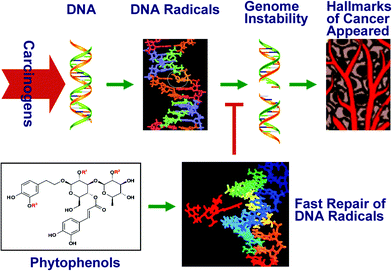 | ||
| Fig. 4 Graphical illustration of the fast repair activity suppressing the hallmarks of cancer. | ||
Almost all of the above-mentioned phytophenols (except one unique case: angoroside C, for which no report of its antitumor activity has been found), with fast repair activity are able to suppress the hallmarks of cancer (Table 2), involving genomic instability, mutation, proliferative signaling, immortality, telomerase activity, telomere maintenance, angiogenesis, invasion and metastasis, which have been described in the above two papers.
| Phytophenols | Suppressed hallmarks of cancer | Reference |
|---|---|---|
| a Note: although rosmarinic acid and pedicularioside G have not been measured for fast repair activity, their structures are very similar to that of verbascoside. | ||
| Verbascoside | Inhibited the proliferation of human leukemia HL-60 cells | 39 |
| Induced human leukemia HL-60 cell redifferentiation into monocytes | 39 | |
| Inhibited lung metastasis of melanoma cells in mice and prolonged survival time significantly | 40 | |
| Decreased human gastric adenocarcinoma cell growth curve, mitotic index and cell surface charge, reduced microvilli on the surface, delayed doubling time | 41 | |
| Decrease tumorigencity (75%) when inoculated subcutaneously in nude mice | 41 | |
| Induced gastric carcinoma cell apoptosis by telomerase inhibition and telomere shortening | 43 | |
| Significantly inhibited tumor-induced angiogenesis and down-regulated MMPs expression, which are necessary for blood vessel invasion of the tumor tissue. | 44 | |
| An antiestrogen in breast cancer cells | 46b | |
| The antitumor activity of L-1210 cells may be due to inhibition of protein kinase C | 46a | |
| Inhibited PMA-induced invasion and migration of human fibrosarcoma cellsviaCa2+-dependent CaMK/ERK and JNK/NF-κB-signaling pathways | 45 | |
| Rosmarinic acid (analog of verbascoside)a | Inhibited human umbilical vein endothelial cell proliferation, migration, adhesion and tube formation | 49 |
| Reduced murine tumorigenesis | 48c | |
| Pedicularioside G (isomer of verbascoside)a | Inhibited hepatoma cell growth, cell migration, angiogenesis and tumorigenesis | 47 |
| Pedicularioside A | Against the growth and viability of human hepatoma cells, human pulmonary adenocarcinoma cells and human gastric adenocarcinoma cells | 48d |
| Pedicularioside N | Against the growth and viability of human hepatoma cells, human pulmonary adenocarcinoma cells and human gastric adenocarcinoma cells | 48d |
| Tea phenols including EGCG | Inhibited human prostate carcinoma, chemoprevention of squamous cell carcinoma of the head and neck | 53,54b,55 |
| Inhibited growth of peripheral blood T lymphocytes of adult T-cell leukemia patients | 54b | |
| Suppressed cell proliferation and enhanced apoptosis, inhibited cell invasion, angiogenesis and metastasis | 54a,58 | |
| Suppressed angiogenesis in lung tumorigenesis in mice | 57a | |
| Reduced cell proliferation, surface charge and microvilli, and DNA damage in human hepatoma cells | 57b | |
| Inhibited tumorigenesis in a variety of organs, including skin, lung, oral cavity, esophagus, stomach, small intestine, colon, liver, pancreas, ovary, and breast | 51 | |
| Suppressed various tumor promotion and mitogenic signals, and blocked signal transduction | 52 | |
| Inhibited telomerase activity and led to telomerase fragmentation | 56 | |
| Cistanoside C | Inhibited growth of human hepatoma cells, human lung adenocarcinoma cells and human gastric adenocarcinoma cells | 48a |
| Martynoside | Decreased the growth and viability of some cancer cells, but did not affect normal cells | 48c |
| Echinocoside | An antiestrogen in breast cancer cells | 45,48c |
| Leucosceptoside A | Against the growth and viability of human hepatoma cells, human pulmonary adenocarcinoma cells and human gastric adenocarcinoma cells | 48d |
| Decreased the growth and viability of some cancer cells, but did not affect normal cells | 48c | |
| Quercetin | Chemoprevention of human colorectal cancer | 60 |
| Inhibited azoxymethane-induced colorectal carcinogenesis in rats | 62 | |
| Decreased pancreatic cancer growth, increased apoptosis and prevented metastasis in nude mice | 63 | |
| Inhibited proliferation and mutant p53 protein expression of human breast cancer cells | 64 | |
| Induced human leukemia HL-60 cell apoptosis | 65 | |
| Rutin | Chemoprevention of human colorectal cancer, increased apoptotic index of colonic crypts, reduced the number of focal areas of dysplasia in mice | 60–61 |
| Reduced the formation of metastasis nodules of melanoma cells | 66 | |
| Silybin | Inhibited invasiveness of human hepatoma cells. Silybin is now being tested in clinical trials to treat prostate cancer, ovarian cancer, and skin cancer | 67–68,73 |
| Caffeic acid | Suppressed the growth and metastasis of human hepatoma cells in nude mice | 69 |
| Ferulic acid | Antiproliferation and apoptosis of human breast cancer cells | 70 |
| Induced apoptosis of human hepatoma cells | 71 | |
| Preventing cancer induced by carcinogenic compounds in mice | 72a |
4.1 Phenylpropanoid glycosides: verbascoside, pedicularioside, cistanosideetc.
Verbascoside (compound 1) inhibited the proliferation of human promyelocytic HL-60 leukemia cells in a concentration- and time-dependent manner, and induced HL-60 redifferentiation into monocytes.39 Verbascoside showed a suppressive effect on lung metastasis of melanoma cells in mice and prolonged survival time significantly, it is thus to be regarded as a possible antimetastatic agent.40Results of our study showed that verbascoside decreased the cell growth curve, mitotic index and cell surface charge, and delayed the doubling time of human gastric adenocarcinoma cells. The microvilli on the surface of the treated cells were reduced significantly (Fig. 5). There was a 75% decrease of the tumorigenicity for the treated cells compared with the untreated cells inoculated subcutaneously in nude mice. It confirmed that verbascoside could reverse gastric adenocarcinoma cells' malignant phenotypic characteristics.41
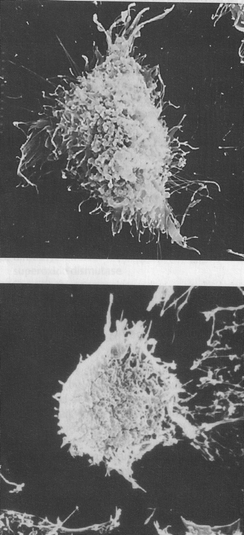 | ||
| Fig. 5 Reverse transformation of human gastric adenocarcinoma cell line MGc80-3 (upper) induced by 20 μmol l−1 verbascoside for 7 days, the abundant amounts of microvilli and microfilaments were reduced significantly (lower). Reproduced by permission of Georg Thieme Verlag KG Publishers. | ||
Suppression of telomerase activity leading to telomere shortening has been rationalized as a crucial anticancer defence.38 Although telomerase inhibiting activity is not a universal mechanism for all antitumor drugs, it is just one of the several possible mechanisms.42 We found that verbascoside inhibited the telomerase activity of human gastric carcinoma cells MKN45 with an IC50 of 17.8 μg ml−1, shortened the average telomere length and displayed a cell cycle sub-G0/G1 peak and G2/M arrest, implying that verbascoside may induce tumor cell apoptosis by telomerase inhibition and telomere shortening.43
Tumors cannot grow beyond a few millimetres without blood supply from the host tissue. Tumors require nutrients and oxygen as well as an ability to evacuate metabolic waste and carbon dioxide. The tumor-associated neovasculature, generated by the process of angiogenesis, addresses these needs. Inhibition of angiogenesis would deprive the growing tumor of nutrients and oxygen supplied by the host circulation and consequently would retard or even abolish tumor growth. Verbascoside significantly inhibited tumor-induced angiogenesis and down-regulated MMPs expression, which are necessary for blood vessel invasion of the tumor tissue.44 Verbascoside inhibited PMA-induced invasion and migration of human fibrosarcoma cellsviaCa2+-dependent CaMK/ERK and JNK/NF-κB-signaling pathways.45 There are 2 other references related to this topic.46
Pedicularioside G (Ped G, compound 9) is an isomer of verbascoside (compound 1), with only a conformational nuance between both: the link of the caffeyl-C4 of glucose (Fig. 2, red circle) in verbascoside is an axial bond, while in Ped G it is an equatorial bond.
Ped G inhibited tumor growth, cell migration, angiogenesis and tumorigenesis.47
Ped G inhibited vascular formation of solid tumors in vivo. A suspension of human hepatoma cells was inoculated onto a chicken embryo chorioallantoic membrane; after 3 days, grafted cells yielded solid tumors that were vascularized. The distribution of vessels radiated around the solid tumor. Ped G treatment for another 3 days evidently suppressed the tumor growth and vessel density, and the radiating vessels disappeared. After solid tumor formation, no chick embryos died, which means that Ped G is not toxic to the chicken embryo chorioallantoic membrane itself. Moreover, the malignant hepatoma cells could be reverse transformed by Ped G. Intact human hepatoma cells were inoculated onto the chorioallantoic membranes of chick embryos. They yielded a light colored disc of tumor lump, whereas in cells pre-incubated with Ped G for 24 h, the tumorigenic ability decreased remarkably in a concentration-dependent manner (Fig. 6).
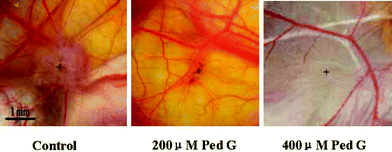 | ||
| Fig. 6 After pretreatment with pedicularioside G, low tumorigenicity and angiogenic activity of human hepatoma cells transplanted in chicken embryo chorioallantoic membranes is exhibited. + represents the centre of a tumor lump, which is a visible light colored disc, but the disc is very thin and small in the Ped G treated groups. Reproduced by permission of Wiley-Blackwell Publishing. | ||
Cistanoside C (compound 4) inhibited the growth of human hepatoma cells, human lung adenocarcinoma cells and human gastric adenocarcinoma cells.45
The effects of martynoside (compound 5), echinocoside (compound 6), and leucosceptoside A (compound 7)48 are listed in Table 2.
4.2 Analogs of verbascoside: rosmarinic acid
Rosmarinic acid (compound 8), a very interesting phytophenolic compound, contains two arms, conjugated and non-conjugated chains, the same as those of verbascoside, but without the saccharide backbone between the two arms (Fig. 2). Rosmarinic acid inhibited several important steps of angiogenesis including cell proliferation, migration, adhesion and tube formation of human umbilical vein endothelial cells in a concentration-dependent manner.49Rosmarinic acid markedly reduced tumorigenesis in a murine, two-stage skin carcinogenesis model by two independent effects: anti-inflammatory activities and inhibition of DNA damage.504.3 Tea polyphenols
Tea polyphenols have been proved to be the main effective components in tea for cancer prevention.51 The suppression of various tumor promotion and mitogenic signals, and the blockage of signal transduction, induction of apoptosis in cancer cells, reverse transformation of carcinogenesis prior to invasion and metastasis, and inhibition of angiogenesis in animal models by tea polyphenols have been demonstrated.52 The green tea polyphenols, particularly EGCG (epigallocatechin gallate, compound 10), possess remarkable potential for the chemoprevention of squamous cell carcinoma of the head and neck,53 inhibit growth of the peripheral blood T lymphocytes of adult T-cell leukemia patients by suppressing HTLV-1 pX gene expression and inducing apoptosis cell death.54EGCG inhibited the proliferation in both androgen sensitive and insensitive human prostate carcinoma cells, and this effect was mediated by deregulation in the cell cycle and induction of apoptosis.55 EGCG has also been shown to inhibit telomerase activity and lead to telomerase fragmentation.56 There is considerable evidence that tea polyphenols result in the suppression of cell proliferation and enhancement of apoptosis, as well as the inhibition of cell invasion, angiogenesis and metastasis.57 The suppression of angiogenesis by treatment with green tea in a lung tumorigenesis model has been observed.58 We found that tea polyphenols reduce cell proliferation, surface charge and microvilli, and DNA damage in human hepatoma cells.57b As mentioned above, tea polyphenols are able to fast repair DNA radicals;26 coincidentally, the physical binding of tea polyphenols to DNA was observed.594.4 Flavonoids: quercetin, rutin and silybin
Quercetin (Q, compound 11) and rutin (R, compound 12) have been reported as the most predominant polyphenols in the human diet. R is a glycoside of Q. Both Q and R have demonstrated chemopreventive activity in a variety of laboratory animal models. The chemopreventive action of Q and R in human colorectal cancer has been reviewed.60 Supplementary dietary Q and R markedly increased the apoptotic index of colonic crypts and reduced the number of focal areas of dysplasia in azoxymethane-treated mice.61 Q, but not R, inhibited azoxymethane-induced colorectal carcinogenesis in rats.62 Q decreased primary tumor growth, increased apoptosis and prevented metastasis in a model of pancreatic cancer by inducing mitochondrial dysfunction, resulting in cytochrome c release, caspase activation and apoptosis.63 Q inhibited human breast cancer cells and the expression of the mutant p53 protein.64 Q is able to induce apoptosis in human leukemia HL-60 cells by stimulation of the release of mitochondrial cytochrome c into cytosol, procaspase-9 processing, and through a caspase-3-dependent mechanism.65Rutin reduced the formation of metastasis nodules of melanoma cells.66Silybin (compound 13) is now being tested in clinical trials to treat prostate cancer67, ovarian cancer,67a and skin cancer.67bSilybin inhibited MMP-2,-9 release as well as invasiveness of HepG2 cells.68 MMP-9 (matrix metalloproteinase 9) is the angiogenic enzyme, directly involved in human hepatic tumorigenesis, invasion and metastasis.
4.5 Hydroxycinnamic acid : caffeic acid and ferulic acid
Caffeic acid (compound 14) being administered subcutaneously or orally, suppressed the growth and metastasis of HepG2 tumor xenografts in nude mice. Caffeic acid is a strong and specific inhibitor of MMP-9 activity and transcription. Thus caffeic acid represents the best candidate for achieving tumor regression.69Ferulic acid (compound 15), a hydroxycinnamic acid, is biosynthesized from caffeic acid. Animal and in vitro studies suggest that ferulic acid may have direct antitumor activity against breast cancer70 and liver cancer,71 and may be effective at preventing cancer induced by exposure to carcinogenic compounds.72
5. Conclusions and perspective
Enhancement of the fast repair mechanism to lower levels of DNA damage may be required for cancer prevention, while elimination of the fast repair mechanism to aggravate DNA damage may be required during chemical therapy and/or radiation therapy. Therefore, the fast repair of DNA damage would be closely related to all strategies of prevention and intervention of cancer .It is worth noting that the correlation between fast repair and cancer prevention must be carefully interpreted, as correlation does not imply causation. If such causation exists, substantial studies would be needed in future.
Besides, the DNA radicals per se may induce mutation and cancer directly, but also may undergo intramolecular rearrangement or react with proteins and lipids, and in this case, DNA radicals are considered to play a causative role in aging and several degenerative diseases, such as Alzheimer's disease, Parkinson's disease, cognitive dysfunction, heart disease, cataracts, Xeroderma pigmentosum, ataxia telangiectasia, Bloom's syndrome, Cockayne syndrome and various types of inflammatory disease. There seems to be little doubt that the fast repair mechanism could be valuable to protect against these chronic and stubborn diseases, and to develop a broad pharmacological action potential. Unfortunately, knowledge of the fast repair of DNA radicals in many diseases is still in infancy. More in-depth understanding of its physiological and pathophysiological significance is awaited. Simpler and easier detection methods for DNA radicals in cells are anxiously expected, so that the fast repair mechanism may attract more scientists from many disciplines and develop at a faster pace.
Acknowledgements
The National Natural Sciences Foundation of China grants (29972017, 39870902 and 39270193), The China–France Cooperation Research sponsored by the Ministry of Education of China (001230), The China–France Advanced Research Program of the Ministry of Sciences and Technology of China (SI 00-05) provided support for this study.We thank Professors Nianyun Lin, Side Yao and Wenfeng Wang (Laboratory of Radiation Chemistry, Shanghai Institute of Applied Physics, Chinese Academy of Sciences, Shanghai, China) for the valuable guide on experiments into fast repair activity. The late Professor Botao Fan (ITODYS, CNRS UMR7086, Université Paris 7, Paris, France) contributed to the guide on the theoretical studies of phytophenol docking into DNA. He regrettably died of liver cancer in 2007. We also thank Dr Changjun Lin for the excellent illustration of the graphical abstract.
The authors declare no potential conflict of interest relevant to this article.
References
- J. H. J. Hoeijmakers, Nature, 2001, 411, 366 CrossRef CAS.
- H. Kasai, Mutat. Res., Rev. Mutat. Res., 1997, 387, 147 CrossRef CAS.
- M. F. Rajewsky, J. Engelbergs, J. Thomale and T. Schweer, Mutat. Res., Rev. Mutat. Res., 2000, 462, 101 CrossRef CAS.
- D. M. Wilson, 3rd, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 12754 CrossRef.
- C. Bernstein, H. Bernstein, C. M. Payne and H. Garewal, Mutat. Res., Rev. Mutat. Res., 2002, 511, 145 CrossRef CAS.
- (a) L. A. Aaltonen, P. Peltomaki, F. S. Leach, P. Sistonen, L. Pylkkanen, J. P. Mecklin, H. Jarvinen, S. M. Powell, J. Jen and S. R. Hamilton, Science, 1993, 260, 812 CrossRef CAS; (b) E. C. Friedberg, Nature, 2003, 421, 436 CrossRef.
- J. E. Cleaver, Nature, 1968, 218, 652.
- T. Gansler, P. A. Ganz, M. Grant, F. L. Greene, P. Johnstone, M. Mahoney, L. A. Newman, W. K. Oh, C. R. Thomas, M. J. Thun, A. J. Vickers, R. C. Wender and O. W. Brawley, Ca-Cancer J. Clin., 2010, 60, 345 Search PubMed.
- J. H. J. Hoeijmakers, N. Engl. J. Med., 2009, 361, 1475 CrossRef CAS.
- T. A. Yap, S. K. Sandhu, C. P. Carden and J. S. de Bono, Ca-Cancer J. Clin., 2011, 61, 31 Search PubMed.
- S. Steenken, Chem. Rev., 1989, 89, 503 CrossRef CAS.
- S. E. Gomez-Mejiba, Z. Zhai, H. Akram, L. J. Deterding, K. Hensley, N. Smith, R. A. Tomer, K. B. Tomer, R. P. Mason and D. C. Ramirez, Free Radical Biol. Med., 2009, 46, 853 CrossRef CAS.
- (a) P. Jaruga and M. Dizdaroglu, Nucleic Acids Res., 1996, 24, 1389 CrossRef CAS; (b) F. M. Yakes and B. Van Houten, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 514 CrossRef CAS.
- D. Schulte-Frohlinde, G. Behrens and A. Onal, Int. J. Radiat. Biol., 1986, 50, 103 CAS.
- R. L. Zheng, Y. M. Shi, Z. J. Jia, C. Y. Zhao, Q. Zhang and X. R. Tan, Chem. Soc. Rev., 2010, 39, 2827 RSC.
- (a) W. Y. Li, R. L. Zheng, B. N. Su, Z. J. Jia, H. C. Li, Y. Jiang, S. Yao and N. Y. Lin, Int. J. Radiat. Biol., 1996, 69, 481 CrossRef CAS; (b) Y. Shi, W. Lin, B. Fan, Z. Jia, S. Yao, J. Kang, W. Wang and R. Zheng, Biochim. Biophys. Acta, Gen. Subj., 1999, 1472, 115 CrossRef CAS.
- L. A. Loeb, Cancer Res., 1989, 49, 5489.
- V. Gorbunova, A. Seluanov, Z. Mao and C. Hine, Nucleic Acids Res., 2007, 35, 7466 CrossRef CAS.
- (a) K. Gao, B. T. Fan, N. El Fassia, K. Zakrzewska, Z. J. Jia, R. L. Zheng, A. Panaye, T. Couesnon and J. P. Doucet, QSAR Comb. Sci., 2003, 22, 18 CrossRef CAS; (b) O. Sperandio, B. T. Fan, K. Zakrzewska, Z. J. Jia, R. L. Zheng, A. Panaye, J. P. Doucet and N. El Fassi, SAR QSAR Environ. Res., 2002, 13, 243 Search PubMed.
- J. Boultwood, C. Fidler, P. Shepherd, F. Watkins, J. Snowball, S. Haynes, R. Kusec, A. Gaiger, T. J. Littlewood, A. J. Peniket and J. S. Wainscoat, Am. J. Hematol., 1999, 61, 5 CrossRef CAS.
- O. Delalande, K. Gao, B. T. Fan, K. Zakrzewska, N. El Fassi, Z. J. Jia, R. L. Zheng, A. Panaye and J. P. Doucet, SAR QSAR Environ. Res., 2002, 13, 675 Search PubMed.
- R. L. Willson, P. Wardman and K. D. Asmus, Nature, 1974, 252, 323 CAS.
- (a) P. O'Neill, Radiat. Res., 1983, 96, 198; (b) P. O'Neill and P. W. Chapman, Int. J. Radiat. Biol., 1985, 47, 71 CrossRef.
- (a) R. F. Anderson, C. Amarasinghe, L. J. Fisher, W. B. Mak and J. E. Packer, Free Radical Res., 2000, 33, 91 CrossRef CAS; (b) R. F. Anderson, L. J. Fisher, Y. Hara, T. Harris, W. B. Mak, L. D. Melton and J. E. Packer, Carcinogenesis, 2001, 22, 1189 CrossRef CAS; (c) R. F. Anderson and T. A. Harris, Free Radical Res., 2003, 37, 1131 CrossRef CAS.
- Z. H. Zou, S. D. Yao, H. C. Lee, W. Z. Lin, J. S. Zhang, N. Y. Lin, JAERI-Conf, Waseda University, Tokyo, Japan, 1995 Search PubMed.
- Y. Jiang, S.-D. Yao and N.-Y. Lin, Radiat. Phys. Chem., 1997, 49, 447 CrossRef CAS.
- Y. M. Li, Z. H. Han, S. H. Jiang, Y. Jiang, S. D. Yao and D. Y. Zhu, Acta Pharmacol. Sin., 2000, 21, 1125 Search PubMed.
- H. Y. Fu, Y. S. Katsumura, M. Z. Lin, K. K. Hata, Y. Muroya and Y. Hatano, J. Radiat. Res., 2008, 49, 609 Search PubMed.
- (a) Z. J. Jia, Z. M. Liu and C. Z. Wang, Phytochemistry, 1991, 30, 3745 CrossRef CAS; (b) Z. J. Jia and J. J. Gao, Phytochemistry, 1993, 34, 1188 CrossRef CAS.
- Q. Zhang, J. Pan, C. Zhao, Y. Wang, Z. Jia and R. Zheng, Cell Biol. Int., 2008, 32, 654 CrossRef CAS.
- X. Tan, C. Zhao, J. Pan, Y. Shi, G. Liu, B. Zhou and R. Zheng, Cell Biol. Int., 2009, 33, 690 CrossRef CAS.
- (a) C. Y. Huang, Y. M. Shi, W. F. Wang, Z. J. Jia, Y. Wang, S. D. Yao, N. Y. Lin and R. L. Zheng, Pharmazie, 2003, 58, 664 CAS; (b) W. Li, Z. Zou, R. Zheng, C. Wang, Z. Jia, S. Yao and N. Lin, Radiat. Phys. Chem., 1997, 49, 429 CrossRef CAS; (c) Y. Shi, W. Wang, Y. Shi, Z. Jia, S. Yao, W. Lin, Z. Han and R. Zheng, Sci. China, Ser. C: Life Sci., 1999, 42, 621 Search PubMed; (d) C. Zhao, Y. Shi, W. Wang, W. Lin, B. Fan, Z. Jia, S. Yao and R. Zheng, Radiat. Phys. Chem., 2002, 63, 137 CrossRef CAS.
- H. Kasai and S. Nishimura, in Oxidative Stress: Oxidants and Antioxidants, ed. H. Sies, Academic Press, London, 1991 Search PubMed.
- (a) Y. Shi, J. Kang, W. Lin, P. Fan, Z. Jia, S. Yao, W. Wang and R. Zheng, Biochim. Biophys. Acta, Gen. Subj., 1999, 1472, 279 CrossRef CAS; (b) Y. Shi, W. Wang, C. Huang, Z. Jia, S. Yao and R. Zheng, Mutagenesis, 2008, 23, 19 CAS; (c) C. Zhao, Y. Shi, W. Lin, W. Wang, Z. Jia, S. Yao, B. Fan and R. Zheng, Mutagenesis, 2001, 16, 271 CrossRef CAS; (d) C. Zhao, Y. Shi, W. Wang, Z. Jia, S. Yao, B. Fan and R. Zheng, Sci. China, Ser. C: Life Sci., 2001, 44, 610 Search PubMed.
- M. G. Simic, Mutat. Res., Fundam. Mol. Mech. Mutagen., 1988, 202, 377 CrossRef CAS.
- (a) W. Li, R. Zheng, Z. Jia, Z. Zou and N. Lin, Biophys. Chem., 1997, 67, 281 CrossRef CAS; (b) Y. Shi, W. Lin, P. Fan, Z. Jia, S. Yao, J. Kang, W. Wang and R. Zheng, Radiat. Phys. Chem., 2000, 58, 131 CrossRef CAS; (c) Y. Shi, W. Wang, B. Fan, Z. Jia, S. Yao and R. Zheng, Biochim. Biophys. Acta, Gen. Subj., 2000, 1474, 383 CrossRef CAS; (d) C. Zhao, Y. Shi, W. Wang, Z. Jia, S. Yao, B. Fan and R. Zheng, Biochem. Pharmacol., 2003, 65, 1967 CrossRef CAS.
- (a) P. J. Boon, P. M. Cullis, M. C. R. Symons and B. W. Wren, J. Chem. Soc., Perkin Trans. 2, 1984, 1393 RSC; (b) R. Teoule, in Effects of Ionizing Radiation on DNA, ed. J. Huttermann, Springer, Berlin, 1978 Search PubMed.
- (a) D. Hanahan and R. A. Weinberg, Cell, 2000, 100, 57 CAS; (b) D. Hanahan and R. A. Weinberg, Cell, 2011, 144, 646 CrossRef CAS.
- K. W. Lee, H. J. Kim, Y. S. Lee, H. J. Park, J. W. Choi, J. Ha and K. T. Lee, Carcinogenesis, 2007, 28, 1928 CrossRef CAS.
- T. Ohno, M. Inoue, Y. Ogihara and I. Saracoglu, Biol. Pharm. Bull., 2002, 25, 666 CrossRef CAS.
- J. Li, Y. Zheng, H. Zhou, B. Su and R. Zheng, Planta Med., 1997, 63, 499 CrossRef CAS.
- C. M. Wang, Z. J. Jia and R. L. Zheng, Planta Med., 2007, 73, 180 CrossRef CAS.
- F. Zhang, Z. Jia, Z. Deng, Y. Wei, R. Zheng and L. Yu, Planta Med., 2002, 68, 115 CrossRef CAS.
- M. Wartenberg, P. Budde, M. de Marees, F. Grunheck, S. Y. Tsang, Y. Huang, Z. Y. Chen, J. Hescheler and H. Sauer, Lab. Invest., 2003, 83, 87 Search PubMed.
- Y.P. Hwang, H.G. Kim, J.H. Choi, B.H. Park, M.H. Jeong, T.C. Jeong and H. G. Jeong, Mol. Nutr. Food Res., 2011, 55, S103–S116 CrossRef.
- (a) J. M. Herbert, J. P. Maffrand, K. Taoubi, J. M. Augereau, I. Fouraste and J. Gleye, J. Nat. Prod., 1991, 54, 1595 CrossRef CAS; (b) Z. Papoutsi, E. Kassi, S. Mitakou, N. Aligiannis, A. Tsiapara, G. P. Chrousos and P. Moutsatsou, J. Steroid Biochem. Mol. Biol., 2006, 98, 63 CrossRef CAS.
- P. Mu, X. Gao, Z. J. Jia and R. L. Zheng, Basic Clin. Pharmacol. Toxicol., 2008, 102, 30 Search PubMed.
- (a) J. Li, R. L. Zheng, Z. M. Liu and Z. J. Jia, Chin. Pharma. J., 1995, 30, 269 Search PubMed; (b) W. Li, R. Zheng, S. Zhao, Y. Jiang and N. Lin, Sci. China, Ser. C, 1996, 544 CAS; (c) I. Saracoglu, M. Inoue, I. Calis and Y. Ogihara, Biol. Pharm. Bull., 1995, 18, 1396 CrossRef CAS; (d) R. L. Zheng and Z. J. Jia, International Symposium on Natural Antioxidants Molecular Mechanisms and Health Effects, AOCS Press, Champaign, IL, USA, 1995, p. 64 Search PubMed.
- S. S. Huang and R. L. Zheng, Cancer Lett., 2006, 239, 271 CrossRef CAS.
- N. Osakabe, A. Yasuda, M. Natsume and T. Yoshikawa, Carcinogenesis, 2004, 25, 549 CAS.
- U.-L. Lee and S.-W. Choi, ISRN Oncology, 2011, 2011 Search PubMed.
- J.-K. Lin, in Tea and Tea Products: Chemistry and Health-Promoting Properties, ed. C.-T. Ho, J.-K. Lin and F. Shahidi, CRC Press, Boca Raton, 2009 Search PubMed.
- J. W. Kim, A. R. M.R. Amin and D. M. Shin, Cancer Prev. Res., 2010, 3, 900 Search PubMed.
- (a) A. Kazi, D. M. Smith, K. Daniel, S. Zhong, P. Gupta, M. E. Bosley and Q. P. Dou, In Vivo, 2002, 16, 397 Search PubMed; (b) H. C. Li, S. Yashiki, J. Sonoda, H. Lou, S. K. Ghosh, J. J. Byrnes, C. Lema, T. Fujiyoshi, M. Karasuyama and S. Sonoda, Jpn. J. Cancer Res., 2000, 91, 34 CrossRef CAS.
- S. Gupta, N. Ahmad, A. L. Nieminen and H. Mukhtar, Toxicol. Appl. Pharmacol., 2000, 164, 82 CrossRef CAS.
- I. Naasani, H. Seimiya and T. Tsuruo, Biochem. Biophys. Res. Commun., 1998, 249, 391 CrossRef CAS.
- (a) J. Liao, G. Y. Yang, E. S. Park, X. F. Meng, Y. H. Sun, D. X. Jia, D. N. Seril and C. S. Yang, Nutr. Cancer, 2004, 48, 44 Search PubMed; (b) B. Zhou, J. Pan, F. Dai, C. Y. Zhao, L. P. Zhang, Q. Y. Wei, L. Yang, R. L. Zheng and Z. L. Liu, Res. Chem. Intermed., 2004, 30, 627 CAS.
- C. S. Yang, X. Wang, G. Lu and S. C. Picinich, Nat. Rev. Cancer, 2009, 9, 429 CrossRef CAS.
- T. Kuzuhara, Y. Sei, K. Yamaguchi, M. Suganuma and H. Fujiki, J. Biol. Chem., 2006, 281, 17446 CrossRef CAS.
- M. Lipkin, B. Reddy, H. Newmark and S. A. Lamprecht, Annu. Rev. Nutr., 1999, 19, 545 CrossRef CAS.
- K. Yang, S. A. Lamprecht, Y. H. Liu, H. Shinozaki, K. H. Fan, D. Leung, H. Newmark, V. E. Steele, G. J. Kelloff and M. Lipkin, Carcinogenesis, 2000, 21, 1655 CrossRef CAS.
- A. A. Dihal, V. C. J. de Boer, H. van der Woude, C. Tilburgs, J. P. Bruijntjes, G. M. Alink, I. M. C. M. Rietjens, R. A. Woutersen and R. H. Stierum, J. Nutr., 2006, 136, 2862 CAS.
- M. Mouria, A. S. Gukovskaya, Y. Jung, P. Buechler, O. J. Hines, H. A. Reber and S. J. Pandol, Int. J. Cancer, 2002, 98, 761 CrossRef CAS.
- M. A. Avila, J. A. Velasco, J. Cansado and V. Notario, Cancer Res., 1994, 54, 2424 CAS.
- I. K. Wang, S. Y. Lin-Shiau and J. K. Lin, Eur. J. Cancer, 1999, 35, 1517 CrossRef CAS.
- C. Martinez Conesa, V. Vicente Ortega, M. J. Yanez Gascon, M. Alcaraz Banos, M. Canteras Jordana, O. Benavente-Garcia and J. Castillo, J. Agric. Food Chem., 2005, 53, 6791 CrossRef CAS.
- (a) D. Gallo, S. Giacomelli, C. Ferlini, G. Raspaglio, P. Apollonio, S. Prislei, A. Riva, P. Morazzoni, E. Bombardelli and G. Scambia, Eur. J. Cancer, 2003, 39, 2403 CrossRef CAS; (b) R. P. Singh and R. Agarwal, Eur. J. Cancer, 2005, 41, 1969 CrossRef CAS.
- A. Huber, P. Thongphasuk, G. Erben, W. D. Lehmann, S. Tuma, W. Stremmel and W. Chamulitrat, Biochim. Biophys. Acta, Gen. Subj., 2008, 1780, 837 CrossRef CAS.
- T. W. Chung, S. K. Moon, Y. C. Chang, J. H. Ko, Y. C. Lee, G. Cho, S. H. Kim, J. G. Kim and C. H. Kim, FASEB J., 2004, 18, 1670 CrossRef CAS.
- M. Kampa, V. I. Alexaki, G. Notas, A. P. Nifli, A. Nistikaki, A. Hatzoglou, E. Bakogeorgou, E. Kouimtzoglou, G. Blekas, D. Boskou, A. Gravanis and E. Castanas, Breast Cancer Res., 2004, 6, R63 CrossRef CAS.
- Y. S. Lee, Arch. Pharmacal Res., 2005, 28, 1183 Search PubMed.
- (a) P. Lesca, Carcinogenesis, 1983, 4, 1651 CrossRef CAS; (b) H. Mori, K. Kawabata, N. Yoshimi, T. Tanaka, T. Murakami, T. Okada and H. Murai, Anticancer Res., 1999, 19, 3775 CAS.
- (a) R. P. Singh and R. Agarwal, Curr. Cancer Drug Targets, 2004, 4, 1 Search PubMed; (b) T. W. Flaig, D. L. Gustafson, L. J. Su, J. A. Zirrolli, F. Crighton, G. S. Harrison, A. S. Pierson, R. Agarwal and L. M. Glode, Invest. New Drugs, 2007, 25, 139 CAS.
| This journal is © The Royal Society of Chemistry 2011 |