Synthesis of diverse spiroisoxazolidinohydantoins by totally regio- and diasteroselective 1,3-dipolar cycloadditions†
Maria
Cristina Bellucci
a,
Tommaso
Marcelli
b,
Leonardo
Scaglioni
a and
Alessandro
Volonterio
*b
aDipartimento di Chimica Agroalimentare, Università degli Studi di Milano, via Celoria 2, 20133 Milano, Italy
bDepartment of Chemistry, Materials and Chemical Engineering “Giulio Natta”, Politecnico di Milano, via Mancinelli 7, 20131 Milano, Italy. E-mail: alessandro.volonterio@polimi.it; Fax: +39 0223993080; Tel: +39 0223993136
First published on 22nd September 2011
Abstract
5-Carboxymethylene hydantoins have been synthesized in high yield and under very mild conditions (20 °C, dichloromethane) through a highly (often totally) regio- and diastereoselective domino process involving readily accessible carbodiimides and acetylenedicarboxylic acid monoesters. Such compounds could be considered very intriguing intermediates for the preparation of spirohydantoins and other hydantoin derivatives. For instance, they react smoothly with either acyclic and cyclic nitrones to form spiroisoxazolidinohydantoins in high yield and with total control of the regio- and diastereoselectivity. Starting from an enantiomerically pure chiral pyrroline nitrone, the reaction was also enantiospecific. Both processes have been studied in detail and the diastereoselectivities seen have been rationalized upon examination of the alternative transition states and by density functional theory calculations of the cycloaddition step.
Introduction
Spiro compounds, having cyclic structures fused at a central carbon, are important naturally occurring substances with highly pronounced biologically properties and are of recent interest also due to their interesting conformational features and their structural implications on biological systems.1 For instance, the almost mutually orthogonal position of the two-ring system linked to the spiro carbon, has been used to mimic structural motifs found in proteins, thus increasing the probability of interactions with biological systems.2 In fact, it is well known that the difference in activity and receptor selectivity of drugs might be explained by the conformation of the contained privileged structure. Generation of semi-rigid drugs facilitates the study of their interaction with the receptors, may lead to more selective interactions with fewer side effects, and permits the rational design of more potent and selective drugs in the future.3 In particular, spirohydantoins have recently attracted much attention because they exhibit various biological activities. For instance, spirohydantoins I (Fig. 1) can act as new psychotropic agents (antidepressants, anxiolytics, antipsychotics),4 compounds II show antidiabetic and antiepileptic activities5 while compounds III and IV are potent antagonists of the melanin-concentrating hormone receptor-1 (MCH-R1)6 and of the leukocyte function associate antigen-1 (LFA-1), respectively.7 | ||
| Fig. 1 Examples of bioactive spirohydantoins. | ||
Although spirohydantoins are considered privileged structures, making them attractive for the preparation of compound libraries with the potential for diverse biological activity, very few methods for their synthesis and modification have been reported to date.
Within the scope of a project aimed at developing new domino processes for the synthesis of small heterocycles,8 we became interested in the synthesis of libraries of 5-carboxymethylene hydantoins which could be suitable starting materials for the construction of spirohydantoins and other hydantoin derivatives. In particular, herein we wish to report the synthesis of spiroisoxazolidinohydantoins 4 by means of a two step synthetic procedure: a domino process involving readily accessible carbodiimides 1 and acetylenedicarboxylic acid monoesters 2 producing 5-carboxymethylene hydantoins 3 with high regio- and diastereoselectivity in most cases, followed by a totally regio- and diastereoselective 1,3-dipolar cycloaddition reaction with nitrones (Scheme 1).9
 | ||
| Scheme 1 Retrosynthetic analysis for the synthesis of spiroisoxalidinohydantoins. | ||
It is worth noting that the spiroisoxalidinohydantoins 4 described herein, in contrast to the previous reported spirohydantoins prepared through dipolar cycloadditions, possess a carboxyl moiety, which could be further functionalized to modulate the structure of the final compounds.
Results
We recently have demonstrated that carbodiimides, when treated with suitable carboxylic acids, such as activated α,β-unsaturated acids, are useful reagents for the straightforward synthesis of 1,3,5-trisubstituted hydantoins under very mild conditions, by means of a regiospecific domino process consisting of a condensation step between the two reactants leading to the formation of O-acyl isourea intermediates which undergoes nucleophilic aza-Michael reaction, followed by final N→O acyl migration step.8a,b,g We thus decided to apply this strategy for the synthesis of 5-carboxymethylene hydantoins 3 starting with acetylenedicarboxylic acid monoesters 2 (see Scheme in Table 1).|
|
|||||
|---|---|---|---|---|---|
| Entry | X | Acid | Carbodimide | Product | Yields (%)a |
a Overall yields.
b Reaction performed with 1 equiv. of TMP.
c An almost 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of regioisomers.
d A 1.5:1 mixture in favor of the E isomer.
e A 2:1 mixture in favor of the Z-isomer.
f A 4:1 mixture in favor of the Z-isomer. :1 mixture of regioisomers.
d A 1.5:1 mixture in favor of the E isomer.
e A 2:1 mixture in favor of the Z-isomer.
f A 4:1 mixture in favor of the Z-isomer.
|
|||||
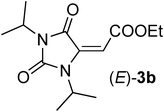
| 1 | Et | 2a |

|

|
88 |
| 2 | Et | 2a |

|
|
89 |
| 3b | t-Bu | 2b |

|

|
85 |
| 4 | Et | 2a |

|

|
91 |
| 5 | Et | 2a |

|

|
79 |
| 6 | Et | 2a |

|

|
77 |
| 7 | Et | 2a |

|

|
80c |
| 8 | Et | 1a |

|

|
85 |
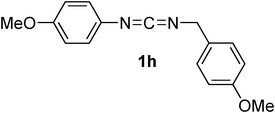
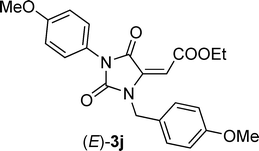
| 9 | Et | 2a |
|
|
82 |
| 10 | Et | 1a |

|

|
83d |
| 11 | Et | 2a |

|

|
74e |
| 12 | Et | 2a |

|

|
75f |
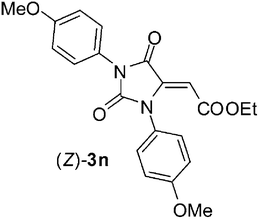
| 13 | Et | 2a |

|
|
70 |

Commercially available DCC 1a and DIC 1b reacted smoothly with acetylenedicarboxylic acid monoethylester 2a in DCM at rt affording hydantoins (E)-3a,b, respectively, in high yields and as the only diastereoisomers (entries 1 and 2, Table 1). The stereochemistry of the double bond of derivative 3b was assessed by NOE experiment which evidenced a close proximity between the vinylic proton and the methyl groups belonging to the iso-propyl group (see ESI†). Moreover, the olefinic proton of both (E)-3a,b resonated around 5.6 ppm which is a typical value for (E) stereochemistry in 2-aminofumarate-like systems, being the same protons for (Z)-2-aminomaleate-like systems resonating between 6 and 7 ppm (see below).10 Also acetylenedicarboxylic acid mono-tert-butylester 2b reacted efficiently with carbodiimide 1a producing hydantoin (E)-3c as the only stereoisomer (entry 3, Table 1). However, in this case, to achieve good yields, the reaction was performed in the presence of 1 equivalent of 2,4,6-trimethylpyridine (TMP) probably because the sterically hindered tert-butyl group renders the acid less reactive. As expected, by performing the reaction with “weakly” asymmetric carbodiimides,8bi.e.carbodiimides bearing two alkyl substituents are very different in terms of steric hindrance, we obtained a complete regiospecific process. Indeed, N-t-butyl, N’-benzyl carbodiimide 1c and N-trityl, N’-n-butyl carbodiimide 1d were produced, when reacted with acid 2a, only the regio- and diastereoisomers (E)-3d,e, respectively (entries 4 and 5, Table 1), which arise from the nucleophilic attack of the less congested nitrogen bearing the primary alkyl substituent. It is worth noting that, as expected,11 the N-trityl moiety is so sterically congested that also N-trityl, N’-aryl carbodiimides, such as 1e, gave the formation of the only hydantoin (E)-3f by nucleophilic attack of the less reactive aniline moiety (entry 6, Table 1). These derivatives are intriguing because, as already demonstrated, the t-butyl and the trityl groups could be easily cleaved rendering such compound precursors of both N-alkyl and N-aryl 1,5 disubstituted hydantoins.11 When “weakly” asymmetric N,N’-dialkyl carbodiimides bear similar substituents in terms of steric bulkiness, such as t-butyl and phenetyl in carbodiimide 1f, we obtained the formation of an equimolar ratio of both the regioisomers (E)-3g,h (entry 7, Table 1), with total diasterocontrol in both cases (only E-double bond is formed).12 “Strongly” asymmetric carbodiimides,8bi.e.carbodiimides bearing two N-substituents that are very different in terms of electronic features, such as N-aryl, N’-alkyl carbodiimides 1g,h, when reacted with acid 2a gave rise to the formation of the only regioisomers (E)-3i,j arising from the nucleophilic attack of the nitrogen bearing the alkyl substituent in the aza-Michael step (entries 8 and 9, Table 1). However, to our surprise, when the alkyl substituent is small, such as a methyl group in carbodiimide 1i, the reaction remained completely regioselective but in favor of the nucleophilic attack of the aniline moiety leading to the formation of a 1.5:1 mixture of the two stereoisomers (E)-3k/(Z)-3k (entry 10, Table 1). The regiochemistry of both hydantoins 3k was assessed by 1H, 13C Heteronuclear Multiple Bond Coherence (HMBC) NMR experiments which evidenced a close proximity between the N-methyl protons of both compounds (E)/(Z)-3k and the two carbonyls belonging to the hydantoin ring. Moreover, by performing long range proton–carbon coupling experiments, the two carbonyls belonging to the hydantoin ring resulted in a quartet, coupling with the N-methyl protons, and a double quartet, having an additional coupling with the vinylic proton. Concerning the stereochemistry (E)-(Z), a first assignment was done considering the NMR chemical shifts of the vinylic protons (5.53 ppm for the (E)-stereoisomer and 6.04 ppm for the (Z)-stereoisomer).
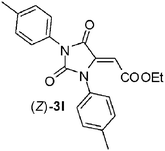
To confirm such assignments, we performed NOE experiments which evidenced a close proximity between the vinylic and the aromatic protons in the compound having the (E) geometry (2% NOE effect), while a very small NOE was detected for the (Z)-hydantoin (0.2% NOE effect). Finally, also less reactive symmetric N,N’-diaryl carbodiimides1j-l reacted smoothly with acid 1a in the same conditions, namely DCM at rt, giving rise to the formation, in most cases, of an unbalanced mixture of two hydantoin (Z)- and (E)-diastereoisomers. Surprisingly the diastereoselection, which ratio depends on the nucleophilicity of the aniline moiety, was always in favor of the (Z)-diastereoisomer, (entries 11–13, Table 1).13 In fact, less nucleophilic N,N’-diphenyl carbodiimide 1j and N,N’-di-para-tolyl carbodiimide 1k produced, in high yields, a 2:1 and a 4:1 mixture of (Z)-3l/(E)-3l and (Z)-3m/(E)-3m, respectively, while more nucleophilic N,N’-di-para-metoxyphenyl carbodiimide 1l gave rise to the formation of the only diastereoisomer (Z)-3n.
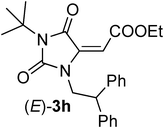
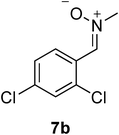
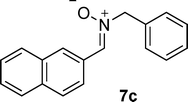
Since 1,3-dipolar cycloadditions provide a powerful means for the synthesis of a wide range of heterocycles,14 we decided to investigate the use of 5-carboxymethylene hydantoins 3 for the synthesis of densely functionalized spiroisoxazolidinohydantoins 4, which could be valuable molecules in the biomedical field, as well as intermediates for other bio-important compounds by reductive cleavage of the N–O bond (Table 2). To our surprise, compounds (E)-3a reacted with C,N-diphenyl nitrone 7a producing spirohydantoin 4a, in high yield, as the only product (entry 1, Table 2). The total regio-15 and diastereoselectivity of the process was confirmed by reacting (E)-3c with nitrone 7b. Also in this case we obtained the formation of only 4b as a single regio- and diastereoisomer in high yield (entry 2, Table 2). These results are surprising because while it is known that the 4,5 stereochemistry reflect the geometry of the carbon-carbon double bond of the dipolarophile, the 3,4 stereochemistry (endo/exo selectivity) is difficult to control in a selective way (the numbering is referred on the isoxazolidine ring).14 Moreover, the 3,4 stereochemistry generally is affected by a possible (E)/(Z) tautomerization that occur when acyclic nitrones are used as dipolarophiles. The relative configuration of the new stereocenters formed in the reaction was determined considering the following data: (1) the coupling constants between the methynic protons H3-H4 are always bigger than 10 Hz, typical values for an anti disposition in five member cyclic systems, and (2) NOE experiments on derivative 4b clearly evidenced a proximity between H4 and the aromatic proton in ortho respect to the chlorine atoms, whereas no NOE was observed between H3 and H4 (see ESI†). The total stereocontrol could be ascribed to the steric interaction between the nitrone and the particularly hindered 5-carboxymethylene hydantoins. In particular, the steric interaction between the ester moiety, rather than the N-substituents of the 5-carboxymethylene hydantoins, and the nitrone seems to play a key role in controlling the reactivity affecting also the yields of the process. In fact, more sterically congested tert-butyl ester (E)-3c reacted with C-naphthyl-N-benzyl nitrone 7c producing 4c in lower yields (50%, entry 3, Table 2) and (E)-3a did not react at all with nitrone 7d (entry 4, Table 2) bearing a bulky t-butyl substituent at the nitrogen atom. At the same time, regioisomers (E)-3g,h, bearing hindered substituents at the nitrogen atoms, such as t-butyl and phenethyl, reacted effectively with nitrone 7b producing spirohydantoins 4d,e, respectively, in high yields (entries 5 and 6, Table 2).
|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | R1 | R2b | Xc | Hydantoin | R3 | Ar | Nitrone | Producta | Yield (%)b |
| a Only regio- and diastereoisomer obtained. b Overall yields. c No reaction occurred. | |||||||||
| 1 | c-Hex | c-Hex | Et |

|
Ph | Ph |

|

|
75 |
| 2 | c-Hex | c-Hex | t-Bu |

|
Me | 2,4-diCl-Ph |

|

|
79 |
| 3 | c-Hex | c-Hex | t-Bu |

|
Bn | naphtyl |

|

|
50 |
| 4 | c-Hex | c-Hex | Et |

|
t-Bu | Ph |

|
/ | n.r.c |
| 5 | (Ph)2CHCH2 | t-Bu | Et |

|
Me | 2,4-diCl-Ph |

|

|
71 |
| 6 | t-Bu | (Ph)2CHCH2 | Et |
|
Me | 2,4-diCl-Ph |
|

|
78 |
| 7 | Ph | Bn | Et |

|
Bn | naphtyl |
|

|
72 |
| 8 | Me | Ph | Et |

|
Me | 2,4-diCl-Ph |

|

|
75 |
| 9 | Me | Ph | Et |

|
Me | 2,4-diCl-Ph |

|
|
73 |
| 10 | p-Tol | p-Tol | Et |

|
Ph | Ph |

|
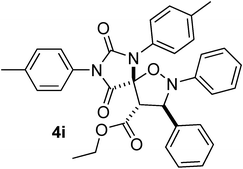
|
78 |
| 11 | p-Tol | p-Tol | Et |

|
Ph | Ph |

|

|
81 |
| 12 | p-Tol | p-Tol | Et |

|
Me | 2,4-diCl-Ph |

|

|
76 |
| 13 | p-Tol | p-Tol | Et |
|
Me | 2,4-diCl-Ph |
|

|
77 |

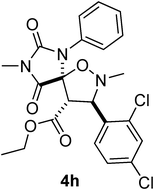
Moreover, when C-naphthyl,N-benzyl nitrone 7c was reacted with the ethylester derivative (E)-3j, we obtained the formation of 4f in high yield (entry 6, Table 1). The total regio- and diastereocontrol of the cycloaddition is not affected by the stereochemistry of the exocyclic carbon–carbon double bond of the dipolarophile. In fact, both steroisomers (E)- and (Z)-3j reacted with 7b producing only one product, respectively 4g and 4h in both cases in high yields (entries 8 and 9, Table 2). Moreover, also N,N’-di-para-tolyl-5-carboxymethylene hydantoin (E)- and (Z)-3k reacted smoothly with nitrones 7a and 7b producing the corresponding spirohydantoins 4i–l, respectively, in high yields and with total regio- and stereocontrol (entries 10–13, Table 2). It is worth noting that spirohydantoins arising from reactions between the same nitrone and (E)/(Z) diastereoisomeric hydantoins always lead to the formation of epimers at the quaternary stereocenter, having anti relative configuration at the new tertiary stereocenters formed in the isoxazolidine ring.16
Then, we decided to examine the reactivity of cyclic nitrones. Since selectivity in cycloadditions with cyclic nitrones are often much higher than with acyclic nitrones due to the absence of (E)-(Z) isomerization in the dipole,14 we expected, all the more, a complete stereoselective process. Indeed, nitrone 7d17 when reacted with hydantoins (E)-3a and (E)-3k gave rise again to a complete regio- and diastereoselective process producing spirohydantoins 4m,n, respectively, in high yields (Scheme 2). Also with cyclic nitrones, the stereoselectivity of the process is not affected by the stereochemistry of the carbon–carbon double bond of the dipolarophile. Accordingly, the reaction between hydantoin (Z)-3k and 7d lead to the formation of only one product in very high yields, the spiroadduct 4o having the two isoxazolidine methynic protons in anti disposition, such as in 4m,n. Again, the relative 3,4 stereochemistry for compounds 4n,o was determined by the coupling constants between H3 and H4 that were calculated between 11.0 and 12.0 Hz, typical values for an anti disposition. However, this was not the case for spirohydanotin 4m, showing a coupling constant of 4.0 Hz. Nevertheless, ROESY experiments showed a strong NOE between H3 and a methylenic proton belonging to the ethyl ester moiety and a very weak NOE with H4, suggesting a syn disposition for H3 and the ester group and an anti disposition for H3 and H4 (see ESI†).
 | ||
| Scheme 2 Reaction with cyclic nitrone. | ||
The excellent results obtained in term of stereocontrol of the cycloaddition lead us to investigate the possibility to obtain a complete regio-, diastereo- and enantioselective process by using enantiomerically pure nitrones. In this regard, chiral pyrrolidine-N-oxides represent five-member cyclic nitrones able to undergo highly regio- and diastereoselective 1,3-dipolar cycloaddition. In fact, such compounds have been widely used for the asymmetric synthesis of pyrrolizidine and indolizidine alkaloids.18 Gratifyingly, when enantiomerically pure pyrroline-N-oxide 7e19 was reacted with (E)-3b in the same conditions shown above (toluene, sealed tube at 80 °C) we obtained the formation of the only isomer (+)-4p in very high yields, showing that this strategy could be effectively used for the synthesis of enantiomerically pure spiroisoxazolidinohydantoins (Scheme 3).
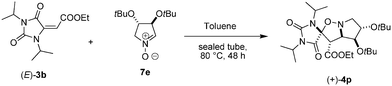 | ||
| Scheme 3 Synthesis of enantiomerically pure (+)-4p | ||
To asses the stereochemistry of (+)-4p we have firstly assigned all the proton NMR signals by COSY, HSQC, and long range proton-carbon coupling experiments (HMBC, see ESI†). Then, we calculated the coupling constant between the methynic protons H3-H4 which resulted to be 10.4 Hz (Fig. 2), typical value for an anti disposition, while no coupling has been detected between the methynic protons H3-H5.
 | ||
| Fig. 2 Determination of the stereochemistry of (+)-4p | ||
However, by performing 2D NOESY experiments, we clearly detected a NOE between H3 and H6 (medium interaction), while no NOEs were detected between H3 and either H4 and H5, confirming an anti disposition between H3 and H5.
Discussion
The reaction sequence with asymmetric carbodiimides is portrayed in Scheme 4. In the case of symmetric carbodiimides (R1 = R2) only two products can be formed, namely hydantoins (E)- and (Z)-3, while for asymmetric carbodiimides the reaction could provide four different products, i.e. two diastereoisomeric couple of regioisomers (E)/(Z)-3 and (E)/(Z)-3’. | ||
| Scheme 4 Reaction sequence with asymmetric carbodiimides. | ||
The first step involves addition of the carboxylic acid 1 to the carbodiimide 2 to form a reactive intermediate O-acylisourea, existing in two prototropic tautomeric forms 5 and 5’ when R1 ≠ R2.20 The regioselectivity of the subsequent intramolecular aza-Michael addition depends mainly on the difference of nucleophilicity between the two incipient amine moieties N-R1 and N-R2 on the carbodiimide. It is therefore not surprising that when “weakly” asymmetric carbodiimides bearing two alkylic substituents which are very different in terms of steric hindrance, such in 1c and 1d, the reaction results totally regioselective in favor of the formation of the regioisomer arising from the nucleophilic attack of the less sterically congested amine moiety (see entries 4 and 5, Table 1), while when the substituent are similar we obtained the formation of an equimolecular mixture of both regioisomers (entry 7, Table 1). With “strongly” asymmetric carbodiimides we expected a completely regioselective outcome of the reaction. Indeed, carbodiimides 1g,h gave rise to the formation of only regioisomers arising from the aza-Michael addition of the more nucleophilic N-alkyl compared to the N-aryl moiety. Quite surprisingly, when the N-alkyl substituent is a small methyl group, as in 1i, we again obtained a completely regioselective process but in favor of the unexpected nucleophilc attack of the N-aryl moiety, suggesting a more nucleophilic behavior of the N-phenyl compared to the N-methyl (entry 10, Table 1). However, a similar behavior has been already observed on the condensation of N-methyl, N’-phenyl urea to unsymmetrical 1,2-ketones.21
Also the diastereoselectivity of the reaction seems to be dependent on the nature of the carbodiimideN-substituents. Accordingly, when the nucleophilic attack on the aza-Michael step arise from an N-alkyl substituent we always obtained a totally diastereoselective process leading to the formation of the (E) diasteroeoisomer. The selectivity achieved could be attributed to a steric interaction between the activating ester group and the alkyl group attached to the nitrogen nucleophile, as depicted in the late transition state TS II in Scheme 5, which renders the formation of the (Z)-hydantoin less favorable. However, when the nucleophilic attack occurs from a N-aryl moiety, a π-staking interaction between the delocalized system consisting of the electron poor carbon–carbon double bond and the carbonyl group, with the electron rich aniline moiety should be taken in consideration (TS IV, Scheme 5).22 Effectively, the reaction of N,N’-diaryl carbodiimides with acetylenedicarboxylic acid monoesters leads to the formation of an unbalanced mixture of (E)/(Z) isomers, with the (Z)-isomer always as the major one. Moreover, the more electron rich the aniline moiety is, the better the diastereoselectivity achieved (when the aniline moiety is an anisidine, we even obtained a diastereospecific reaction). The only exception to this trend was obtained with N-trityl, N’-p-methoxyphenyl carbodiimide 1e which, when reacted with acid 2a, produced only the diastereoisomer (E)-3f which occurred from the nucleophilic attack of the anisidine moiety (entry 6, Table 1). However, we believe that in this case the exceptional hindrance of the trityl group renders TS IV less favorable with respect to TS III where the ester group is far away.
 | ||
| Scheme 5 Rationalization of the stereochemical outcome. | ||
In comparison to the Diels–Alder reaction, the 1,3-dipolar cycloaddition of nitrones with olefins generally exhibits lower levels of regio- and stereocontrol (endo/exo selectivity), due to significant contributions by both LUMO (dipole)-HOMO (dipolarophile) and HOMO (dipole)-LUMO (diparophile) interactions, further complicated by the possibility of interconversion of the nitrone geometry in the case of acyclic nitrones.23 For this reason, it is generally very difficult to obtain the selective formation of only one diastereoisomer. However, when either acyclic and cyclic nitrones were reacted with 5-carboxymethylene hydantoins, independently from the (E)/(Z) stereochemistry of the carbon–carbon double bond, we always achieved a total regio- and diastereoisomeric process, affording the formation of only the exo diastereoisomer.
In order to shed light on the observed diastereoselectivity, we performed a computational analysis of the 1,3-dipolar cycloaddition step. We considered the reaction of hydantoinA with nitroneB, yielding spiroisoxazolidinohydantoin C (Scheme 6), as a suitable computational model for the formation of compounds 4k and 4l. In order to get a complete stereochemical description of this reaction, we considered the reaction of (E) and (Z) isomers of compounds A and B leading to the formation of both exo and endo adducts, for a total of eight isomeric transition states (Fig. 3).
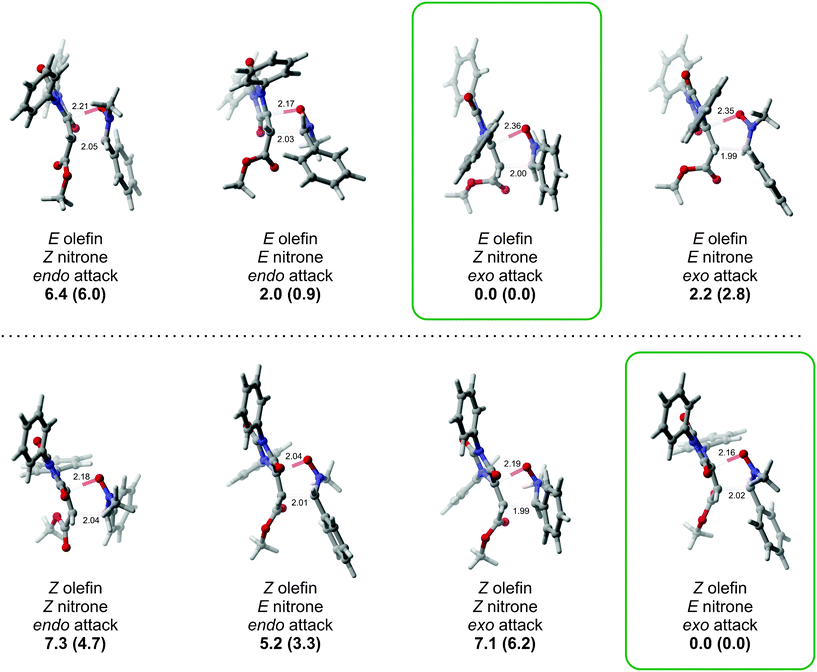 | ||
| Fig. 3 Calculated transition states for the 1,3 dipolar cycloaddition. B3LYP (M06-2X) relative energies are expressed in kcal mol−1, distances are in Angstroms. | ||
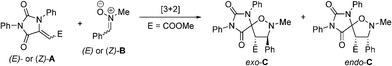 | ||
| Scheme 6 Computational model. | ||
The calculations predict the formation of the exo adduct to be kinetically favored for both (E) and (Z) isomers of compound A, in qualitative agreement with the experimentally observed selectivity. However, while in the case of the Eolefin, the lowest energy transition state corresponds to the cycloaddition of nitrone (Z)-B, this trend is reversed for the reaction of hydantoin (Z)-A. With respect to the origin of the diastereoselectivity, for the reaction of (E)-A, the only transition state clearly displaying severe intermolecular steric interactions is the endo attack of the Znitrone, with short contacts (≥3.0 Å) between the phenyl substituent of the nitrone and the hydantoin ester moiety. Therefore, we interpret the energy differences between the other transition states to arise mainly from the relative stability of the nitrone isomers, with (Z)-B being favored. The computational results for the reaction of hydantoin (Z)-A indicate a more substantial preference for the formation of the exo adduct. In this case, the observed selectivity seems to mainly arise from unfavorable interactions between either the methyl, (E)-Bendo attack and (Z)-Bexo attack, or phenyl, (Z)-Bendo attack, substituents of the nitrone and one of the phenyl rings of hydantoin (Z)-A (in the background in Fig. 3). While we note that the calculated energy differences between the isomeric transition states do not quantitatively account for the formation of a single diastereoisomer at 80 °C for compound (E)-A, B3LYP and M06-2X yield the same qualitative results, in agreement with the experimental findings for both geometric isomers of the hydantoin. These results strongly suggest that the (E)/(Z) tautomerism of the nitrones plays a crucial role in the process. C-Alkyl and C-aryl acyclic nitrones are usually assumed to undergo cycloadditions as the most stable (Z) isomer, in contrast to cyclic nitrones, which must react in the only available (E) form. Indeed, this seems the case when the reaction is carried out starting with (E)-hydantoins, generating two TS having almost 6 Kcal mol−1 difference, thus predicting a high degree of exo/endo selectivity. However, with hydantoins having (Z) configuration at the carbon–carbon double bond, the less stable (E)-nitrone tautomer seems operative, leading to the formation of the exo diastereoisomer through a TS which is more than 7 Kcal mol−1 less energetic over the TSs involving the (Z)-nitrone. This hypothesis is also corroborated by the fact that performing the reaction between nitrone 7b and hydantoins (E)- and (Z)-3k in the presence of a catalytic amount of benzoic acid, which is known to have a marked catalytic effect on the rate of (E)/(Z) nitrone interconverstion,24 we obtained the same result of the reaction without additives, namely a complete stereocontrolled process.
In order to prove that the spiroisoxazolidinohydantoins obtained—beside being very intriguing compounds themselves—could also be further functionalized ad hoc for specific tasks, we tried to selectively functionalize the ester moiety. Accordingly, treatment of 4g with methanolic NaOH leads to the formation of the corresponding acid that could be coupled with both alkyl or aryl amines, such as benzyl amine and 2-methyl-5-methoxyaniline, leading to the formation of amides 8 and 9, respectively, in high overall yield (Scheme 7).
 | ||
| Scheme 7 Synthesis of amides 8 and 9. | ||
Conclusions
In summary, we have developed a general, straightforward method for the preparation of highly diverse spiroisoxazolidinohydantoins in good to excellent yields by means of a two highly stereoselective step synthetic pathway. The first step consists of a domino reaction between carbodiimides and acetylenedicarboxylic acid monoesters, which takes place in very mild conditions and allowed us to obtain a large array of structurally diverse 5-carboxymethylene hydantoins. Both the regio- and the diastereoselectivity of the process are generally completely controlled by an appropriate choice of the carbodiimideN-substituents. The resulting 5-carboxymethylene hydantoins reacted smoothly with different nitrones, either cyclic and acyclic, giving rise to the formation of the target spiroisoxazolidinohydantoins with total regio- and diastereocontrol and in high yields. The stereochemical outcome of the [3+2] cycloaddition was studied using DFT calculations and the computational results are in agreement with the experimentally observed selectivity. A large array of structurally diverse spiroisoxazolidinohydantoins, which can be further functionalized at the ester moiety, can be synthesized through this method. The method also looks particularly suitable for solid phase/combinatorial chemistry. This issue is currently in progress in our laboratory.Experimental section
General mthods
Commercially available reagent-grade solvents were employed without purification. Carbodiimides were prepared by dehydration of the corresponding urea derivatives25, while acetylenedicarboxylic acid monoesters by carboxylation of the corresponding propiolates.261H NMR spectra were run on spectrometers 250, 400 or 500 MHz. Chemical shifts are expressed in ppm (δ), using tetramethylsilane (TMS) as internal standard for 1H and 13C nuclei (δH and δC = 0.00). High-resolution MS spectra were recorded with a FT-ICR (Fourier Transform Ion Ciclotorn Resonance) instrument, equipped with an ESI source, or a standard MS instrument, equipped with an EI source.Computational methods
All calculations were carried out using the Gaussian 0327 and Gaussian 0928 packages. The transition states were fully optimized using the B3LYP29 functional with the 6-31+G* basis set. For all transition states, vibrational analysis carried out at the same level of theory yielded one single imaginary vibration. In order to get more accurate energies, single point calculations were carried out on all the stationary points with both B3LYP and M06-2X30 functionals using the larger 6-311+G** basis set. Considering that the cycloaddition reactions take place in a very apolar solvent (toluene), the effect of solvation was not taken into account. Hence, the final gas-phase energies were calculated by adding the zero-point vibrational correction to the single-point energies obtained with the large basis set.Synthesis of 5-carboxymethylene hydantoins 3. General procedure
To a stirred solution of acetylenedicarboxylic acid monoesters 2 (1 equiv.) in DCM (0.1 M solution) 1.1 equiv. of carbodiimide 1 was added. After total consumption of stating material (TLC monitoring) the organic solvent was evaporated under reduced pressure and the crude purified by flash chromatography.:20); FTIR (microscope) ν 1778, 1743, 1714 cm−1; 1H-NMR (300 MHz, CDCl3) δ 5.61 (s, 1H), 4.24 (q, J = 7.2 Hz, 2H), 3.83 (m, 1H), 3.66 (m, 1H), 1.48–2.15 (m, 20H), 1.28 (t, J = 7.2 Hz, 3H); 13C-NMR (75.5 MHz, CDCl3) δ 164.9, 160.3, 150.2, 134, 101.5, 61.7, 53.8, 51.9, 29.0, 25.6, 25.1, 24.0; ESI (m/z) 349.1 [M++H, (18)], 371.2 [M++Na, (100)]; HRMS calcd for [C19H28N2O4] 348.2049, found 348.2056.
:20); FTIR (microscope) ν 1780, 1732 cm−1; 1H-NMR (250 MHz, CDCl3) δ 5.63 (s, 1H), 4.36 (m, 1H), 4.32 (q, J = 7.5 Hz, 2H), 4.28 (m, 1H), 1.44 (d, J = 5,0 Hz, 6H), 1.41 (d, J = 5.0 Hz, 6H), 1.35 (t, J = 7.5 Hz, 3H); 13C-NMR (62.9 MHz, CDCl3) δ 164.9, 153.0, 134.1, 101.7, 61.6, 45.0, 44.1, 19.6, 19.4, 14.0; ESI (m/z) 269.1 [M++H, (11)], 291.1 [M++Na, (100)]; HRMS calcd for [C13H20N2O4] 268.1423, found 268.1431.
:20); FTIR (microscope) ν 1775, 1731, 1717 cm−1; 1H-NMR (300 MHz, CDCl3) δ 5.58 (s, 1H), 3.99 (m, 1H), 3.67 (m, 1H), 1.54–2.14 (m, 16H), 1.51 (s, 9H), 1.05–1.36 (m, 4H); 13C-NMR (75.5 MHz, CDCl3) δ 163.9, 160.0, 153.0, 133.1, 103.2, 82.1, 53.3, 51.6, 29.3, 29.2, 28.0, 26.1, 25.9, 25.2, 25.1; ESI (m/z) 377.1 [M++H, (4)], 399.1 [M++Na, (100)]; HRMS calcd for [C21H32N2O4] 376.2362, found 376.2350.
:20); FTIR (microscope) ν 1773, 1753, 1722 cm−1; 1H-NMR (300 MHz, CDCl3) δ 7.28 (m, 15H), 5.73 (s, 1H), 4.22 (q, J = 7.5 Hz, 2H), 3.26 (m, 2H), 1.67 (m, 2H), 1.23 (m, 2H), 1.08 (t, J = 7.5 Hz, 3H), 0.90 (t, J = 6.8 Hz, 3H); 13C-NMR (75.5 MHz, CDCl3) δ 170.3, 166.9, 165.6, 145.1, 142.4, 128.6, 127.3, 126.5, 96.4, 74.0, 62.3, 41.6, 29.0, 19.4, 13.9, 13.5; ESI (m/z) 483.1 [M++H, (18)], 505.1 [M++Na, (100)]; HRMS calcd for [C30H30N2O4] 482.2206, found 482.2214.
:20); FTIR (microscope) ν 1777, 1743 cm−1; 1H-NMR (300 MHz, CDCl3) δ 7.34 (d, J = 10.5 Hz, 2H), 7.30–7.15 (m, 14H), 7.01 (d, J = 10.5 Hz, 2H), 5.79 (s, 1H), 3.88(s, 3H), 3.75 (q, J = 7.5 Hz, 2H), 1.01 (t, J = 7.5 Hz, 3.H); 13C-NMR (75.5 MHz, CDCl3) δ 163.5, 160.5, 159.3, 146.2, 139.7, 132.1, 128.8, 127.9, 114.0, 97.4, 73.0, 61.9, 61.0, 55.5, 13.8; ESI (m/z) 533.1 [M++H, (9)], 555.1 [M++Na, (100)]; HRMS calcd for [C33H28N2O5] 532.1998, found 532.2007.
:20); FTIR (microscope) ν 1768, 1733, 1710 cm−1; 1H-NMR (400 MHz, CDCl3) δ 7.18 (m, 10H), 5.79 (s, 1H), 4.58 (t, J = 8.4 Hz, 1H), 4.22 (q, J = 7.2 Hz, 2H), 4.03 (d, J = 8.4 Hz, 2H), 1.43 (s, 9H), 1.25 (t, J = 7.2 Hz, 3H); 13C-NMR (100.6 MHz, CDCl3) δ 165.7, 160.7, 153.4, 140.8, 133.5, 128.5, 128.2, 126.9, 105.7, 61.7, 58.8, 48.0, 43.1, 29.0, 13.9; ESI (m/z) 421.1 [M++H, (8)], 443.1 [M++Na, (100)]; HRMS calcd for [C25H28N2O4] 420.2049, found 420.2057.
:20); FTIR (microscope) ν 1754, 1722 cm−1; 1H-NMR (400 MHz, CDCl3) δ 7.18 (m, 10H), 5.31 (s, 1H), 4.32 (t, J = 8.4 Hz, 1H), 4.20 (q, J = 7.2 Hz, 2H), 4.09 (d, J = 8.4 Hz, 2H), 1.39 (s, 9H), 1.25 (t, J = 7.2 Hz, 3H); 13C-NMR (100.6 MHz, CDCl3) δ 164.3, 160.3, 153.7, 140.5, 135.3, 128.6, 128.0, 127.1, 100.5, 61.3, 58.5, 47.3, 44.6, 28.4, 14.0; ESI (m/z) 421.2 [M++H, (4)], 443.2 [M++Na, (100)]; HRMS calcd for [C25H28N2O4] 420.2049, found 420.2042.
:20); FTIR (microscope) ν 1778, 1732, 1720 cm−1; 1H-NMR (400 MHz, CDCl3) δ 7.41–7.24 (m, 10H), 5.52 (s, 1H), 4.80 (s, 2H), 4.19 (q, J = 8.0 Hz, 2H), 1.22 (t, J = 8.0 Hz, 3H); 13C-NMR (100.6 MHz, CDCl3) δ 164.0, 158.8, 152.9, 134.5, 134.0, 129.1, 128.4, 128.3, 127.3, 125.9, 103.8, 61.6, 44.3, 13.9; ESI (m/z) 351.1 [M++H, (21)], 373.1 [M++Na, (100)]; HRMS calcd for [C20H18N2O4] 350.1267, found 350.1271.
:20); FTIR (microscope) ν 1788, 1753, 1722 cm−1; 1H-NMR (400 MHz, CDCl3) δ 7.41 (d, J = 16.0 Hz, 2H), 7.17 (d, J = 16.0 Hz, 2H), 6.94 (d, J = 16.0 Hz, 2H), 6.87 (d, J = 16.0 Hz, 2H), 6.08 (s, 1H), 4.75 (s, 2H), 3.83 (s, 3H), 3.81 (s, 3H), 3.71 (q, J = 12.0 Hz, 2H), 1.00 (t, J = 12.0 Hz, 3H); 13C-NMR (100.6 MHz, CDCl3) δ 163.6, 162.4, 159.6, 159.4, 154.1, 135.3, 130.5, 127.7, 127.4, 127.1, 114.3, 114.1, 100.3, 61.1, 55.5, 55.3, 42.7, 13.8; ESI (m/z) 411.2 [M++H, (100)]; HRMS calcd for [C22H22N2O6] 410.1478, found 410.1461.
:20); FTIR (microscope) ν 1790, 1763, 1734 cm−1; 1H-NMR (400 MHz, CDCl3) δ 7.44 (m, 3H), 7.24 (m, 2H), 5.47 (s, 1H), 4.21 (q, J = 6.4 Hz, 2H), 3.11 (s, 3H), 1.25 (t, J = 6.4 Hz, 3H); 13C-NMR (100.6 MHz, CDCl3) δ 164.3, 159.9, 153.0, 137.0, 132.0, 130.1, 129.4, 127.7, 103.8, 61.7, 25.1, 14.1; ESI (m/z) 275.1 [M++H, (100)]; HRMS calcd for [C14H14N2O4] 274.0954, found 274.0963.
:20); FTIR (microscope) ν 1753, 1734 cm−1; 1H-NMR (400 MHz, CDCl3) δ 7.36 (m, 3H), 7.19 (m, 2H), 6.04 (s, 1H), 3.58 (q, J = 8.1 Hz, 2H), 3.11 (s, 3H), 0.88 (t, J = 8.1 Hz, 3H); 13C-NMR (75.5 MHz, CDCl3) δ 163.4, 162.7, 154.2, 135.1, 134.6, 129.1, 128.4, 126.4, 100.6, 60.9, 25.4, 13.7; ESI (m/z) 275.2 [M++H, (100)]; HRMS calcd for [C14H14N2O4] 274.0954, found 274.0943.
:20); FTIR (microscope) ν 1781, 1751, 1718 cm−1; 1H-NMR (400 MHz, CDCl3) δ 7.42 (m, 10H), 5.58 (s, 1H), 4.22 (q, J = 7.2 Hz, 2H), 1.24 (t, J = 7.2 Hz, 3H); 13C-NMR (75.5 MHz, CDCl3) δ 164.2, 158.8, 151.7, 135.9, 131.9, 130.9, 129.4, 128.5, 127.6, 126.4, 125.8, 104.2, 61.7, 13.9; ESI (m/z) 337.2 [M++H, (100)]; HRMS calcd for [C19H16N2O4] 336.1110, found 336.1121.
:20); FTIR (microscope) ν 1765, 1733, 1722 cm−1; 1H-NMR (300 MHz, CDCl3) δ 7.40–7.15 (m, 8H), 6.20 (s, 1H), 3.70 (q, J = 7.5 Hz, 2H), 2.41 (s, 6H), 1.01 (t, J = 7.5 Hz, 3H); 13C-NMR (75.5 MHz, CDCl3) δ 164.2, 158.8, 152.0, 139.7, 138.5, 136.4, 130.0, 129.0, 128.1, 127.3, 126.0, 104.1, 61.9, 21.5, 14.2; ESI (m/z) 365.2 [M++H, (100)]; HRMS calcd for [C19H16N2O4] 336.1110, found 336.1121.
:20); FTIR (microscope) ν 1767, 1741 cm−1; 1H-NMR (300 MHz, CDCl3) δ 7.42–7.21 (m, 4H), 5.62 (s, 1H), 4.29 (q, J = 7.8 Hz, 2H), 2.41 (s, 3H), 2.39 (s, 3H), 1.31 (t, J = 7.8 Hz, 3H); 13C-NMR (75.5 MHz, CDCl3) δ 164.0, 159.0, 152.0, 139.7, 138.7, 136.5, 130.9, 130.0, 129.6, 129.3, 128.2, 127.5, 126.3, 126.0, 103.9, 61.5, 21.2, 14; ESI (m/z) 365.1 [M++H, (10)], 287.1 [M++Na, (100)]; HRMS calcd for [C21H20N2O4] 364.1423, found 364.1432.
:20); FTIR (microscope) ν 1780, 1754, 1721 cm−1; 1H-NMR (300 MHz, CDCl3) δ 7.40–7.15 (m, 8H), 6.20 (s, 1H), 3.70 (q, J = 7.5 Hz, 2H), 2.41 (s, 6H), 1.01 (t, J = 7.5 Hz, 3H); 13C-NMR (75.5 MHz, CDCl3) δ 164.2, 158.8, 152.0, 139.7, 138.5, 136.4, 130.0, 129.0, 128.1, 127.3, 126.0, 104.1, 61.9, 21.5, 14.2; ESI (m/z) 365.2 [M++H, (100)]; HRMS calcd for [C21H20N2O4] 364.1423, found 364.1413.
:20); FTIR (microscope) ν 1766, 1723, 1712 cm−1; 1H-NMR (300 MHz, CDCl3) δ 7.40 (d, J = 12.0 Hz, 4H), 7.17 (d, J = 12.0 Hz, 4H), 6.06 (s, 1H), 3.83 (s, 3H), 3.82 (s,3H), 3.30 (q, J = 7.8 Hz, 2H)1.01 (t, J = 7.8 Hz, 3H); 13C-NMR (75.5 MHz, CDCl3) δ 163.5, 162.3, 159.5,159.3, 155.2, 130.5, 127.6, 127.1, 114.2, 100.3, 61.2, 55.2, 13.8; ESI (m/z) 397.1 [M++H, (16)], 419.1 [M++Na, (100)]; HRMS calcd for [C21H20N2O6] 396.1321, found 396.1316.
Synthesis of spiroisoxalidinohydantoins 4. General procedure
A stirred solution of 5-carboxymethylene hydantoins 3 (1 equiv.) and nitrone 7 (1.5 equiv.) in toluene (0.1 M solution) was heated at 80 °C in a sealed tube. After 48 h the solution was cooled to rt, the organic solvent removed under reduced pressure and the crude purified by flash chromatography.:20); FTIR (microscope) ν 1765, 1712 cm−1; 1H-NMR (400 MHz, CDCl3) δ 7.58 (m, 2H), 7.37 (m, 3H), 7.18 (m, 2H), 7.05 (m, 2H), 5.18 (d, J = 16.4 Hz, 1H), 4.17 (m, 1H), 4.00 (m, 1H), 3.95 (m, 1H), 3.84 (d, J = 16.4 Hz, 1H), 3.70 (m, 1H), 2.27–1.68 (m, 16H), 1.30 (m, 4H), 1.17 (t, J = 11.6 Hz, 3H); 13C-NMR (100.6 MHz, CDCl3) δ 168.8, 166.8, 154.3, 148.4, 137.1, 128.9, 128.6, 128.0, 125.2, 119.9, 91.7, 77.2, 71.9, 61.7, 59.9, 53.7, 51.5, 31.0, 30.7, 29.3, 29.2, 26.5, 26.4, 25.8, 25.2, 25.0, 13.8; ESI (m/z) 568.2 [M++Na, (100)]; HRMS calcd for [C32H39N3O5] 545.2890, found 545.2882.
:20); FTIR (microscope) ν 1765, 1733, 1724 cm−1; 1H-NMR (300 MHz, CDCl3) δ 7.47 (m, 2H), 7.30 (m, 1H), 5.08 (d, J = 15.0 Hz, 1H), 3.87 (m, 1H), 3.73 (d, J = 15.0 Hz, 1H), 3.40 (m, 1H), 2.65 (s, 3H), 2.30–1.55 (m, 20H), 1.27 (s, 9H); 13C-NMR (75.5 MHz, CDCl3) δ 169.9, 165.6, 154.2, 135.9, 134.8, 132.2, 130.2, 130.1, 127.7, 92.4, 83.2, 69.0, 58.2, 53.1, 51.7, 43.8, 31.0, 30.8, 29.6, 27.8, 26.6, 26.4, 26.0, 25.3, 25.2; ESI (m/z) 602.1 [M++Na, (100)], 604.1 [M++Na + 2, (66)], 606.1 [M++Na + 4, (11)]; HRMS calcd for [C29H39Cl2N3O5] 579.2267, found 579.2275.
:20); FTIR (microscope) ν 1770, 1741, 1716 cm−1; 1H-NMR (500 MHz, CDCl3) δ 8.03 (s, 1H), 7.90 (m, 3H), 7.68 (m, 1H), 7.53 (m, 2H), 7.27 (m, 5H), 4.71 (d, J = 10.0 Hz, 1H), 4.02 (d, J = 15.0 Hz, 1H), 3.88 (m, 2H), 3.76 (m, 2H), 2.27 (m, 1H), 2.10 (m, 3H), 1.84–1.57 (m, 13H), 1.36 (s, 9H), 1.29 (m, 3H); 13C-NMR (100.6 MHz, CDCl3) δ 169.0, 166.4, 154.4, 137.5, 133.4, 128.7, 128.5, 128.04, 128.01, 127.9, 127.8, 127.1, 126.4, 125.0, 92.1, 82.8, 72.8, 60.8, 59.7, 53.7, 51.6, 30.9, 30.8, 29.6, 29.5, 27.9, 26.8, 26.5, 25.9, 25.3, 25.2; ESI (m/z) 638.2 [M++H, (100)], 660.1 [M++Na, (72)]; HRMS calcd for [C39H47N3O5] 637.3516, found 637.3502.
:20); FTIR (microscope) ν 1768, 1713 cm−1; 1H-NMR (500 MHz, CDCl3) δ 7.45 (m, 3H), 7.27 (m, 10H), 5.36 (d, J = 15.0 Hz, 1H), 4.66 (t, J = 7.5 Hz, 1H), 4.09 (m, 3H), 3.95 (m, 1H), 3.86 (m, 1H), 2.62 (s, 2H), 1.60 (s, 9H), 1.04 (t, J = 7.5 Hz, 3H); 13C-NMR (100.6 MHz, CDCl3) δ 170.6, 166.5, 155.3, 141.1, 141.0, 136.4, 129.8, 128.54, 128.50, 128.3, 128.2, 127.7, 126.8, 93.4, 68.5, 61.5, 57.2, 48.3, 43.7, 43.0, 29.7, 29.5, 28.6, 13.7; ESI (m/z) ESI (m/z) 646.1 [M++Na, (100)], 648.1 [M++Na + 2, (66)], 650.1 [M++Na + 4, (11)]; HRMS calcd for [C33H35Cl2N3O5] 623.1954, found 623.1945.
:20); 1H-NMR (300 MHz, CDCl3) δ 7.37–7.22 (m, 13H), 4.88 (d, J = 10.7 Hz, 1H), 4.73 (dd, J = 11.0 and 4.0 Hz, 1H), 4.04 (m, 2H), 3.85 (m, 1H), 2.95 (d, J = 10.7 Hz, 1H), 2.65 (s, 3H), 1.60 (s, 9H), 1.09 (t, J = 7.0 Hz, 3H); 13C-NMR (100.6 MHz, CDCl3) δ 170.2, 166.6, 157.0, 142.7, 142.0, 135.9, 134.8, 130.0, 129.7, 128.8, 128.6, 127.9, 127.2, 126.7, 91.6, 61.5, 58.3, 49.3, 46.4, 43.2, 28.6, 13.6; ESI (m/z) 646.1 [M++Na, (100)], 648.1 [M++Na + 2, (66)], 650.1 [M++Na + 4, (11)]; HRMS calcd for [C33H35Cl2N3O5] 623.1954, found 623.1946.
:20); 1H-NMR (400 MHz, CDCl3) δ 7.73 (m, 4H), 7.41 (m, 10H), 7.28 (m, 8H), 5.09 (d, J = 16 Hz, 1H) ,4.63 (m, 2H), 3.97 (m, 2H), 3.82 (m, 3H), 0.98 (t, J = 8.0 Hz, 3H); 13C-NMR (100.6 MHz, CDCl3) δ 168.3, 167.0, 154.9, 137.1, 136.9, 131.5, 133.5, 133.2, 132.5, 131.5, 128.4, 128.1, 127.8, 127.7, 127.6, 127.4, 127.3, 126.4, 126.3, 125.7, 125.2, 91.9, 73.1, 61.6, 60.2, 59.6, 43.3, 13.7; ESI (m/z) 612.2 [M++H, (23)], 634.1 [M++Na, (100)]; HRMS calcd for [C38H33N3O5] 611.2420, found 611.2434.
:20); FTIR (microscope) ν 1788, 1753, 1725 cm−1; 1H-NMR (500 MHz, CDCl3) δ 7.63 (d, J = 7.5 Hz, 2H), 7.37 (m, 4H), 7.09 (dd, J = 8.5 and 1.5 Hz, 1H), 6.90 (d, J = 8.5 Hz, 1H), 5.02 (d, J = 11.0 Hz, 1H), 4.05 (q, J = 7.0 Hz, 2H), 4.74 (d, J = 11.0 Hz, 1H), 3.16 (s, 3H), 2.69 (s, 3H), 1.13 (t, J = 7.0 Hz, 3H); 13C-NMR (100.6 MHz, CDCl3) δ 169.2, 166.6, 154.3, 135.9, 134.8, 133.4, 131.5, 129.7, 129.6, 129.4, 128.2, 127.6, 127.5, 93.4, 69.3, 61.8, 57.9, 43.6, 25.0, 13.7; ESI (m/z) 502.0 [M++Na + 2, (74)], 500.0 [M++Na, (100)]; HRMS calcd for [C22H21Cl2N3O5] 477.0858, found 477.0861.
:20); FTIR (microscope) ν 1778, 1727 cm−1; 1H-NMR (400 MHz, CDCl3) δ 7.38 (m, 7H), 7.18 (m, 1H), 4.20 (d, J = 11.6 Hz, 1H), 4.13 (d, J = 11.6 Hz, 1H), 3.97 (m, 1H), 3.87 (m, 1H), 3.11 (s, 3H), 2.49 (s, 3H), 1.00 (t, J = 7.2 Hz, 3H); 13C-NMR (100.6 MHz, CDCl3) δ 170.1, 165.6, 154.7, 134.9, 134.6, 130.7, 129.6, 129.2, 129.1, 128.4, 127.7, 126.4, 100.6, 66.5, 61.6, 42.7, 13.6; ESI (m/z) 502.0 [M++Na + 2, (32)], 500.0 [M++Na, (100)]; HRMS calcd for [C22H21Cl2N3O5] 477.0858, found 477.0867.
:20); FTIR (microscope) ν 1781, 1745, 1716 cm−1; 1H-NMR (400 MHz, CDCl3) δ 7.55 (d, J = 8.4 Hz, 2H), 7.29–7.05 (m, 16H), 5.04 (d, J = 10.8 Hz, 1H), 4.01 (m, 2H), 3.83 (d, J = 10.8 Hz, 1H), 2.38 (s, 3H), 2.32 (s, 3H), 1.07 (t, J = 7.2 Hz, 3H); 13C-NMR (100.6 MHz, CDCl3) δ 167.9, 166.8, 153.4, 148.4, 138.4, 138.3, 130.0, 129.6, 128.6, 128.5, 128.2, 127.7, 125.7, 125.5, 120.3, 92.7, 72.6, 61.8, 58.8, 21.2, 21.1, 13.8; ESI (m/z) 562.2 [M++H, (3)], 584.1 [M++Na, (100)]; HRMS calcd for [C34H31N3O5] 561.2264, found 561.2269.
:20); FTIR (microscope) ν 1777, 1756, 1733 cm−1; 1H-NMR (400 MHz, CDCl3) δ 7.47 (d, J = 6.8 Hz, 2H), 7.33 (d, J = 6.8 Hz, 2H), 7.23 (m, 7H), 7.03 (m, 3H), 6.92 (m, 2H), 6.76 (d, J = 6.8 Hz, 2H), 4.39 (s, 2H), 4.15 (m, 1H), 4.02 (m, 1H), 2.33 (s, 3H), 2.23 (s, 3H), 1.13 (t, J = 7.2 Hz, 3H);1H-NMR (400 MHz, acetone-d6) δ 7.47 (d, J = 6.8 Hz, 2H), 7.23 (m, 14H), 6.79 (d, J = 6.8 Hz, 2H), 4.47 (d, J = 11.6 Hz, 1H), 4.41 (d, J = 11.6 Hz, 1H), 4.26 (m, 1H), 4.11 (m, 1H), 2.36 (s, 3H), 2.27 (s, 3H), 1.17 (t, J = 7.2 Hz, 3H); 13C-NMR (100.6 MHz, CDCl3) δ 168.1, 165.8, 153.8, 138.9, 138.7, 136.7, 129.9, 129.5, 129.2, 128.8, 128.5, 128.3, 127.9, 126.9, 126.3, 125.7, 123.9, 92.3, 67.6, 62.3, 61.8, 61.0, 21.1, 13.9; ESI (m/z) 584.1 [M++Na, (100)], 600.2 [M++K, (5)]; HRMS calcd for [C34H31N3O5] 561.2264, found 561.2255.
:20); FTIR (microscope) ν 1781, 1751, 1719 cm−1; 1H-NMR (400 MHz, CDCl3) δ 7.46 (d, J = 8.4 Hz, 2H), 7.29 (m, 3H), 7.21 (m, 5H), 7.04 (dd, J = 8.4 and 2.0 Hz, 1H), 6.89 (d, J = 8.4 Hz, 1H), 4.01 (d, J = 11.2 Hz, 1H), 4.04 (m, 1H), 3.93 (m, 1H), 3.73 (d, J = 11.2 Hz, 1H), 2.64 (s, 3H), 2.31 (s, 3H), 1.04 (t, J = 7.2 Hz, 3H); 13C-NMR (100.6 MHz, CDCl3) δ 168.4, 166.7, 153.4, 138.3, 135.8, 134.7, 131.7, 130.5, 130.0, 129.6, 128.6, 127.6, 127.5, 125.8, 93.1, 69.1, 61.9, 58.0, 43.7, 21.2, 13.7; ESI (m/z) 590.1 [M++Na, (100)], 592.1 [M++Na + 2, (66)], 594.1 [M++Na + 4, (11)]; HRMS calcd for [C29H27Cl2N3O5] 567.1328, found 567.1335.
:20); FTIR (microscope) ν 1758, 1722 cm−1; 1H-NMR (400 MHz, CDCl3) δ 7.36–7.29 (m, 11H), 4.31 (s, 2H), 4.11 (m, 1H), 4.04 (m, 1H), 2.56 (s, 3H), 2.41 (s, 3H), 1.13 (t, J = 6.8 Hz, 3H);1H-NMR (400 MHz, acetone-d6) δ 7.57 (d, J = 8.8 Hz, 2H), 7.34 (m, 9H), 4.37 (d, J = 12.0 Hz, 1H), 4.27 (d, J = 12.0 Hz, 1H), 4.10 (m, 1H), 4.06 (m, 1H), 2.48 (s, 3H), 2.38 (s, 3H), 1.08 (t, J = 7.2 Hz, 3H); 13C-NMR (100.6 MHz, CDCl3) δ 169.2, 165.7, 153.7, 139.2, 138.4, 134.9, 130.3, 129.8, 129.7, 129.6, 128.8, 127.7, 126.3, 125.7, 125.6, 100.8, 66.5, 61.7, 61.0, 42.8, 21.1, 13.8; ESI (m/z) 590.1 [M++Na, (100)], 592.1 [M++Na + 2, (66)], 594.1 [M++Na + 4, (11)]; HRMS calcd for [C29H27Cl2N3O5] 567.1328, found 567.1331.
:20); 1H-NMR (400 MHz, CDCl3) δ 7.18 (m, 4H), 5.39 (br s, 1H), 4.21 (m, 1H), 4.01 (m, 1H), 3.78 (m, 1H), 3.58 (m, 1H), 3.40 (m, 1H), 3.20 (m, 1H), 3.20 (m, 1H), 2.55 (br d, J = 16 Hz, 1H), 2.18 (m, 1H), 2.00 (m, 3H), 1.75–1.58 (m, 10H), 1.19 (t, J = 8.0 Hz, 3H), 1.21–1.17 (m, 4H), 0.94 (m, 1H), 0.55 (m, 2H); 13C-NMR (100.6 MHz, CDCl3) δ 169.4, 168.3, 154.4, 134.4, 134.2, 128.7, 127.3, 127.1, 93.0, 62.6, 62.0, 59.7, 53.3, 51.2, 49.3, 30.8, 30.3, 29.4, 29.3, 25.8, 25.0, 24.9, 23.1, 14.0; ESI (m/z) 496.1 [M++1, (100)]; HRMS calcd for [C28H37N3O5] 495.2733, found 495.2744.
:30); FTIR (microscope) ν 1784, 1754, 1730 cm−1; 1H-NMR (400 MHz, CDCl3) δ 7.44–7.35 (m, 6H), 7.04 (m, 3H), 4.60 (d, J = 12.0 Hz, 1H), 4.20 (m, 1H), 4.04 (d, J = 12.0 Hz, 1H), 4.02 (m, 1H), 4.44 (m, 2H), 3.11 (s, 3H), 3.04 (m, 1H), 2.82 (br d, J = 12.0 Hz, 1H), 1.16 (t, J = 8.1 Hz, 3H); 13C-NMR (100.6 MHz, CDCl3) δ 170.2, 166.4, 154.5, 133.4, 133.3, 131.7, 128.9, 128.6, 128.3, 128.2, 127.7, 126.3, 96.7, 63.6, 61.7, 59.1, 51.2, 28.9, 25.4, 14.0; ESI (m/z) 444.1 [M++Na, (100)], 422.2 [M++H, (41)]; HRMS calcd for [C23H23N3O5] 421.1638, found 421.1645.
:20); FTIR (microscope) ν 1778, 1751, 1721 cm−1; 1H-NMR (400 MHz, acetone-d6) δ 7.32 (m, 5H), 7.02 (m, 3H), 5.29 (br s, 1H), 4.23 (m, 1H), 4.17 (m, 1H), 3.54 (d, J = 12.0 Hz, 1H), 3.08 (m, 1H), 3.03 (s, 3H), 2.71 (br m, 3H), 1.21 (t, J = 8.1 Hz, 3H); 13C-NMR (100.6 MHz, acetone-d6) δ 174.2, 172.9, 159.6, 138.8, 138.3, 138.0, 135.2, 134.4, 134.0, 133.3, 132.3, 131.5, 101.5, 68.3, 66.7, 61.1, 56.0, 31.8, 29.2, 18.6; ESI (m/z) 444.1 [M++Na, (100)], 422.2 [M++H, (23)]; HRMS calcd for [C23H23N3O5] 421.1638, found 421.1625.
:20); [α]25D: + 24.5 (c = 0.4, CHCl3); FTIR (microscope) ν 1773, 1738, 1724 cm−1; 1H-NMR (400 MHz, CDCl3) δ 4.41 (d, J = 10.4 Hz, 1H), 4.17 (m, 2H), 4.02 (m, 2H), 3.87 (d, J = 10.4 Hz, 1H), 3.84 (d, J = 4.4 Hz, 1H), 3.69 (m, 1H), 3.57 (dd, J = 15.2 and 4.4 Hz, 1H), 3.19 (d, J = 15.2 Hz, 1H), 1.36–1.30 (m, 15H), 1.17–1.14 (m, 18 H); 13C-NMR (100.6 MHz, CDCl3) δ 170.1, 168.2, 154.5, 93.5, 80.3, 79.6, 74.8, 74.6, 74.2, 65.2, 61.6, 49.5, 43.9, 43.4, 28.8, 28.33, 28.31, 28.2, 20.6, 20.2, 19.6, 19.5, 14.1, ; ESI (m/z) 520.1 [M++Na, (100)], 498.2 [M++H, (5)]; HRMS calcd for [C25H43N3O7] 497.3101, found 497.3113.
Synthesis of amide derivatives 8 and 9. General procedure
To a stirred solution of spirohydantoin 4 g (1 equiv.) in MeOH (0.1 M solution) 2 equiv. of a 1N aqueous solution of NaOH was added at rt and the mixture stirred over night. The organic solvent was removed under reduced pressure, the aqueous acidified with a 1N aqueous solution of HCl and extracted with AcOEt. The combined organic layers were dried over anhydrous Na2SO4, filtered, concentrated under vacuum. The resulting acid was dissolved in DMF (0.1 M solution), treated with the corresponding amine (1.1 equiv.), EDCI (1.1 equiv.) and TMP (2 equiv.) and the resulting solution was stirred at rt over-night. The organic solvent was removed under reduced pressure, the crude dissolved in a 1N aqueous solution of HCl and extracted with AcOEt. The combined organic layers were dried over anhydrous Na2SO4, filtered, concentrated under vacuum and the crude purified by flash chromatography.:40); FTIR (microscope) ν 1788, 1758 cm−1; 1H-NMR (400 MHz, CDCl3) δ 7.85 (s, 1H), 7.57 (m, 2H), 7.38 (m, 2H), 7.31 (m, 1H), 7.18 (m, 4H), 6.98 (m ,1H), 6.95 (m, 3H), 5.33 (bs, 1H), 4.98 (d, J = 12.0 Hz, 1H), 4.23 (dd, J = 16.0 and 8.0 Hz, 1H), 4.11 (dd, J = 16.0 and 8.0 Hz, 1H), 3.55 (d, J = 12.0 Hz, 1H), 3.05 (s, 3H), 2.61 (s, 3H); 13C-NMR (100.6 MHz, CDCl3) δ 169.7, 164.9, 154.3, 136.7, 135.7, 134.9, 133.6, 130.7, 129.9, 129.3, 128.7, 128.5, 127.9, 127.7, 127.6, 127.3, 93.8, 68.4, 58.9, 43.8, 43.6, 25.0; ESI (m/z) 561.1 [M++Na, (100)]; HRMS calcd for [C27H24Cl2N4O4] 538.1175, found 538.1172.
:40); FTIR (microscope) ν 1777, 1761, 1733 cm−1; 1H-NMR (400 MHz, CDCl3) δ 8.59 (s, 1H), 7.40 (d, J = 4.1 Hz, 1H), 7.32 (d, J = 4.1 Hz, 1H), 7.18 (m, 3H), 7.05 (d, J = 7.8 Hz, 1H), 6.98 (dd, J = 7.8 and 4.1 Hz, 1H), 6.92 (m, 2H), 6.73 (m, 2H), 6.35 (dd, J = 7.8 and 4.1 Hz, 1H), 5.48 (d, J = 5.2 Hz, 1H), 4.03 (d, J = 5.2 Hz, 1H), 3.61 (s, 3H), 3.10 (s, 3H), 2.96 (s, 3H), 1.84 (s, 3H); 13C-NMR (100.6 MHz, CDCl3) δ 182.3, 172.8, 169.3, 147.6, 145.9, 143.9, 141.7, 141.5, 141.1, 140.4, 140.0, 139.9, 139.2, 138.5, 127.9, 120.9, 115.0, 112.4, 103.6, 80.9, 70.6, 66.7, 56.0, 36.6, 28.1; ESI (m/z) 591.1 [M++Na, (100)]; HRMS calcd for [C28H26Cl2N4O5] 568.1280, found 568.1289.
References
- P. Pradhan, M. Patra, A. K. Behera, B. K. Mishra and R. K. Behera, Tetrahedron, 2006, 62, 779–828 CrossRef CAS and references cited therein..
- (a) H. Bittermann, F. Böckler, E. Jürgen and P. Gmeiner, Chem.–Eur. J., 2006, 12, 6315–6322 CrossRef CAS; (b) H. Bittermann and P. Gmeiner, J. Org. Chem., 2006, 71, 97–102 CrossRef CAS; (c) G. Lesma, A. Colombo, A. Sacchetti and A. Silvani, Tetrahedron Lett., 2008, 49, 7423–7425 CrossRef CAS; (d) A. Sacchetti, A. Silvani, G. Lesma and T. Pilati, J. Org. Chem., 2011, 76, 833–839 CrossRef CAS.
- (a) D. L. Boger, M. A. Labroli and T. H. Marsilje, Bioorg. Med. Chem., 2000, 8, 1075–1086 CrossRef CAS; (b) V. J. Hruby, Life Sci., 1982, 31, 189–199 CrossRef CAS.
- (a) E. Naydenova, N. Pencheva, J. Popova, N. Stoyanov, M. Lazarova and B. Aleksiev, Farmaco, 2002, 57, 189–194 CrossRef CAS; (b) H. Byrtus, M. Pawlowski, B. Duszynska, A. Wesolowska, E. Chojnaka-Wojcik and A. Bojarski, Polym. J. Pharm., 2001, 53, 395–401 Search PubMed.
- (a) R. M. Schelkum, P. Yuen, K. Serpa, L. T. Meltzer, L. D. Wise, E. R. Whittemore and R. M. Woodward, J. Med. Chem., 2000, 43, 1892–1897 CrossRef CAS; (b) H. U. Stilz, W. Guba, B. Jablonka, M. Just, O. Klinger, W. König, V. Wehner and G. Zoller, J. Med. Chem., 2001, 44, 1158–1176 CrossRef CAS.
- M. W. Rowbottom, T. D. Vickers, J. Tamiya, M. Zhang, B. Dyck, J. Grey, D. Schwarz, C. E. Heise, M. Hedrick, J. Wen, H. Tang, H. Wang, A. Fisher, A. Aparicio, J. Saunders and V. S. Goodfllow, Bioorg. Med. Chem. Lett., 2007, 17, 2171–2178 CrossRef CAS.
- S. C. Watterson, Z. Xiao, D. S. Dodd, D. R. Tortolani, W. Vaccaro, D. Potin, M. Launay, D. K. Stetsko, S. Skala, P. M. Davis, D. Lee, X. Yang, K. W. McIntyre, P. Balimane, K. Patel, Z. Yang, P. Marathe, P. Kadiyala, A. J. Tebben, S. Sheriff, C. Y. Y. Chang, T. Ziemba, H. Zhang, B. –C. Chen, A. J. DelMonte, N. Aranibar, M. McKinnon, J. C. Barrish, S. J. Suchard and T. G. Murali Dhar, J. Med. Chem., 2010, 53, 3814–3830 CrossRef CAS.
- (a) A. Volonterio and M. Zanda, Tetrahedron Lett., 2003, 44, 8549–8551 CrossRef CAS; (b) A. Volonterio, C. Ramirez de Arellano and M. Zanda, J. Org. Chem., 2005, 70, 2161–2170 CrossRef CAS; (c) F. Olimpieri, A. Volonterio and M. Zanda, Synlett, 2008, 3016–3020 CAS; (d) A. Volonterio and M. Zanda, Org. Lett., 2007, 9, 841–844 CrossRef CAS; (e) A. Volonterio and M. Zanda, J. Org. Chem., 2008, 73, 7486–7497 CrossRef CAS; (f) F. Olimpieri, S. Fustero, A. Volonterio and M. Zanda, Synthesis, 2010, 4, 651–660; (g) T. Marcelli, F. Olimpieri and A. Volonterio, Org. Biomol. Chem., 2011, 9, 5156 RSC.
- (a) While preparing the manuscript, a paper describing the synthesis of spiroisoxalidinohydantoins starting from 5-methylenehydantoins has appeared in literature: A. Bahy, Y. Kacem, B. Ben Hassine, Synth. Commun: 2010, 40, 1377-1390. For some other papers dealing with the synthesis of spiro-heterocycles by 1,3-dipolar cycloadditions, see: R. D. Carpenter, P. B. DeBerdt, J. B. Holden, K. A. Milinkevich, T. Min, D. Willenbring, J. C. Fettinger, D. J. Tantillo and M. J. Kurth, J. Comb. Chem., 2008, 10, 225–229 Search PubMed; (b) K. G. Guggenheim, J. D. Butler, P. P. Painter, B. A. Lorsbach, D. J. Tantillo and M. J. Kurth, J. Org. Chem., 2011, 76, 5803–5812 CrossRef CAS.
- (a) R. Huisgen, K. Herbig, A. Siegl and H. Huber, Chem. Ber., 1966, 99, 2526–2445 CAS; (b) M-. J. Fan, G-. Q. Li and Y-. M. Liang, Tetrahedron, 2006, 62, 6782–6791 CrossRef CAS; (c) Q. Zhu, H. Jiang, J. Li, M. Zhang, X. Wang and C. Qi, Tetrahedron, 2009, 65, 4604–4613 CrossRef CAS.
- F. Olimpieri, M. C. Bellucci, A. Volonterio and M. Zanda, Eur. J. Org. Chem., 2009, 6179–6188 CrossRef CAS.
- The regio- and diastereochemistry of (E)-3f,g were assessed by NOE experiments (see Supporting Information).
- The stereochemistry of the E/Z isomers was assessed considering the chemical shift of the vinilic protons and by NOE experiments (see Supporting Information).
- (a) For some recent reviews dealing with general 1,3-dipolar cycloadditions, see: S. Karlsson and H.-E. Högberg, Org. Prep. Proced. Int., 2001, 33, 103–172 Search PubMed; (b) G. Broggini, G. Molteni, A. Terraneo and G. Zecchi, Heterocycles, 2003, 59, 823–858 CrossRef CAS; (c) K. V. Gothelf and K. A. Jørgensen, Chem. Rev., 1998, 98, 863–909 CrossRef CAS; (d) V. Nair and T. D. Suja, Tetrahedron, 2007, 63, 12247–12275 CrossRef CAS; (e) S. Kobayashi, K. A. Jørgensen, Cycloaddition Reaction in Organic Synthesis, Wiley-VCH, Weinheim, 2002 Search PubMed; (f) M. Pineiro and M. V. Pinho e Melo, Eur. J. Org. Chem., 2009, 5287–5307 CrossRef CAS; (g) C. Najera and J. M. Sansano, Org. Biomol. Chem., 2009, 7, 4567–4581 RSC; (h) For some reviews on nitrones and their cycloaddition reactions, see: M. Frederickson, Tetrahedron, 1997, 53, 403–425 Search PubMed; (i) H. M. I. Osborn, N. Gemmell and L. M. Harwood, J. Chem. Soc., Perkin Trans. 1, 2002, 2419–2438 RSC; (j) K. Rück-Braun, T. H. E. Freysoldt and F. Wierschem, Chem. Soc. Rev., 2005, 34, 507–516 RSC.
- The regiochemistry was determined by comparison with the reactivity of related 5-cyanomethylene hydantoins with nitrile oxides (see U. Groselj, A. Drobnic, S. Recnik, J. Svete, B. Stanovnik, A. Golobic, N. Lah, I. Leban, A. Meden, S. Golic-Grdadolnik, Helv. Chim. Acta, 2001, 84, 3403-3417). Moreover the spirocarbon of all adducts resonances between 90 and 95 ppm as expected for a spirocarbon bonded to the nitorne oxigen.
- The relative anti configuration of compounds arising from the reaction of (Z)-3k,l has been determinate by calculating the coupling constants between the isoxazolidine methynic protons which were always in the range of 11 Hz. However, in the case of spiroisoxazolidinohydantoins 4j,l it was necessary to run the NMR spectra in acetone-d6 because in CDCl3 the two protons resonated at the same ppm leading to a singlet integrating for two protons.
- S. Cicchi, M. Corsi and A. Goti, J. Org. Chem., 1999, 64, 7243–7245 CrossRef CAS.
- (a) For some recent reviews, see: J. Revuelta, S. Cicchi, A. Goti and A. Brandi, Synthesis, 2007, 485–504 Search PubMed; (b) A. Brandi, F. Cardona, S. Cicchi, F. M. Cordero and A. Goti, Chem.–Eur. J., 2009, 15, 7808–7821 CrossRef CAS.
- S. Cicchi, I. Höld and A. Brandi, J. Org. Chem., 1993, 58, 5274–5275 CrossRef CAS.
- It should be noted that, in comparison with the previous study on the reaction between carbodiimides and activated α,β-insaturated carboxylic acids (Ref. 8b), O-acylisurea 5 and 5’ could rearrange through an O→N acyl migration mechanism to form the corresponding N-acylurea derivatives. However, in the present work, we never observed the formation of N-acylureas, rendering the reaction with acetylenedicarboxylic acid monoesters completely chemoselective.
- (a) J. R. Goodell, B. Leng, T. K. Snyder, A. B. Beeler and J. A. Porco, Jr, Synthesis, 2010, 2254–2270 CAS.
- (a) A π-staking interaction was previously invoked to explain the selectivity found in both inter- and intramolecular aza-Michael reaction: F. Dumas, B. Mezrhab, J. d’Angelo, C. Riche and A. Chiaroni, J. Org. Chem., 1996, 61, 2293–2304 Search PubMed; (b) S. Fustero, S. Monteagudo, M. Sanchez-Rossello, S. Flores, P. Barrio and C. del Pozo, Chem.–Eur. J., 2010, 16, 9835–9845 CrossRef CAS.
- (a) For theoretical investigations on 1,3-dipolar cycloadditions, see: A. Milet, Y. Gimbert and A. E. Greene, J. Comput. Chem., 2006, 27, 157–162 Search PubMed; (b) M. L. Kuznetsov and V. Y. Kukushkin, J. Org. Chem., 2006, 71, 582–592 CrossRef CAS; (c) E. Borsini, G. Broggini, A. Contini and G. Zecchi, Eur. J. Org. Chem., 2008, 2808–2816 CrossRef CAS; (d) L. Meng, S. C. Wang, J. C. Fettinger, M. J. Kurth and D. J. Tantillo, Eur. J. Org. Chem., 2009, 1578–1584 CrossRef CAS; (e) T. K. Das, S. Salampuria and M. Banerjee, THEOCHEM, 2010, 959, 22–29 CrossRef CAS; (f) E. Frank, Z. Mucsi, M. Szecsi, I. Zupko, J. Wölfling and G. Schneider, New J. Chem., 2010, 34, 2671–2681 RSC; (g) N. Acharjee and A. Banerji, J. Computational Chem., 2011, 967, 50–58 Search PubMed; (h) K. Alimohammadi, Y. Sarrafi, M. Tajbakhsh, S. Yeganegi and M. Hamzehloueian, Tetrahedron, 2011, 67, 1589–1597 CrossRef CAS.
- J. Bjørgo, D. R. Boyd and D. C. Neill, J. Chem. Soc., Chem. Commun., 1974, 478–479 RSC.
- P. Molina, E. Aller and A. Lorenzo, Synthesis, 1998, 26, 283–287 CrossRef.
- M. Schmitt, J. Bourguignon J.-, C. –G. Wermuth, D. Schott, B. Rousseau and J.-P. Beaucourt, J. Labelled Compd. Radiopharm., 1989, 27, 23–33 CrossRef CAS.
- M. J. Frisch et al. Gaussian 03, Revision C.02, Gaussian Inc., Wallingford CT, 2004 Search PubMed.
- M. J. Frisch et al. Gaussian 09, Revision A.02, Gaussian Inc., Wallingford CT, 2009 Search PubMed.
- (a) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785–789 CrossRef CAS; (b) A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS; (c) A. D. Becke, J. Chem. Phys., 1992, 96, 2155–2160 CrossRef CAS; (d) A. D. Becke, J. Chem. Phys., 1992, 97, 9173–9177 CrossRef CAS; (e) A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- (a) Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 CrossRef CAS; (b) Y. Zhao and D. G. Truhlar, Acc. Chem. Res., 2008, 41, 157–167 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available: Copies of the 1H and 13C NMR spectra for all new compounds and NOE experiments. Full ref. 24 and 25. Cartesian coordinates and energies for all stationary points. See DOI: 10.1039/c1ra00573a/ |
| This journal is © The Royal Society of Chemistry 2011 |