C(naphthyl)–H bond activation by rhodium: isolation, characterization and TD-DFT study of the cyclometallates†
Achintesh
Narayan Biswas
a,
Purak
Das
a,
Sandip
Sengupta
a,
Amitava
Choudhury
b and
Pinaki
Bandyopadhyay
*a
aDepartment of Chemistry, University of North Bengal, Raja Rammohunpur, Siliguri, 734013, India. E-mail: pbchem@rediffmail.com; Fax: 91 353 2699001; Tel: 91 353 2776381
bDepartment of Chemistry, Missouri S & T, Rolla, MO 65409, USA
First published on 22nd September 2011
Abstract
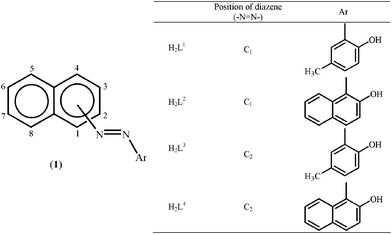
The C1(naphthyl)–H, C2(naphthyl)–H, C3(naphthyl)–H and C8(naphthyl)–H bonds of the naphthyl group present in a group of naphthylazo–2′–hydroxyarenes (H2L) have been activated by [Rh(PPh3)3Cl] in a toluene medium. Here the cyclometallation is accompanied by metal centered oxidation [Rh(I)→Rh(III)]. All the resulting cyclometallates [Rh(PPh3)2(L)Cl] (2–5) have been isolated in a pure form. The characterization of the cyclometallates [Rh(PPh3)2(L)Cl] have been done on the basis of spectral (IR, UV–vis, and FAB mass) data. The structures of the representative cyclometallates 2a, 3a, 4a, 4b and 5b have been determined by X-ray diffraction. In all the cyclometallates, rhodium(III) is coordinated to naphthylazo–2′–hydroxyarenes via terdentate C(naphthyl), N(diazene), O(phenolato/ naphtholato) donor centers & one chloride ion in a plane along with two axial transPPh3 molecules. Intermolecular association in the solid state is observed due to C–H⋯π and π⋯π interactions. Compounds show an oxidative response within 0.93 to 1.11 V (vs.SCE) and a reductive response at ∼ −1.0 V (vs.SCE). Both the responses are based on the coordinated diazene function and are irreversible in nature, indicating limited stability of the oxidized and reduced species. The electronic structures of selected cyclometallates have been calculated using a TD-DFT model and the simulated spectra are consistent with the observed spectra of those cyclometallates.
Introduction
The activation of C–H bonds in organic compounds promoted by transition metal complexes has experienced a rapid growth in view of its diverse synthetic potential.1 In this regard, the cyclometallation reaction, probably the mildest route for activating the C–H bond, has received increased attention.2Cyclometalation reactions have proven to be a powerful synthetic route, especially in the activation of C–H bonds, which are otherwise difficult to access.3In general, the metal centres, precoordinated to a Lewis basic heteroatom group of the organic substrate, are brought in the vicinity of C–H bonds to be activated, which ultimately leads to the formation of a metal-carbon bond.2a The presence of more than one potential C–H activation site in a molecule often poses an interesting challenge regarding the selectivity of the process. The issue of regioselectivity can be addressed by attuning the hetroatom coordination to direct the metal ion to a site proximal to the selected C–H bond.4
The present work stems from our interest in C–H bond activation by transition metal complexes.5 Herein, C(naphthyl)–H bond activation of a group of napthylazo–2′–hydroxyarenes (H2L, 1) by rhodium has been reported. Wilkinson’s catalyst [Rh(PPh3)3Cl] has been specifically chosen as the metal precursor for its known ability to promote cyclometalation following an oxidative addition pathway,6 thereby accommodating terdentate dianionic diazene substrates (H2L, 1).7 The preferential C(naphthyl)–H bond activation by rhodium has been explored by varying the position of the primary donor (diazene group) attached to the naphthyl group. Furthermore, an additional or auxiliary donor has been incorporated in the substrate molecule to examine its influence on the selectivity of the C(naphthyl)–H bond activation. The isolation, properties and molecular structure of the resulting cyclometallates have been described. The electronic structures of cyclometallates have been calculated using time dependent-density functional theory (TD-DFT).
Results and discussions
C(Naphthyl)–H bond activation by rhodium
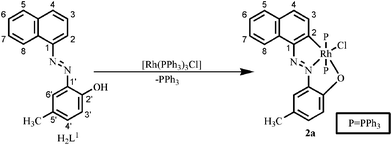
Napthylazo–2′–hydroxyarenes (H2L1–H2L4) with diazene as the directing group offer quasi-equivalent metalation sites. When the primary donor (diazene) is attached to the C1of a naphthyl group, it offers two potentials sites for metallation, at C2 (ortho) and C8 (peri) leading to the formation of two isomeric cyclometallates. On the other hand, C3 and C1 are the probable sites of metalation when the directing diazene group is at C2 position.Driven by the aim to examine the consequences of the substrate modifications on C(naphthyl)–H bond activation, reactions of the naphthylazoarenes (H2L1–H2L4) have been carried out with [Rh(PPh3)3Cl] in refluxing toluene. The reaction proceeded smoothly and afforded the cyclorhodates (2–5) in decent yields. Elemental analysis and 1H NMR spectral data are found to be consistent with the formation of rhodium(III) cyclometallates via metal based two-electron oxidation. In the case of 1–(2′–hydroxy–5′–methylphenylazo) naphthalene (H2L1), the reaction was found to be regiospecific in nature, affording a dark blue cyclometallate 2a as the sole product (Scheme 1). Structure determination of 2a by X-ray crystallography (Fig. 1) revealed that H2L1 is coordinated to rhodium(III) in a dianionic terdentate fashion viaC2(naphthyl), N(diazene) and O(phenolato) donors. The C,N,O–coordinated ligand along with a chloride ion constitute an equatorial plane around the metal center, while the two PPh3 occupy the remaining two axial trans positions. The Rh–C, Rh–N, Rh–O, Rh–P and Rh–Cl distances are in agreement with reported values.7
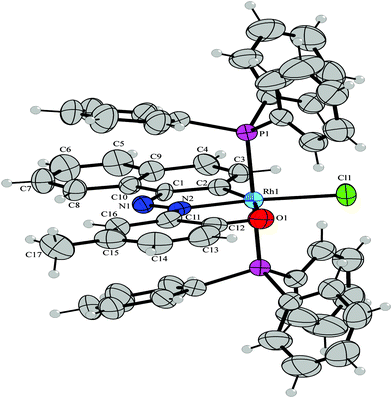 | ||
| Fig. 1 ORTEP diagram of 2a. Selected bond distances (Å): Rh1–C2, 1.904(6); Rh1–N2, 1.984(5); Rh1–O1, 2.288(5); Rh1–Cl1, 2.3673(10); Rh1–P1, 2.3835(6), N1–N2, 1.270(5). Selected angles (°): C2–Rh1–N2, 80.3(2); N2–Rh1–O1, 77.20(19); O1–Rh1–Cl1, 87.22(12); C2–Rh1–P1, 91.50(14); N2–Rh1–P1, 89.52(12); O1–Rh1–P1, 88.56(11); Cl1–Rh1–P1, 90.086(12). | ||
 | ||
| Scheme 1 Formation of the cyclorhodate 2a. | ||
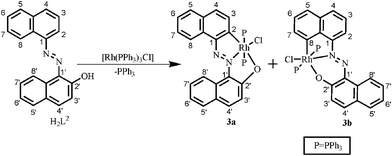
Thus, the complex [Rh(PPh3)3Cl] regiospecifically activates the C2–H bond of the naphthyl group of H2L1 where the primary donor (–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–) is at C1 of the naphthyl ring and the auxiliary donor is the phenolic functional groups. To examine the effect of naphthol as an auxiliary donor on the activation of the C(naphthyl)–H bond by rhodium(I), 1–(2′–hydroxynaphthylazo)naphthalene (H2L2) was chosen as substrate. Interestingly, the reaction of H2L2 with [Rh(PPh3)3Cl] afforded a mixture of blue (3a) and pink (3b) compounds as products (Scheme 2). On the basis of elemental analysis and mass spectral data it is established that compounds (3a & 3b) are isomeric. The molecular structure of the blue cyclometallate (3a) has been shown in Fig. 2. The compound 3a shows a rhodium(III) center bonded to C2 of the naphthyl ring, N2 of the diazene functionality, O1 of the naphtholato fragment of the terdentate donor system and Cl1, along with two mutually trans triphenylphosphines in a distorted octahedral geometry.
N–) is at C1 of the naphthyl ring and the auxiliary donor is the phenolic functional groups. To examine the effect of naphthol as an auxiliary donor on the activation of the C(naphthyl)–H bond by rhodium(I), 1–(2′–hydroxynaphthylazo)naphthalene (H2L2) was chosen as substrate. Interestingly, the reaction of H2L2 with [Rh(PPh3)3Cl] afforded a mixture of blue (3a) and pink (3b) compounds as products (Scheme 2). On the basis of elemental analysis and mass spectral data it is established that compounds (3a & 3b) are isomeric. The molecular structure of the blue cyclometallate (3a) has been shown in Fig. 2. The compound 3a shows a rhodium(III) center bonded to C2 of the naphthyl ring, N2 of the diazene functionality, O1 of the naphtholato fragment of the terdentate donor system and Cl1, along with two mutually trans triphenylphosphines in a distorted octahedral geometry.
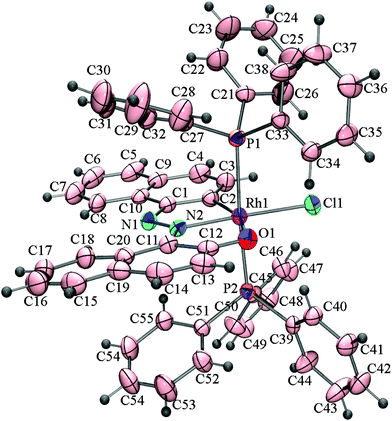 | ||
| Fig. 2 ORTEP diagram of 3a drawn at 50% probability. The solvent molecule has been omitted for clarity. Selected bond distances (Å): Rh1–C2, 2.002(5); Rh1–N2, 1.987(4); Rh1–O1, 2.189(3); Rh1–Cl1, 2.3895(13); Rh1–P1, 2.3719(14), Rh1–P2, 2.3874(13), N1–N2, 1.291(5). Selected angles (°): C2–Rh1–N2, 80.3(2); N2–Rh1–O1, 78.73 (14); N2–Rh1–P1, 92.93(11); C2–Rh1–P1, 89.91(14); O1–Rh1–P1, 88.83(9); N2–Rh1–P2, 91.78(11); C2–Rh1–P2, 90.33(14); O1–Rh1–P2, 92.65(9); C2–Rh1–P1, 89.91(14); P1–Rh1–P2, 175.25(5). | ||
 | ||
| Scheme 2 Formation of the cyclorhodates 3a and 3b. | ||
Single crystals, suitable for X-ray crystallographic analysis of the peri-isomer 3b could not be grown. The presence of the naphthol group as an auxiliary donor, thus, ensures activation of both the C2(naphthyl)–H & C8(naphthyl)–H bonds by rhodium, whereas rhodium regiospecifically activates the C2(naphthyl)–H bond when the phenolic group is an auxiliary donor. The formation of isomeric cyclorhodates, 3a & 3b, can be rationalized considering the well known azo-hydrazo tautomerism displayed by naphthylazonaphthols in solution. The tautomeric behaviour of naphthylazonaphthol is well known8 and it seems to play an important role in the selectivity of C–H bond activation. The azo-enol form of naphthylazonaphthol initially binds the metal center in the complex [Rh(PPh3)3Cl] via oxidative insertion of rhodium into the O–H bond with concomitant dissociation of one triphenylphosphine, generating a reactive intermediate which activates the C2(naphthyl)–H bond only with simultaneous elimination of H2 to afford the cyclometallate 3a. On the other hand, the hydrazo-keto form of H2L2 binds the metal center in the complex [Rh(PPh3)3Cl] via oxidative insertion of rhodium into N–H bond with simultaneous dissociation of one triphenylphosphine and produces a reactive intermediate, which activates the C8(naphthyl)–H bond and undergoes cyclometallationvia elimination of H2 resulting in the cyclometallate 3b with a five-membered metallocarbocycle. The exclusive formation of the orthometallate 2a occurs only in case of H2L1, which exists in azo-enolic form predominantly in solution.
The effect of the directing diazene group at the C2 position on the activation of C(naphthyl)–H bonds by rhodium(I) has also been examined. The reactions of [Rh(PPh3)3Cl] with 2–(2′–hydroxy–5′–methylphenylazo)naphthalene (H2L3) and 2–(2′–hydroxynapthylazo)naphthalene (H2L4) under identical conditions affords mixtures of cyclorhodates, 4a & 4b, and 5a & 5b, respectively. X-ray crystallographic analysis of the cyclorhodates 4a, 4b and 5b have confirmed the regioselective activation of C1(naphthyl)–H and C3(naphthyl)–H bonds in the these complexes. The molecular structures of 4a, 4b and 5b are shown in Fig. 3–5.
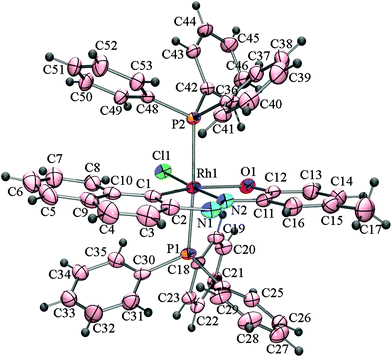 | ||
| Fig. 3 ORTEP diagram of 4a drawn at 50% probability. Selected bond distances (Å): Rh1–C1, 2.040(4); Rh1–N2, 1.938(3); Rh1–Cl1, 2.3755(12); Rh1–O1, 2.184(3); N1–N2, 1.283(3). Selected angles (°): C1–Rh1–N2, 78.82(16); N2–Rh1–O1, 80.19(13); P1–Rh1–Cl1, 87.93(4); O1–Rh1–Cl1, 93.91(8); O1–Rh1–P2, 93.41(7); C1–Rh1–P1, 90.44(10); N2–Rh1–P2, 93.16(9); P1–Rh1–P2, 174.11(4). | ||
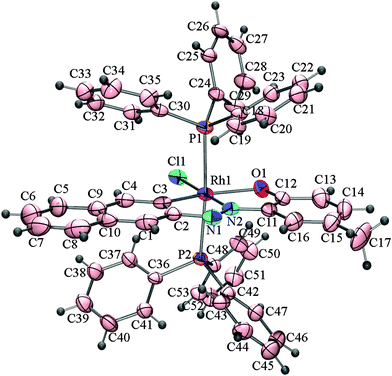 | ||
| Fig. 4 ORTEP diagram of 4b drawn at 50% probability. Selected bond distances (Å): Rh1–C3, 1.960(5); Rh1–N2, 1.961(4); Rh1–Cl1, 2.3824(11); Rh1–O1, 2.196(3); N1–N2, 1.280(4). Selected angles (°): C3–Rh1–N2, 79.9(2); N2–Rh1–O1, 80.51(17); P1–Rh1–Cl1, 88.74(4); O1–Rh1–Cl1, 98.80(9); O1–Rh1–P2, 94.16(8); C3–Rh1–Cl1, 100.76(16); C3–Rh1–P1, 88.94(12); N2–Rh1–P2, 92.24(10); P1–Rh1–P2, 174.81(5). | ||
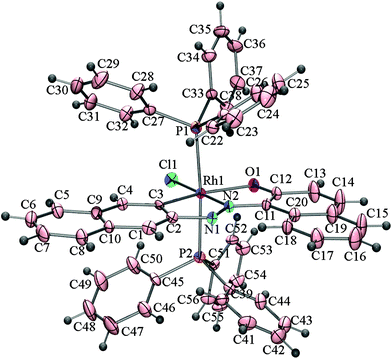 | ||
| Fig. 5 ORTEP diagram of 5b drawn at 50% probability. Selected bond distances (Å): Rh1–C3, 1.993(6); Rh1–N2, 1.968(5); Rh1–Cl1, 2.3731(18); Rh1–O1, 2.161 (5); N1–N2, 1.298(7). Selected angles (°): C3–Rh1–N2, 80.6(3); N2–Rh1–O1, 80.20(2); P1–Rh1–Cl1, 89.51(7); O1–Rh1–Cl1, 97.82(15); O1–Rh1–P2, 92.49(15); C3–Rh1–Cl1, 101.4(2); C3–Rh1–P1, 88.5(2); N2–Rh1–P2, 91.5(17); P1–Rh1–P2, 174.86(7). | ||
The probable steps behind the formation of the cyclometallates 4a & 4b are envisaged as follows. The azo-enol form of H2L3 binds [Rh(PPh3)3Cl] via the oxidative insertion of rhodium into the O–H bond with the concomitant dissociation of one triphenylphosphine, generating a reactive intermediate, which has an equal chance to activate either the C1(naphthyl)–H or the C3(naphthyl)–H bond and undergoes cyclometallationvia elimination of H2 to afford a five membered metallocarbocycle, resulting in the formation of 4a and 4b. Similarly, the azo-enol form of H2L4 follows the same route, resulting in cyclometallates 5a and 5b. The hydrazo-keto form of H2L3 and H2L4 fails to provide any stable five-membered metallocarbocycles.
All the rhodium(III) cyclometallates uniformly display strong bands near 520, 690 and 750 cm−1 which are attributed to vibrations arising from the trans-Rh(PPh3)2 moiety. It is known that the trans-M(PPh3)2 fragments display such vibrations.9 All the rhodium(III) cyclometallates exhibit various non-covalent interactions in the solid state. C–H⋯π, π⋯π and π stacking interactions between the aromatic rings have all been observed. Geometric parameters of all the interactions are compiled in the ESI.† Descriptions of the crystal packing have also been provided in the supporting information.
Electrochemistry
The electron transfer properties of the [Rh(PPh3)2(L)Cl] complexes have been studied in an acetonitrile![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :dichloromethane (9:1) solution with the supporting electrolyte, NBu4ClO4 (0.1M), by cyclic voltammetry. All the complexes show an oxidative response to more positive potentials with respect to the SCE and a reductive response to negative ones. A representative voltammogram is shown in Fig. 6 and the voltammetric data is presented in Table S4, ESI.† Both responses are believed to be centered on the coordinated azo ligand.7 The oxidative response, observed within 0.93 to 1.11 V vs.SCE, is irreversible in nature, as evidenced by the fact that the cathodic peak current (ipc) is less than the anodic peak current (ipa). The one-electron nature of this oxidation has been verified by comparing its current height (ipa) with that of the standard ferrocene–ferrocenium couple under identical experimental conditions. The reductive response, displayed at around −0.9 V vs.SCE, is also irreversible in nature. The cyclic voltammetric studies thus show that these rhodium(III) organometallics are quite stable, while the oxidized and reduced species undergo rapid decomposition.
:dichloromethane (9:1) solution with the supporting electrolyte, NBu4ClO4 (0.1M), by cyclic voltammetry. All the complexes show an oxidative response to more positive potentials with respect to the SCE and a reductive response to negative ones. A representative voltammogram is shown in Fig. 6 and the voltammetric data is presented in Table S4, ESI.† Both responses are believed to be centered on the coordinated azo ligand.7 The oxidative response, observed within 0.93 to 1.11 V vs.SCE, is irreversible in nature, as evidenced by the fact that the cathodic peak current (ipc) is less than the anodic peak current (ipa). The one-electron nature of this oxidation has been verified by comparing its current height (ipa) with that of the standard ferrocene–ferrocenium couple under identical experimental conditions. The reductive response, displayed at around −0.9 V vs.SCE, is also irreversible in nature. The cyclic voltammetric studies thus show that these rhodium(III) organometallics are quite stable, while the oxidized and reduced species undergo rapid decomposition.
![Cyclic voltammogram of [Rh(PPh3)2(L1)Cl], 4a in an acetonitrile : dichloromethane (9 : 1, v/v) solution (0.1M NBu4ClO4) at scan rate 50 mV s−1.](/image/article/2011/RA/c1ra00572c/c1ra00572c-f6.gif) | ||
| Fig. 6
Cyclic voltammogram of [Rh(PPh3)2(L1)Cl], 4a in an acetonitrile:dichloromethane (9:1, v/v) solution (0.1M NBu4ClO4) at scan rate 50 mV s−1. | ||
TD-DFT study
The rhodium(III) cyclometallates (2a–5b) are soluble in common organic solvents. The electronic spectra of the cyclometallates, recorded in dichloromethane show several intense absorptions in the visible and ultraviolet regions. Electronic spectra of the rhodium(III) cyclometallates are shown in Fig. 7. Spectral data are presented in the Experimental Section. The absorption bands observed in the visible and ultraviolet region with a high molar extinction coefficient have been reported as metal–to–ligand charge-transfer transitions (MLCT) and intraligand charge-transfer transitions respectively.7 The absorptions in the ultra-violet region were attributed to the usual n→π* and π→π* transitions occurring within the ligand orbitals.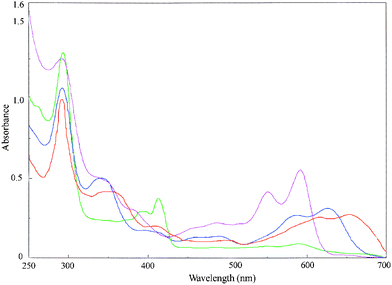 | ||
| Fig. 7 Electronic spectra of the cyclorhodates 4a (orange), 4b (blue), 5a (green) and 5b (pink) in dichloromethane. | ||
The time-dependent DFT (TD-DFT) calculations of the representative rhodium(III) cyclometallates (4a & 4b) has been done to gain further insight into the nature of the absorptions. Only the singlet excited states have been calculated. Excitation energies and oscillator strengths for the various absorption bands have been reported together with the composition of the solution vectors in terms of most relevant transitions (Table 1).
| Code | State | Energy (eV) | λ cal (nm) | λ exp (nm) | Oscillator strength (f) | Composition | Character |
|---|---|---|---|---|---|---|---|
| 4a | S1 | 1.8090 | 686 | 627 | 0.075 | HOMO→LUMO (93.5%) | ILCT+LLCT |
| S14 | 3.0267 | 410 | – | 0.072 | HOMO−5→LUMO (55.5%) | LLCT+MLCT | |
| HOMO→LUMO+7 (12.4%) | |||||||
| HOMO−4→LUMO (12.2%) | |||||||
| S44 | 3.5650 | 348 | 340 | 0.194 | HOMO−5→LUMO+1 (33.8%) | LLCT+LMCT | |
| HOMO−4→LUMO+1 (33.5%) | |||||||
| S59 | 3.8183 | 325 | 292 | 0.068 | HOMO−6→LUMO+1 (51.9%) | LLCT+MLCT | |
| HOMO−3→LUMO+4 (27.5%) | |||||||
| 4b | S1 | 1.8920 | 656 | 668 | 0.027 | HOMO→LUMO (61.7%) | ILCT+LLCT |
| HOMO−2→LUMO (27.1%) | |||||||
| S3 | 2.2337 | 556 | – | 0.088 | HOMO−2→LUMO (61.1%) | LLCT+ILCT | |
| HOMO→LUMO (22.5%) | |||||||
| S5 | 2.5167 | 493 | – | 0.041 | HOMO−3→LUMO (78.7%) | LLCT+MLCT | |
| HOMO−4→LUMO (10.4%) | |||||||
| S12 | 3.0187 | 411 | – | 0.072 | HOMO−5→LUMO (44.2%) | LLCT+ILCT+MLCT | |
| HOMO→LUMO+5 (21.1%) | |||||||
| HOMO−4→LUMO (10.6%) | |||||||
| S49 | 3.6229 | 343 | 340 | 0.125 | HOMO→LUMO+14 (24.8%) | LLCT | |
| HOMO−5→LUMO+1 (23.0%) | |||||||
| HOMO−17→LUMO (20.7%) | |||||||
It is revealed that the HOMO is delocalized over the naphthylazophenolato fragment (Fig. 8 and Tables S5 and S6, ESI,†) for both 4a & 4b. The HOMO−1 has significant contributions from the rhodium d orbitals in 4a & 4b (41.75 and 44.69% respectively). The lowest unoccupied molecular orbitals (LUMO) are almost entirely localized over the ligand moiety. The isomers have been found to differ in the energy of the HOMO, with a lower energy in the case of 4b than that for 4a. The HOMO–LUMO gaps have been computed to be 1.44 and 1.56 eV respectively. The calculated lowest energy transition wavelengths of 4a & 4b are 686 nm and 656 nm respectively. The results for these absorptions agree with those determined experimentally by UV–vis spectroscopy (Table 1). In 4a, the lowest energy transition is HOMO→LUMO (93.5%), which corresponds to an intraligand charge transfer transition (LLCT). In 4b, the main transitions in the lowest energy region are HOMO→LUMO (61.7%) and HOMO−2→LUMO (27.1%). The HOMO–LUMO gap is computed to be 1.44 eV. The most intense transition is observed in 4a in the UV region (ca. 325 nm) and involves excitation from HOMO and HOMO−3 to LUMO+1 and LUMO+3 respectively, with predominantly intraligand π–π* character and a small admixture of MLCT. Similarly, in the case of 4b, the same absorption band appears at 340 nm and involves excitation from HOMO, HOMO−5 and HOMO−17 to LUMO+14, LUMO+1 and LUMO respectively, with predominantly intraligand π–π* character and a small admixture of MLCT.
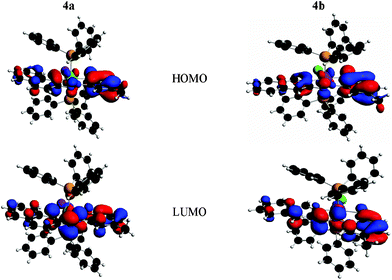 | ||
| Fig. 8 Partial molecular orbital diagram of the cyclorhodates 4a and 4b. | ||
Conclusions
The activation of different C(naphthyl)–H bonds viz. C1–H, C2–H, C3–H and C8–H bonds in naphthylazo–2′–hydroxyarenes (H2L, 1) have been achieved by rhodium(I) following an oxidative addition pathway affording rhodium(III) complexes. The different positions of the directing primary donor (the diazene functional group) and the incorporation of suitable auxiliary donors govern the selectivity of the C–H bond activation. The azo-hydrazo tautomerism of naphthylazo–2′–hydroxyarenes also plays an important role in the observed selectivity. Regiospecific C2(naphthyl)–H activation has been achieved by rhodium(I) when the primary donor (diazene) is at C1 and the auxiliary donor is phenol, whereas the presence of naphthol as the auxiliary donor leads to the regioselective activation of C2(naphthyl)–H & C8(naphthyl)–H bonds. Regioselective activation of C1(naphthyl)–H & C3(naphthyl)–H bonds have been achieved by rhodium(I), where the primary donor (diazene) is at C2 and the auxiliary donor is phenol or naphthol. The origin of the electronic transitions in the cyclorhodates has been investigated by TD-DFT. The calculations corroborate the experimental results. Based on the TD-DFT calculations, the lowest energy transitions in cyclorhodates have been shown to be intra-ligand charge transfer transitions occurring in the terdentate dianionic naphthylazo–2′–hydroxyarenes ligands.Experimental Section
Materials
RhCl3.nH2O was obtained from Arora-Matthey, Kolkata. Complex [Rh(PPh3)3Cl] was prepared following a reported method.10Solvents and chemicals used for the syntheses were of analytical grade. Tetrabutylammonium perchlorate for the electrochemical work was synthesized following a reported method.11Physical measurements
Elemental microanalyses (C, H and N) were performed by either a Perkin-Elmer (Model 240C) or a Heraeus Carlo Erba 1108 elemental analyzer. The IR and Electronic spectra were recorded on a Jasco 5300 FT-IR spectrophotometer and a JASCO V-500 spectrophotometer respectively. NMR spectra were obtained by a Bruker DPX 300 NMR Spectrometer. Electrochemical measurements were performed by a computer controlled PAR model (VERSASTAT II) electrochemical instrument. A platinum disc working electrode, a platinum wire auxiliary electrode and an aqueous saturated calomel reference electrode (SCE) were used in the cyclic voltammetry experiments. All electrochemical experiments were performed under a dinitrogen atmosphere at 298 K in 1:9 dichloromethane/acetonitrile using [nBu4N][ClO4] (TBAP) as the supporting electrolyte. The reported potentials are uncorrected for the junction potential.
Synthesis of the ligands
Compounds (H2L1–H2L4) were synthesized by published procedures.12,5cIsolation of rhodium(III) cyclometallates
:1,v/v) as eluant a bluish green band was isolated and extracted with acetonitrile. Evaporation of the solvent afforded the titled compound. Yield: 55%. Anal. Calc. for C53H42N2OClP2Rh: C, 68.95; H, 4.59; N, 3.03. Found: C, 68.80; H, 4.82; N, 2.92%.1H NMR (CDCl3):δ: 1.76 (s,3H, aryl CH3); aromatic protons: 5.89 (s, 1H), 6.19 (d, 1H, J = 6.6 Hz), 6.45 (dd, 1H, J = 6.6 Hz), 6.87 (d, 1H, J = 6.6 Hz), 7.06 (m, 11H), 7.18 (m, 9H), 7.24 (m, 1H), 7.32 (t, 1H), 7.45 (m, 12H), 8.27 (d, 1H, J = 6 Hz). IR (KBr): ν 1402 cm−1 (–NN–). UV–vis (dichloromethane), λmax/nm (ε/M−1 cm−1): 676 (11,950), 627 (9,600), 290(40,300). MS: m/z: 923 [M]+, 398 [M–2PPh3]+.
:1 v/v) has been used as eluent . A blue fraction and a pink–violet fraction were obtained. The bands were separated and extracted with acetonitrile.
3a. Yield 28%. Anal. Calc. for C56H42N2OClP2Rh: C, 70.12; H, 4.41; N, 2.92. Found: C, 69.79; H, 4.60; N, 2.86%.1H NMR (CDCl3):δ: 6.48 (d,1H), 6.99 (m,7H), 7.26 (m, 12H), 7.45 (m, 9H), 7.67 (m, 10H), 7.95 (d, 1H), 8.10 (d, 1H, J = 6 Hz), 8.21 (d, 1H, J = 6 Hz). IR (KBr): ν 1405 cm−1 (–NN–). UV–vis (dichloromethane), λmax/nm (ε/M−1 cm−1): 289 (63,400), 378 (17,200), 404 (13,600), 604 (19,100), 655 (25,600). MS: m/z 958 [M]+.
3b. Yield 45%. C56H42N2OClP2Rh: C, 70.12; H, 4.41; N, 2.92. Found: C, 70.09; H, 4.56; N, 3.02%. 1H NMR (CDCl3):δ: 6.42 (d, 1H, J = 6.3 Hz), 7.02 (m, 12H), 7.21 (m, 12H), 7.37 (m, 13H), 7.55 (m, 2H), 7.81 (d, 1H, J = 6 Hz), 8.12 (d, 1H, J = 7.8 Hz). IR (KBr): ν 1395 cm−1 (–NN–). UV–vis (dichloromethane), λmax/nm (ε/M−1 cm−1): 278 (80,300), 560 (28,200), 602 (32,400). MS: m/z 958 [M]+, 661[M−(PPh3+Cl)]+.
:1 v/v) afforded a sky blue (4a) fraction and a greenish blue (4b) fraction. The bands were separated and extracted with acetonitrile.
4a. Yield 20%. Anal. Calc. for C53H42N2OClP2Rh: C, 68.95; H, 4.59; N, 3.03. Found: C, 68.76; H, 4.62; N, 3.02%.1H NMR (CDCl3):δ: 1.78 (s,3H, aryl CH3), 5.93 (s,1H), 6.25 (d, 1H, J = 6.3 Hz), 6.56 (dd, 1H), 6.94 (t, 5H) 7.05–7.15 (m, 5H), 7.14–7.21 (m, 15H), 7.39–7.43 (m, 10H), 8.4 (d, 1H, J = 8.1 Hz). IR (KBr): ν 1402 cm−1 (–NN–). UV–vis (dichloromethane), λmax/nm (ε/M−1 cm−1): 292 (47,500), 340 (22,300), 584 (12,000), 627 (9,600). MS: m/z 922 [M]+.
4b. Yield 35%. Anal. Calc. for C53H42N2OClP2Rh: C, 68.95; H, 4.59; N, 3.03. Found: C, 69.10; H, 4.69; N, 2.88%.1H NMR (CDCl3): δ: 1.78 (s,3H, aryl CH3), 5.52 (d, 1H), 6.04 (m, 3H), 6.93–7.03 (m, 15H), 7.28–7.67 (m, 20H). IR (KBr): ν (–NN–)1408 cm−1. UV–vis (dichloromethane), λmax/nm (ε/M−1 cm−1): 292 (15,500), 340 (9,500), 624 (3,600), 668 (5,100). MS: m/z 922 [M]+, 660 [M–PPh3]+, 626 [M–(PPh3+Cl)]+.
:1 v/v) afforded a blue (5a) fraction and a purple (5b) fraction. The bands were separated and extracted with acetonitrile.
5a. Yield: 20%. Anal. Calc. for C56H42N2OClP2Rh: C, 70.12; H, 4.41; N, 2.92. Found: C, 70.31; H, 4.45; N, 3.10%.1H NMR (CDCl3):δ: 6.56 (d, 1H, J = 6.0 Hz), 7.12–7.18 (m, 11H), 7.20–7.39 (m, 15H), 7.40 (m, 5H), 7.45–7.54 (m, 8H), 7.64 (s, 1H), 8.54 (d, 1H, J = 6.6 Hz). IR (KBr): ν 1405 cm−1 (–NN–). UV-vis (dichloromethane), λmax/nm (ε/M−1 cm−1): 261 (59,600), 293 (80,500), 391 (18,500), 413 (23,600), 589 (5,800). MS: m/z 958 [M]+.
5b. Yield: 72%. Anal. Calc. for C56H42N2OClP2Rh: C, 70.12; H, 4.41; N, 3.70. Found: C, 70.31; H, 4.45; N, 3.61%. 1H NMR (CDCl3):δ: 6.42 (d, 1H, J = 6.6 Hz), 7.10 (s, 1H), 7.20–7.31 (m, 15H), 7.34–7.51 (m, 18H), 7.71 (m, 6H, J = 8.1 Hz), 8.62 (d, 1H, J = 6.3 Hz). IR (KBr): ν 1395 cm−1 (–NN–). UV–vis (dichloromethane), λmax/nm (ε/M−1 cm−1): 292 (25,200), 348sh (9,400), 384 (5,400), 550 (8,400), 592 (11,600). MS: m/z 958 [M]+.
Crystallography
Single crystals of complexes 2a and 3a were obtained by the slow diffusion of a dichloromethane solution of the complexes into n-hexane, whereas the single crystals of the cyclometallates 4a,4b and 5b were obtained by slow evaporation from the acetonitrile extract of the compounds. Crystallographic and refinement data for the reported structures† are presented in Table 2. Data on the crystals was collected on a Bruker SMART 1000 CCD area-detector diffractometer using graphite monochromated Mo-Kα (λ = 0.71073 Å) radiation by ω scan. The structure was solved by direct methods using SHELXS-9713 and difference Fourier syntheses and refined with SHELXL97 package incorporated in WinGX 1.64 crystallographic collective package.14 All the hydrogen positions for the compound were initially located in the difference Fourier map, and for the final refinement, the hydrogen atoms were placed geometrically and held in the riding mode. The last cycles of refinement included atomic positions for all the atoms, anisotropic thermal parameters for all non-hydrogen atoms and isotropic thermal parameters for all the hydrogen atoms. Full-matrix-least-squares structure refinement against |F2| molecular geometry calculations were performed with PLATON,15 and molecular graphics were prepared using ORTEP-3.16| Compound reference | 3a2a | 3a. CH2Cl2 | 4a | 4b | 5b |
|---|---|---|---|---|---|
| Chemical formula | C53H42ClN2OP2Rh | C57H44Cl3N2OP2Rh | C53H42ClN2OP2Rh | C53H42ClN2OP2Rh | C56H42ClN2OP2Rh |
| Formula Mass | 923.19 | 1044.14 | 923.19 | 923.19 | 959.27 |
| Crystal system | Monoclinic | Monoclinic | Triclinic | Triclinic | Monoclinic |
| a/Å | 24.4771(3) | 17.5179(2) | 11.116(3) | 11.8442(2) | 11.905(3) |
| b/Å | 9.2446(2) | 16.35860(10) | 11.742(4) | 11.9633(3) | 17.478(4) |
| c/Å | 21.62350(10) | 18.225 | 18.141(6) | 17.3738(4) | 22.043(5) |
| α (°) | 90.00 | 90.00 | 108.857(4) | 72.1650(10) | 90.00 |
| β (°) | 116.3650(10) | 109.9790(10) | 99.704(5) | 70.5110(10) | 95.527(4) |
| γ (°) | 90.00 | 90.00 | 100.613(5) | 79.4750(10) | 90.00 |
| Unit cell volume/Å3 | 4384.03(11) | 4908.42(6) | 2135.2(11) | 2200.31(8) | 4565.2(17) |
| T/K | 293(2) | 298(2) | 293(2) | 293(2) | 293(2) |
| Space group | C2/c | P2(l/c |
P![[l with combining macron]](https://www.rsc.org/images/entities/char_006c_0304.gif)
|
P
|
P21/n |
| No. of formula units per unit cell, Z | 4 | 4 | 2 | 2 | 4 |
| No. of reflections measured | 8710 | 20154 | 20530 | 15152 | 42204 |
| No. of independent reflections | 3148 | 7005 | 7501 | 3911 | 7893 |
| R int | 0.0299 | 0.0596 | 0.0430 | 0.0444 | 0.0666 |
| Final R1 values (I > 2σ(I)) | 0.0277 | 0.0459 | 0.0516 | 0.0301 | 0.0967 |
| Final wR(F2) values (I > 2σ(I)) | 0.0644 | 0.1062 | 0.1077 | 0.0548 | 0.1962 |
| Final R1 values (all data) | 0.0372 | 0.0844 | 0.0712 | 0.0489 | 0.0999 |
| Final wR(F2) values (all data) | 0.0684 | 0.1264 | 0.1151 | 0.0619 | 0.1981 |
| Goodness of fit on F2 | 1.048 | 0.991 | 1.048 | 1.009 | 1.172 |
Method and Computational Details
The calculations have been performed within the TDDFT formalism as implemented in ADF2007.17 Two approximations are generally made: one for the XC potential, and one for the XC kernel, which is the functional derivative of the time-dependent XC potential with respect to density. We used LDA (local density approximation) including the VWN parametrization18 in the SCF step and Becke19 and Perdew–Wang20 gradient corrections to the exchange and correlation respectively and Adiabatic local Density Approximation (ALDA) for the XC kernel, in the post-SCF step. TD-DFT calculations have been performed with the uncontracted triple-STO basis set with a polarization function for all atoms. In the calculation of the optical spectra, 70 lowest spin-allowed singlet–singlet transitions have been taken into account. Transition energies and oscillator strengths have been interpolated by a Gaussian convolution with a σ of 0.2 eV. Solvent effects were modelled by the “Conductor-like Screening Model” (COSMO)21 of solvation as implemented in ADF.Acknowledgements
The financial supports from CSIR and Department of Science & Technology (DST), New Delhi are gratefully acknowledged.References
- (a) R.H. Crabtree, J. Chem. Soc., Dalton Trans., 2001, 2437 Search PubMed; (b) J.A. Labinger and J.E. Bercaw, Nature, 2002, 417, 507 CrossRef CAS; (c) I. Omae, Coord. Chem. Rev., 2004, 248, 995 CrossRef CAS; (d) A.E. Shilov, G.B. Shul’pin, Activation and Catalytic Reactions of Saturated Hydrocarbons in the Presence of Metal Complexes, Kluwer Academic Publishers, Dordretch, Germany, 2000 Search PubMed; (e) W.D. Jones, Acc. Chem. Res., 2003, 36, 140 CrossRef CAS; (f) D. Fiedler, D.H. Leung, R.G. Bergman and K.N. Raymond, Acc. Chem. Res., 2005, 38, 351; (g) M. Lersch and M. Tilset, Chem. Rev., 2005, 105, 2471 CrossRef CAS; (h) E. Carmona, M. Paneque, L.L. Santos and V. Salazar, Coord. Chem. Rev., 2005, 249, 1729 CrossRef CAS; (i) R.H. Crabtree, J. Organomet. Chem., 2004, 689, 4083 CrossRef CAS; (j) R. Giri, B.-F. Shi, K.M. Engle, N. Maugel and J.-Q. Yu, Chem. Soc. Rev., 2009, 38, 3242 RSC.
- (a) M. Albrecht, Chem. Rev., 2010, 110, 576 CrossRef CAS; (b) G. Dyker, Angew. Chem., Int. Ed., 1999, 38, 1699 CrossRef CAS; (c) Y. Guari, S. Sabo-Etienne and B. Chaudret, Eur. J. Inorg. Chem., 1999, 1047 Search PubMed; (d) V. Ritleng, C. Sirlin and M. Pfeffer, Chem. Rev., 2002, 102, 1731 CrossRef CAS; (e) A.D. Ryabov, Chem. Rev., 1990, 90, 403 CrossRef CAS; (f) B.A. Arndtsen, R.G. Bergman, T.A. Mobley and T.H. Peterson, Acc. Chem. Res., 1995, 28, 154 CrossRef.
- (a) J. Dupont, C.S. Consorti and J. Spencer, Chem. Rev., 2005, 105, 2527 CrossRef CAS; (b) M.E. van der Boom and D. Milstein, Chem. Rev., 2003, 103, 1759 CrossRef CAS; (c) M. Albrecht and G. van Koten, Angew. Chem., Int. Ed., 2001, 40, 3750 CrossRef CAS.
- (a) W. Clegg, S.H. Dale, E. Hevia, G.W. Honeyman and R.E. Mulvey, Angew. Chem., Int. Ed., 2006, 45, 2370 CrossRef CAS; (b) W. Clegg, S.H. Dale, R.W. Harrington, E. Hevia, G.W. Honeyman and R.E. Mulvey, Angew. Chem., Int. Ed., 2006, 45, 2370 CrossRef CAS.
- (a) D.N. Neogi, P. Das, A.N. Biswas and P. Bandyopadhyay, Polyhedron, 2006, 25, 2149 CrossRef CAS; (b) D.N. Neogi, A.N. Biswas, P. Das, R. Bhawmick and P. Bandyopadhyay, Inorg. Chim. Acta, 2007, 360, 2181 CrossRef CAS; (c) A.N. Biswas, P. Das, V. Bagchi, A. Choudhury and P. Bandyopadhyay, Eur. J. Inorg. Chem., 2011 DOI:10.1002/ejic.
- B. Rybtchinsky, R. Cohen, Y. Ben-David, J.M.L. Martin and D. Milstein, J. Am. Chem. Soc., 2003, 125, 11041 CrossRef CAS.
- Cyclometallation of structurally related 2-(arylazo)phenols with [Rh(PPh3)3Cl] has been reported earlier. See S. Dutta, S.M. Peng, S. Bhattacharya, J. Chem. Soc., Dalton Trans., 2000, 4623 Search PubMed.
- R.A. Cox and E. Buncel, The chemistry of hydrazo, azo and azoxy groups, ed. S. Patai, Wiley, London, 1975, ch. 18 Search PubMed.
- (a) S. Bhattacharya and C.G. Pierpont, Inorg. Chem., 1991, 30, 1511 CrossRef CAS; (b) S. Bhattacharya and C.G. Pierpont, Inorg. Chem., 1991, 30, 2906 CrossRef CAS; (c) A. Das, F. Basuli, S.-M. Peng and S. Bhattacharya, Polyhedron, 1999, 18, 2729 CrossRef CAS.
- J.A. Osborn and G. Wilkinson, Inorg. Synth., 1967, 10, 67 CAS.
- D.T. Sawyer, J.L. Roberts, Jr., Experimental Electrochemistry for Chemists, Wiley, New York, 1974; pp. 167-215 Search PubMed.
- B.S. Furniss, A.J. Hannaford, V. Rogers, P.W.G. Smith, A.R. Tatchell (ed.) Vogel’s Text Book of Practical Organic Chemistry, fouth ed., ELBS and Longman Group Ltd., London, 1978 Search PubMed.
- G.M. Sheldrick, Acta. Cryst., 2008, A64, 112 CrossRef CAS.
- L.J. Farrugia, WinGX Version 1.64, An Integrated System of Windows Programs for the Solution, Refinement and Analysis of Single-Crystal X-ray Diffraction Data, Department of Chemistry, University of Glasgow, 2003 Search PubMed.
- PLATON: A.L. Spek, J. Appl. Crystallogr., 2003, 36, 7 CrossRef CAS.
- L.J. Farrugia, J. Appl. Crystallogr., 1997, 30, 565 CrossRef CAS.
- The ADF program system was obtained from Scientific Computing and Modeling, Amsterdam (www.scm.com/). For a description of the methods used in ADF, see: G. T. Velde, F. M. Bickelhaupt, E. J. Baerends, C. F. Guerra, S. J. A. Van Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, p. 931 Search PubMed.
- S.H. Vosco, L. Wilk and M. Nusair, Can. J. Phys., 1980, 58, 1200 CrossRef CAS.
- A.D. Becke, Phys. Rev., 1988, A38, 3098.
- J.P. Perdew and Y. Wang, Phys. Rev., 1992, B45, 13244.
- (a) A. Klamt and G.J. Schüürmann, J. Chem. Soc., Perkin Trans. 2, 1993, 799 Search PubMed; (b) A. Klamt and V. Jonas, J. Chem. Phys., 1996, 105, 9972 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Full description of the crystal packing of the cyclorhodates (ESI 1), cyclic-voltammetric data (ESI 2), Energies and percentage composition of the Mos (ESI 3). CCDC reference numbers, 772448, 772237, 772044, 772045 and 772238. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1ra00572c |
| This journal is © The Royal Society of Chemistry 2011 |