An electrochemically formed three-dimensional structure of polypyrrole/graphene nanoplatelets for high-performance supercapacitors†
Peng
Si
a,
Shujiang
Ding
b,
Xiong-Wen (David)
Lou
*a and
Dong-Hwan
Kim
*a
aSchool of Chemical and Biomedical Engineering Nanyang Technological University, 70 Nanyang Drive, 637457, Singapore. E-mail: dhkim@ntu.edu.sg, XWLOU@ntu.edu.sg; Fax: +65 6791 6905; Tel: +65 6790 4111
bDepartment of Applied Chemistry, School of Science, Xi'an Jiaotong University, Xi'an, P. R. China 710049
First published on 21st September 2011
Abstract
A novel nanoplatelet-like structure of the composites of polypyrrole (PPy) and graphene (GR) is facilely synthesized by an electrochemical method and is further employed as a supercapacitor. The nanocomposite of PPy/GR shows a porous structure with a specific surface area of as high as 136.5 m2 g−1. As a result, the composite material exhibits a high specific capacitance of 285 F g−1 at a discharge rate of 0.5 A g−1, and excellent cycling stability. Specifically, over 90% of its initial capacitance can be retained after 1000 charge/discharge cycles. With advantageous features, such as facile fabrication process, high specific capacitance and excellent cycle life, this electrochemically synthesized PPy/GR nanocomposite is quite promising for high-performance supercapacitor applications.
1. Introduction
With the fast depletion of fossil energy sources and increasingly severe pollution conditions, there is a growing worldwide demand for the development of energy conversion and storage devices.1 Supercapacitors have attracted considerable attention in recent years due to their capability to rapidly store and efficiently deliver electric energy,2 which is especially promising for electric vehicles. Compared with batteries, supercapacitors have several advantages such as high power density, long operational life, and low maintenance costs.3Electrical double-layer (EDL) capacitance and pseudocapacitance are two mechanisms contributing to the charge storage in supercapacitors.4 Supercapacitors based on carbon materials, such as activated carbon (AC), mesoporous carbon (MC) and carbon nanotubes (CNTs), often belong to EDL capacitors, which utilize their large surface area to accumulate charges at the electrode/electrolyte interface. In addition to EDL capacitance, metal oxides and conducting polymers (CPs) often contribute to pseudocapacitance, where fast Faradaic reactions are involved during the charging and discharging processes.5 For example, CPs with N-containing groups, such as polypyrrole (PPy) and polyaniline (PANI), can give extremely high pseudocapacitance through the redox reactions between the ions of electrolyte and N-containing species, which act as the electron donor.6 Despite the many advantages, chemically prepared CPs have some major drawbacks, such as poor conductivity in their undoped states and instability during the charge/discharge process,7 that limit their application in high-performance supercapacitors. Carbon materials usually exhibit good stability, but the EDL capacitance is relatively low.8 Although it was reported that micro- or meso- porous carbon could improve the capacitance as a result of the increase of surface area,9 the improvement is actually limited. For instance, only 10–20% of the theoretical capacitance could be achieved in microporous ACs, due to the inaccessibility of the small pores for the electrolyte, wetting deficiencies on the electrode surface and difficulties in forming EDL in the micropores.10 For another example, mesoporous AC with a high surface area of 2000 m2 g−1 only displays slight enhancement in capacitance.11 In addition, the capacitance of CNTs is also relatively low.12 Combining the advantages of CPs and carbon materials, it is therefore highly desirable to develop nanocomposite materials. For example, the nanocomposite based on CNTs and PANI has been shown to exhibit high capacitance and improved stability.13
Recently, graphene has become a ‘rising star’ owing to its intriguing two dimensional structure,14 large specific surface area,15 extraordinarily high electrical and thermal conductivities,16 and superior mechanical strength.17 It is shown that chemically modified graphene sheets possess an exceptionally high surface area up to 2675 m2 g−1, excellent conductivity of 7200 S m−1, tensile modulus up to 35 GPa at room temperature, and intrinsic capacitance of 21 μF cm−2 that is superior to other carbon materials.18–19 If all the surface area can be utilized to form EDL, graphene can in principle deliver a capacitance of as high as 550 F g−1. However, the measured capacitance of graphene is generally in the range of tens to 135 F g−1.20 This is mainly caused by the re-stacking or agglomeration of individual graphene sheets during the process of electrode preparation, which limits the accessibility of electrolytes and the formation of EDL charges on the graphene surface.
To improve the capacitive performance of graphene-based materials, nanocomposites of CP-grafted graphene have been studied quite recently. For example, graphene/PANI composite was prepared by Zhang et al. via the chemical polymerization of aniline with graphene oxide (GO) followed by hydrazine reduction and reprotonation.5 Wu et al. synthesized a multilayered graphene/PANI hybrid material through vacuum filtration of the mixture solution of PANI and chemically converted graphene.21 Very recently, a nanocomposite of graphene and PPy has been fabricated by Boseet al. via a surface modification of graphene followed by in situpolymerization.22 However, currently reported methods to prepare graphene/CP composites mostly involve chemical reduction of graphene and chemical polymerization, which could result in low conductivity of materials and poor adhesion to the electrode surface. In addition, the chemical reducing agents used are hazardous to the environment and human health, and may introduce additional chemical groups, which could increase the resistance of graphene sheets due to electron scattering.23 Therefore, “green” and efficient methods are highly favorable for the synthesis of CP/GR nanocomposites. In the present study, we first report an environment-friendly electrochemical strategy to prepare the PPy/GR nanocomposite facilely by elctropolymerization followed by electrochemical reduction. Briefly, the pyrrole monomers are polymerized onto graphene oxide (GO) sheets forming PPy/GO nanoplatelets on the electrode surface under a constant potential. Afterwards, the GO in the nanocomposite is electrochemically reduced to GR. The as-formed PPy/GR composite has a unique three-dimensioanl (3D) structure and exhibits a high surface area up to 136.5 m2 g−1. In addition, the electrochemically-prepared hybrid material also possesses very high electrical conductivity. Benefiting from these features, the PPy/GR nanocomposite is shown to exhibit significantly improved supercapacitive performance.
2. Results and discussion
2.1 Surface morphology study
Fig. 1(a) shows the microstructure of the PPy film, which is composed of cauliflower-like structures with the dimensions on sub-micrometre scale. Fig. 1(b) and (c) display the SEM images of GO and GR films, respectively. Layered GO film demonstrates wrinkled and crimpled structures, which are possibly caused by the electrostatic repulsive interaction between adjacent GO nanosheets that are negatively charged. In contrast, the GR film appears much smoother. The high quality of as-synthesized graphene has also been verified by UV/vis spectra (see Fig. S1, ESI†) and TEM images showing that GO and GR has the dimension of around 1 μm (Fig. S2, ESI†). Fig. 1(d–f) show the surface morphology of the electrochemically synthesized PPy/GO composite and the PPy/GR composite after electrochemical reduction. The PPy/GO composite consists of platelet-like nanostructures that are randomly organized with a porous structure (Fig. 1(d)). After electrochemical reduction, the morphology of the PPy/GR film is barely altered as shown in Fig. 1(e). Both GO and GR in the composites were observed homogeneously encapsulated in the PPy matrix as shown in the TEM images ( Fig. S3, ESI†). In Fig. 1(f), we observe that the obtained PPy/GR composite plate has a thickness of about 50 nm, which is much thicker than that of the single graphene sheet owing to the uniform coating of PPy. The observation is in accordance with the TEM result. The surface area of the as-obtained PPy/GR composite film is measured to be 136.5 m2 g−1 by nitrogen adsorption/desorption method (Fig. S4, ESI†), which is much higher than that of other samples as compared in Table 1 and the reported values of CPs/GR composites listed in Table 2. | ||
| Fig. 1 SEM images of (a) PPy, (b) GO, (c) GR (d) PPy/GO, (e) PPy/GR and (f) high magnification of (e). Scale bar is 1 μm. | ||
| Samples | BET SSA (m2 g−1) | Conductivity (S m−1) |
|---|---|---|
| PPy | 32.8 | 12.3 ± 0.2 |
| GR | 85.6 | 260.4 ± 1.6 |
| PPy /GO | 133.2 | 82.7 ± 0.5 |
| PPy / GR | 136.5 | 142.6 ± 0.8 |
2.2 Electrochemical reduction
GO in the PPy/GO composite is reduced by repeated scanning cyclic voltammetry (CV) in the negative potential window between −1 to 0 V in N2 saturated 0.5 M Na2SO4 solution at a scan rate of 50 mV s−1. Fig. 2(a) shows the CVs for the electrochemical reduction of pure GO film. A notable reduction peak appears at the potential of −0.84 V in the first scanning cycle, and it disappears completely for the next few cycles of scan, indicating irreversibility of the reduction. A similar reduction peak is observed for the PPy/GO composite (Fig. 2(b)) at −0.78 V, indicating that GO in the composite is effectively reduced by the electrochemical method. The reduction potential of GO is consistent with that in previous reports.24–25 | ||
| Fig. 2 Cyclic voltammograms of GO (A) and PPy/GO (B) in N2 saturated 0.5 M Na2SO4 for the first 10 cycles of scan. Scan rate: 50 mV s−1. | ||
2.3 Characterizations of electronic and chemical structures
The reduction of GO in the PPy/GO composite is characterized by X-ray photoelectron spectroscopy (XPS). Fig. 3(a) and (b) show the de-convoluted C1s core-level spectra of the composite before and after electrochemical reduction, respectively. The Lorentzian peaks observed at the binding energies of 284.7, 285.6, 286.7 and 288.3 eV correspond to the chemical bonds of C–C, C–N, C–O and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, respectively.26–27 The relative peak intensities of C–O and CO are substantially reduced after the electrochemical reduction (Fig. 3(b)), evidently showing that the oxygen functionalities on the surface of GO in the composite material have been effectively removed during the electrochemical reduction. Fig. 3(c) shows the wide region scanning spectra of GO, GR, PPy/GO and PPy/GR. The C/O ratios of GO and GR are determined to be 0.53 and 0.81 respectively. The ratio for PPy/GO is found to be 0.76. After electrochemical reduction, the ratio is increased to 1.19, illustrating the successful reduction of GO in the composite. In addition, the appearance of the N1s peak in the spectra of PPy/GO and PPy/GR at around 400 eV indicates the presence of PPy in the composites.
O, respectively.26–27 The relative peak intensities of C–O and CO are substantially reduced after the electrochemical reduction (Fig. 3(b)), evidently showing that the oxygen functionalities on the surface of GO in the composite material have been effectively removed during the electrochemical reduction. Fig. 3(c) shows the wide region scanning spectra of GO, GR, PPy/GO and PPy/GR. The C/O ratios of GO and GR are determined to be 0.53 and 0.81 respectively. The ratio for PPy/GO is found to be 0.76. After electrochemical reduction, the ratio is increased to 1.19, illustrating the successful reduction of GO in the composite. In addition, the appearance of the N1s peak in the spectra of PPy/GO and PPy/GR at around 400 eV indicates the presence of PPy in the composites.
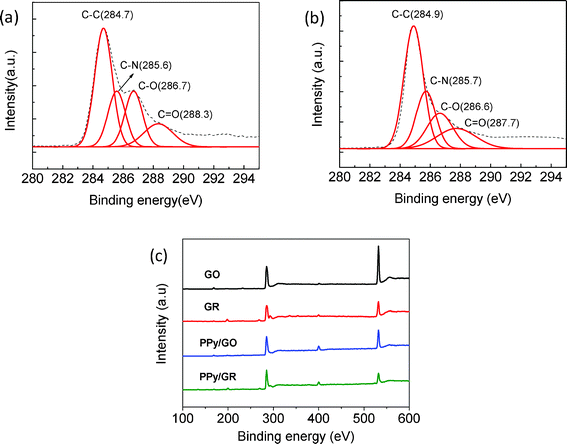 | ||
| Fig. 3 (a) and (b): De-convoluted XPS C1s spectra of the composites of PPy/GO and PPy/GR; (c): wide region scanning XPS spectra of GO, GR, PPy/GO and PPy/GR. | ||
The chemical structures of GO, GR, PPy/GO and PPy/GR is further studied by Raman spectroscopy, which is a sensitive tool to monitor the structural change of carbon-based materials. As shown in Fig. 4, the GO has two bands peaking at 1345 cm−1 (D band) and 1598 cm−1 (G band). The D band indicates the defects or edge planes in the structure while the G band is related to the vibration of sp2 hybridized carbon.28 After chemical reduction of GO, a notable increase of the D/G intensity ratio is observed, reflecting the existence of large amounts of unrepaired defects on the graphene surface due to the removal of oxygen containing groups.29 As for the spectra of PPy/GO and PPy/GR, two additional bands are observed at 934 cm−1 and 1071 cm−1, which correspond to the bipolaron ring deformation and polaron symmetric C–H in-plane bending vibration, respectively.30–31 The appearance of these two bands suggests the presence of PPy in the composite. The D/G intensity ratio for PPy/GO is 0.92, and it is increased to 1.25 after electrochemical reduction, evidently indicating the reduction of GO in the composite.
 | ||
| Fig. 4 Raman spectra of GO, GR, PPy/GO and PPy/GR. | ||
2.4 Thermal behavior and electrical conductivities
The thermal stabilities of GO, PPy, PPy/GO and PPy/GR are investigated by thermo-gravimetric analysis (TGA). As shown in Fig. 5, GO exhibits a complete combustion at around 150 °C, due to the presence of a large amount of oxygen containing groups.32 Meanwhile, PPy exhibits a dynamic mass loss in the temperature range 100–650 °C until complete decomposition at around 700 °C. For the PPy/GO composite, one sharp weight loss of about 20% is observed at around 200 °C, which is likely due to the decomposition of GO. Above 200 °C, PPy/GO exhibits a weight loss trend similar to PPy until complete decomposition at around 600 °C. In contrast, the PPy/GR nanocomposite demonstrates enhanced thermal stability that only starts to decompose around 300 °C. | ||
| Fig. 5 TG curves of GO, PPy, PPy/GO and PPy/GR measured at a heating rate of 10 °C min−1 in air. | ||
The electrical conductivities of PPy, GR, PPy/GO and PPy/GR are measured by a four-probe resistivity system and listed in Table 1. The pure PPy shows a relatively low conductivity of 12.3 S m−1, while as expected GR exhibits a much higher conductivity of 260.4 S m−1, which is similar to the reported value5 and a little lower than that of pristine graphite.20 Surprisingly, in spite of the insulating nature of GO, the PPy/GO nanocomposite shows a high conductivity of 82.7 S m−1, which is approximately six times higher than pure PPy. The enhancement in conductivity could be attributed to the π–π stacking interaction between the aromatic ring plane of GO and the pyrrole backbone in the PPy, which greatly facilitates electron transport. After electrochemical reduction, the conductivity of PPy/GR further increases to 142.6 S m−1, which could contribute to the high conductivity of formed graphene sheets in the composite.
2.5 Electrochemical properties
The electrochemical performances of the materials are evaluated by cyclic voltammetry (CV), galvanostatic charge/discharge technique and electrochemical impedance spectroscopy (EIS) in a three-electrode system. Fig. 6(a) compares the CVs of GR, PPy, PPy/GO and PPy/GR measured in the electrolyte of 0.5 M Na2SO4 with a scan rate of 5 mV s−1. The GR shows a CV curve close to a rectangular shape, revealing good charge propagation and typical EDL capacitive behavior.33 The area of the closed CV loop is however small, implying a relatively low capacitance of the GR due to the agglomeration of GR sheets and the lack of electroactive sites on the surface. On the contrary, PPy shows an oblique and narrow CV loop, reflecting a large interfacial contact resistance with the bulk electrolyte and relatively poor ionic propagation behavior of the CP.4 Interestingly, the PPy/GO nanocomposite exhibits a rectangular CV curve with a closed loop that is much larger than GR, indicating a higher EDL capacitance of the nanocomposite. The high EDL capacitance of PPy/GO largely contributed to the large specific surface area and improved conductivity of the composite film (see Table 1). Upon electrochemical reduction, a further increased area of the rectangular loop with wave-like peaks can be observed, indicating enhanced capacitance for the obtained PPy/GR nanocomposite. This improved capacitive performance could be attributed to the higher conductivity and lower internal resistance of the PPy/GR composite compared to PPy/GO, which may improve the redox activity of PPy and facilitate the charge transport within the electrode.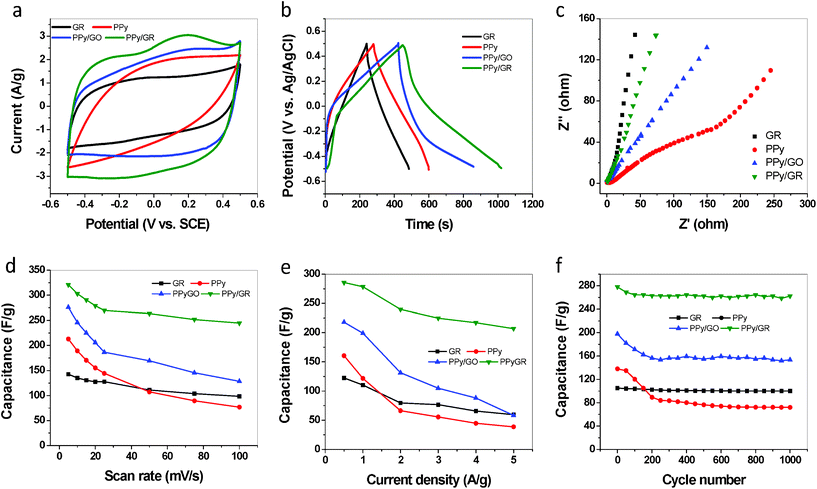 | ||
| Fig. 6 Electrochemical properties of four electrode materials GR, PPy, PPy/GO and PPy/GR: (a) CVs scanned at 5 mV s−1 (b) galvanostatic charge/discharge curves at current density of 0.5 A g−1 (c) Nyquist plots (d) rate performances determined by different scan rates (e) rate performances determined by different current density and (f) cycling performances under the current density of 1 A g−1. | ||
Fig. 6(b) demonstrates the galvanostatic charge/discharge voltage curves for the first cycle at a current density of 0.5 A g−1. The GR and PPy films exhibit large triangular charge/discharge curves, suggesting EDL type of capacitance. The discharging curves of PPy/GO and PPy/GR however display two voltage stages. The first stage ranging from 0.5 to 0.2 V corresponds to a relatively short discharging time and is mainly attributed to the EDL capacitance, whereas the second stage with much longer discharge duration ranging from 0.2 to −0.5 V should be ascribed to the combination of EDL and Faradaic capacitance.34 Although the discharge curves of PPy/GO and PPy/GR are quite similar to each other, discharge time of the PPy/GR is much longer, which extends to 644 s. The PPy/GR demonstrates a high capacitance of 285 F g−1 at this current density, which is superior to previously reported CPs/GR composites as shown in Table 2. In addition, a much larger ‘IR drop’ (overlap of the charge and discharge curve at high potential) is observed for PPy/GO than PPy/GR, suggesting the much higher internal resistance of the former.35 Low internal resistance is desirable for supercapacitor electrodes to reduce the energy loss during the charge/discharge process. Compared with PPy/GO, the electrochemically synthesized PPy/GR nanocomposite is therefore more suitable for the pursuit of high-performance supercapacitors.
Complementary to galvanostatic measurements, EIS is a powerful technique which provides useful information on the electrochemical frequency of the system, and allows for the measurement of redox reaction resistance and equivalent series resistance of the electrodes. The Nyquist plots of GR, PPy, PPy/GO and PPy/GR are shown in Fig. 6(c). The contact resistance (Rc) of electrode materials is determined by the semicircle presented in the lower left portion of the spectra, which corresponds to high frequencies. Large semicircles reflect higher interfacial resistance of the electrode materials with poor charge propagation behavior. While the 45° slopped portion at the low frequency region is the typical Warburg resistance reflecting the ion diffusion behavior of the electrolyte. Smaller Warburg region means lower electronic transport resistance, less obstruction for ion diffusion and better charge propagation behavior. As shown in Figure 6(c), GR exhibits negligible semicircle in high frequency region and a nearly vertical Warburg curve, indicating low contact resistance and good charge-transfer behavior. In contrast, the PPy shows a large semicircle over the low frequency region, indicating high interfacial resistance. The semicircles for PPy/GO and PPy/GR are however not observed, possibly due to the lower impedances of the composites. In addition, PPy/GR exhibits a more vertical Warburg curve than PPy/GO, suggesting better charge propagation behavior and more ideal supercapacitive performance.
Fig. 6(d) depicts the rate performance of the electrode materials measured by the increment of the scan rates in the range 5–100 mV s−1. The capacitance of graphene is relatively low (142 F g−1) at a scan rate of 5 mV s−1. However, 89% and 70% of the capacitance can be retained when the scan rate increased to 20 and 100 mV s−1, respectively. Although the capacitance of PPy is as high as 213 F g−1 at the initial sweep rate of 5 mV s−1, a quick drop in capacitance has been observed with the increase of scan rate. For example, the capacitance of PPy at the scan rate of 50 mV s−1 is only 107 F g−1, which is even smaller than that of GR at the same scan rate. The PPy/GO and PPy/GR also experience quick capacitance drops within the scan rate range of 5–20 mV s−1, but their capacitance at high scan rates is significantly improved compared with PPy. In addition, the PPy/GR exhibits much higher capacitance than PPy/GO at all scan rates. The PPy/GR loses only 12% of the capacitance when the scan rate increases from 20 to 100 mV s−1, whereas the PPy/GO loses 36% of the capacitance in the same range of scan rate. The excellent rate performance of PPy/GR could be understood by considering its porous structure and large surface area that allows easy access for electrolytes, and the high conductivity that improves the charge-propagation behavior. The PPy/GR electrode could therefore maintain a high capacitive performance even at high scanning rates.
The rate performance of the different electrode materials is also measured by the increment of the discharge current densities from 0.5 to 5 A g−1, as shown in Fig. 6(e). The gravimetric specific capacitance (Cg) is calculated using the following equation:
 | (1) |
where I is the discharge current, Δt is the discharge time, ΔV is the potential window, and m is the mass of the active electrode material. The chemically reduced GR has delivered a capacitance of 122 F g−1 at a current density of 0.5 A g−1, while a higher capacitance of 160 F g−1 is obtained for PPy at the same current density. Due to the large surface area of the PPy/GO and PPy/GR, the capacitances of PPy/GO and PPy/GR at the current density of 0.5 A g−1 are 218 and 285 F g−1, respectively, which are much higher than that of PPy and chemically reduced GR. PPy shows a rapid drop in the capacitance with the increment of current density, from 160 F g−1 at 0.5 A g−1 to 38 F g−1 at 5 A g−1, revealing the typical poor rate performance of PPy due to its relatively low conductivity. In contrast, the GR shows superior rate performance, i.e., approximately 50% of the initial capacitance was maintained in the change of current density from 0.5 to 5 A g−1. Similar to PPy, the capacitance of PPy/GO also drops rapidly with the increment of current density. The PPy/GR, however, reveals remarkably improved capacitive responses at high current densities, indicating its excellent rate performance.
The cycling stabilities of the four materials are compared in Fig. 6(f). It is well known that one major drawback of CPs in the application of supercapactiors is the poor cycling stability during the charge/discharge process.21 As demonstrated in Fig. 6(f), the pure PPy film loses 48% of its initial capacitance (from 138 to 72 F g−1) after 1000 charging/discharging cycles at a current density of 1 A g−1. Rapid swelling/shrinkage of the polymer backbone may occur during the charging/discharging process, resulting in the deterioration of the original structure of PPy and lead to the formation of cracks in the electrode film.29PPy/GO also fails to maintain good capacitance retention, with a loss of 17% of the initial capacitance after 200 cycles of charging/discharging. In contrast, the GR is highly stable under the same charge/discharge condition, showing no appreciable capacitance loss observed up to 1000 cycles. As expected, the integration of GR and PPy results in significant enhancement in both the capacitance and the cycling stability. Specifically, the specific capacitance of PPy/GR remains at a high value of above 260 F g−1 after 200 charging/discharging cycles, until the end of 1000 cycles. Over 90% of the initial capacitance has been retained at the end of the cycling. This considerable enhancement could be explained by the twofold role of graphene. Firstly, the graphene nanosheets homogeneously encapsulated in the PPy matrix could serve as the supporting framework that effectively prevents severe structural alterations of the nanocomposite during the cycling process. Simultaneously, the graphene component provides a conduction pathway which favors the charge transport during the charging/discharging cycling.
3. Conclusions
In summary, we have developed a facile electrochemical method to synthesize nanocomposites of PPy and graphene with significantly enhanced supercapacitive performance. The graphene nanosheets are homogeneously encapsulated by PPy forming randomly organized platelet-like nanostructures. The as-obtained PPy/GR nanocomposite exhibits a porous structure with a high surface area up to 136.5 m2 g−1. When evaluated for the potential use in supercapacitors, the PPy/GR nanocomposite manifests a high capacitance of 285 F g−1 at a current density of 0.5 A g−1. Moreover, the PPy/GR nanocomposite exhibits superior rate performance and cycling stability compared with graphene, PPy and PPy/GO. Specifically, the PPy/GR nanocomposite is able to retain above 90% of its initial capacitance (260 F g−1) after 1000 cycles of charging/discharging at a current density of 1 A g−1. The improved supercapacitive performance of the PPy/GR nanocomposite could be explained by the roles of graphene, i.e., enhancing structural stability and electric conductivity. In view of the above advantages, the electrochemically synthesized PPy/GR nanocomposite is a promising candidate for high performance supercapacitors.4. Experimental
Synthesis of GO and GR
GO was synthesized from natural graphite powder by a modified Hummer's method as previously reported.18,36 The as-prepared GO was purified by washing with 10% HCl followed by repeatedly rinsing with copious amount of double-distilled water. The GO powder was obtained by vacuum filtration. Exfoliation was conducted by sonicating the GO aqueous solution (5 mg ml−1) under ambient conditions for 6 h. Then the solution was subjected to centrifugation at 4500 rpm for 10 min to make a stable GO suspension. Reduced graphene (GR) nanosheets were prepared by hydrazine reduction. Briefly, the aqueous solution of GO was first adjusted to a pH of 10–10.5 using ammonia, then hydrazine solution with a weight ratio of 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 to GO was added under vigorous stirring. The GR dispersion was obtained by incubating the mixture in a water bath at 95 °C for 1 h. The GR film was obtained on the pre-polished glassy carbon electrode (GCE) surface by electrophoretic deposition from a 5 mg ml−1graphene aqueous solution at the current density of 5 mA cm−2.
:10 to GO was added under vigorous stirring. The GR dispersion was obtained by incubating the mixture in a water bath at 95 °C for 1 h. The GR film was obtained on the pre-polished glassy carbon electrode (GCE) surface by electrophoretic deposition from a 5 mg ml−1graphene aqueous solution at the current density of 5 mA cm−2.
Synthesis of PPy/GR nanocomposites
Nanocomposites of PPy and GR with the weight ratio of 2.5:1 were prepared from the mixture solution containing 5 mg ml−1 GO and 0.2 M pyrrole monomer. After addition of pyrrole to the GO suspension, the mixture was subjected to vigorous magnetic stirring at room temperature for 10 min and sonication under ambient condition for 5 min to give a homogeneous dispersion of GO and pyrrole. The composite of PPy/GO was directly synthesized on the surface of GCE by electrochemical polymerization using the potentiostatic method. A constant potential of 0.8 V was applied for 900 s to deposit the PPy/GO composite on the electrode. After polymerization, the composite film was carefully rinsed to remove the adsorbed species and dried under a ultrapure nitrogen stream. The GO in the composite was then reduced by scanning CV in a N2 saturated 0.5 M Na2SO4 solution within the potential window of −1–0 V for several cycles until the reduction peak at −0.8 V completely disappeared. The obtained PPy/GR composite film was rinsed again and vacuum dried at room temperature for 1 h before use. Synthesis process of the PPy/GR nanoplatelets is illustrated in Scheme 1.
 | ||
| Scheme 1 Illustration of the electrochemical synthesis process of the PPy/ GR nanoplatelet electrodes. | ||
Material characterizations
The surface morphology of GO, GR, PPy, PPy/GO and PPy/GR was investigated by field emission scanning electron microscopy (FESEM, JEOL JSM-6700F) operating at 5.0 kV. The structure of GO, GR, PPy/GO and PPy/GR was characterized by transmission electron microscopy (TEM, JEOL, JEM-2100F, 200 kV). The electronic and chemical structures of the composite materials were studied by X-ray photoelectron spectroscopy (XPS, ESCALAB MK II spectrometer) and Raman spectroscopy (Renishaw Raman system model 1000 spectrometer), respectively. The surface area of the PPy/GR was measured by Brunauer-Emmett-Teller (BET) plot of nitrogen adsorption–desorption isotherm (Quantachrome Instruments, Autosorb AS-6B). The thermal behavior of the materials was studied by thermogravimetric analysis (TGA) carried out on a TA Instruments 2960 with a heating rate of 10 °C min−1 in air. The electrical conductivity of the composite materials was measured by a conventional four-point-probe with Keithley 2400 electrometer.Electrochemical measurements
The electrochemical properties of the materials were measured by cyclic voltammetry (CV), electrochemical impedance spectroscopy (EIS) and galvanostatic charge–discharge on a CHI 660C workstation (CH Instruments, Inc). A three-electrode cell system, including a modified GCE as the working electrode, platinum wire as the counter electrode and Ag/AgCl as the reference electrode, was employed to conduct the experiment. All the electrochemical measurements were carried out in an electrolyte of 0.5 M Na2SO4 solution at room temperature (approximately 25 °C).Acknowledgements
We gratefully acknowledge the Nanyang Technological University for support of this work. We also thank financial support from the Academic Research Funding Tier 1 (RG65/08) and the National Medical Research Council (NMRC/NIG/00602009) of Singapore.References
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845 CrossRef CAS
.
- J. R. Miller and P. Simon, Science, 2008, 321, 651 CrossRef CAS
- M. Winter and R. J. Brodd, Chem. Rev., 2004, 104, 4245 CrossRef CAS
-
B. Conway, Electrochemical Supercapacitors: scientific fundamentals and technological applications, SpringerUs, 1999 Search PubMed
- K. Zhang, L. L. Zhang, X. S. Zhao and J. Wu, Chem. Mater., 2010, 22, 1392 CrossRef CAS
- D. Hulicova-Jurcakova, M. Kodama, S. Shiraishi, H. Hatori, Z. H. Zhu and G. Q. Lu, Adv. Funct. Mater., 2009, 19, 1800 CrossRef CAS
- J. X. Huang and R. B. Kaner, J. Am. Chem. Soc., 2004, 126, 851 CrossRef CAS
- L. L. Zhang and X. S. Zhao, Chem. Soc. Rev., 2009, 38, 2520 RSC
- J. S. Huang, B. G. Sumpter and V. Meunier, Angew. Chem., Int. Ed., 2008, 47, 520 CrossRef CAS
- C. G. Liu, Z. N. Yu, D. Neff, A. Zhamu and B. Z. Jang, Nano Lett., 2010, 10, 4863 CrossRef CAS
- D. Lozano-Castello, D. Cazorla-Amoros, A. Linares-Solano, S. Shiraishi, H. Kurihara and A. Oya, Carbon, 2003, 41, 1765 CrossRef CAS
- S. W. Lee, B. S. Kim, S. Chen, Y. Shao-Horn and P. T. Hammond, J. Am. Chem. Soc., 2009, 131, 671 CrossRef CAS
- S. R. Sivakkumar, W. J. Kim, J. A. Choi, D. R. MacFarlane, M. Forsyth and D. W. Kim, J. Power Sources, 2007, 171, 1062 CrossRef CAS
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666 CrossRef CAS
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183 CrossRef CAS
- C. Gomez-Navarro, R. T. Weitz, A. M. Bittner, M. Scolari, A. Mews, M. Burghard and K. Kern, Nano Lett., 2007, 7, 3499 CrossRef CAS
- S. Stankovich, D. A. Dikin, G. H. B. Dommett, K. M. Kohlhaas, E. J. Zimney, E. A. Stach, R. D. Piner, S. T. Nguyen and R. S. Ruoff, Nature, 2006, 442, 282 CrossRef CAS
- D. Li, M. B. Muller, S. Gilje, R. B. Kaner and G. G. Wallace, Nat. Nanotechnol., 2008, 3, 101 CrossRef CAS
- J. L. Xia, F. Chen, J. H. Li and N. J. Tao, Nat. Nanotechnol., 2009, 4, 505 CrossRef CAS
- M. D. Stoller, S. J. Park, Y. W. Zhu, J. H. An and R. S. Ruoff, Nano Lett., 2008, 8, 3498 CrossRef CAS
- Q. Wu, Y. X. Xu, Z. Y. Yao, A. R. Liu and G. Q. Shi, ACS Nano, 2010, 4, 1963 CrossRef CAS
- B. Saswata, Nanotechnology, 2011, 22, 295202 CrossRef
- W. Gao, L. B. Alemany, L. J. Ci and P. M. Ajayan, Nat. Chem., 2009, 1, 403 CrossRef CAS
- Y. H. Lin, Y. Y. Shao, J. Wang, M. Engelhard and C. M. Wang, J. Mater. Chem., 2010, 20, 743 RSC
- H. Zhang, Z. J.. Wang, X. Z. Zhou, J. Zhang and F. Boey, J. Phys. Chem. C, 2009, 113, 14071 Search PubMed
- Z. J. Fan, K. Wang, T. Wei, J. Yan, L. P. Song and B. Shao, Carbon, 2010, 48, 1686 CrossRef CAS
- J. Gao, F. Liu, Y. L. Liu, N. Ma, Z. Q. Wang and X. Zhang, Chem. Mater., 2010, 22, 2213 CrossRef CAS
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558 CrossRef CAS
- Y. Zhou, Q. L. Bao, L. A. L. Tang, Y. L. Zhong and K. P. Loh, Chem. Mater., 2009, 21, 2950 CrossRef CAS
- J. Duchet, R. Legras and S. Demoustier-Champagne, Synth. Met., 1998, 98, 113 CrossRef CAS
- Y. Furukawa, S. Tazawa, Y. Fujii and I. Harada, Synth. Met., 1988, 24, 329 CrossRef CAS
- M. J. McAllister, J. L. Li, D. H. Adamson, H. C. Schniepp, A. A. Abdala, J. Liu, M. Herrera-Alonso, D. L. Milius, R. Car, R. K. Prud'homme and I. A. Aksay, Chem. Mater., 2007, 19, 4396 CrossRef CAS
- H. Chen, M. B. Müller, K. J. Gilmore, G. G. Wallace and D. Li, Adv. Mater., 2008, 20, 3557 CrossRef CAS
- L. Li, H. Song, Q. Zhang, J. Yao and X. Chen, J. Power Sources, 2009, 187, 268 CrossRef CAS
- L. Hu, J. Choi, Y. Yang, S. Jeong, F. La Mantia, L. Cui and Y. Cui, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 21490 CrossRef CAS
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS
- S. Biswas and L. T. Drzal, Chem. Mater., 2010, 22, 5667 CrossRef CAS
- D. W. Wang, F. Li, J. P. Zhao, W. C. Ren, Z. G. Chen, J. Tan, Z. S. Wu, I. Gentle, G. Q. Lu and H. M. Cheng, ACS Nano, 2009, 3, 1745 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: See DOI: 10.1039/c1ra00519g |
| This journal is © The Royal Society of Chemistry 2011 |