DOI:
10.1039/C1RA00560J
(Paper)
RSC Adv., 2011,
1, 1310-1317
Silver
cluster-promoted heterogeneous copper catalyst for N-alkylation of amines with alcohols†
Received
5th August 2011
, Accepted 10th August 2011
First published on 27th September 2011
Abstract
A series of metal(M)-loaded Al2O3 catalysts (M/Al2O3) and bimetallic catalysts, CuxAg1−x/Al2O3 with different compositions, Cu0.95Ag0.05/MOx with different supports (MOx), and Cu0.95M′0.05/Al2O3 with different promoter (M′), were prepared by an impregnation method, followed by H2-reduction at 600 °C. For the N-alkylation of amines with alcohols, Al2O3-supported copper–silver bimetallic catalysts with a Cu/Ag molar ratio of 95/5 (Cu0.95Ag0.05/Al2O3) was found to be the most effective heterogeneous catalyst. The alkylation of anilines and aliphatic amines with various alcohols (benzyl and aliphatic alcohols) was achieved with a small amount of the catalyst (1 mol%). Mechanistic studies show that the reaction proceeds through a hydrogen-borrowing mechanism initiated by alcohol dehydrogenation as the rate-limiting step. Structural studies indicate that small Ag nanoclusters are supported on Cu nanoparticles possibly through Ag–O–Cu bonds at the silver–copper boundary. This bimetallic structure can be crucial to an effective promotion of the alcohol dehydrogenation and hydride transfer to the imines.
Introduction
N-Alkyl
amines are important chemicals that find applications as dyes, pharmaceuticals, agrochemicals, surfactants and biologically active compounds. They are typically synthesized by using conventional alkylating agents, such as alkyl halides.1 However, this procedure can be problematic due to over-alkylation and the toxic nature of alkyl halides. Industrial processes applying the reductive amination of carbonyl compounds with heterogeneous catalysts require high temperature and high H2 pressure, and are not always selective for monoalkylation of primary amines.2 A borrowing-hydrogen (or hydrogen-autotransfer) strategy, as an attractive alternative of these methods, employs alcohols as inexpensive and readily available starting materials, which undergo dehydrogenation to provide carbonyl compounds that change to an imine which is reduced to an amine in the presence of complexes of platinum-group metal (PGM), Ru, Rh, Pt, and Ir.3–6 Recently, improved Ru5 and Ir6catalysts with a designed organic ligand were reported to be effective for this one-pot reaction. However, these homogeneous catalysts have problems such as difficulty in the recovery and reuse of expensive catalysts, necessity of special handling of metal complexes, and the indispensable use of co-catalysts such as base and stabilizing ligand. Recyclable heterogeneous precious metal catalysts (Pd7, Ru8, Ag9 and Au10) have also been reported, but some of them suffer from high catalyst loading,8,9 low selectivity to mono-alkylation,7c and need of high pressure (5 atm N2),10 H2 atmosphere,7a high temperature7b or co-catalysts.9 Recent studies have focused on the development of low cost base-metal catalysts which are preferable for industrial application. Homogeneous base-metal catalysts, Cu(OAc)211 and FeBr3,12 were developed, but they need basic co-catalyst (t-BuOK),11 ligand (amino acid),12 or high temperature (160–200 °C).12 Heterogeneous base-metal (Fe13 and Cu14) catalysts generally suffer from a limited scope, high catalyst loading, and need of H2 atmosphere, harsh reaction conditions (high temperatures and pressures), or stoichiometric amounts of basic co-catalyst. Very recently, Heet al.15 showed that supported Cu(OH)x catalysts effectively catalyzed this reaction even in the absence of co-catalyst, though the catalyst suffered from a limited scope; it worked only with activated (aromatic) alcohols. Generally, most of the heterogeneous and/or base-metal catalysts reported were ineffective for less activated systems (e.g. alkyl derivatives).7a,7b,9,11,13,14d,15 From environmental and economic viewpoints, it is desired to accomplish the reaction driven by borrowing-hydrogen mechanism with an inexpensive heterogeneous catalyst. Herein, we present Al2O3-supported metallic Cu catalyst modified with a small amount of Ag (Cu/Ag molar ratio of 95/5) as an inexpensive and versatile heterogeneous catalyst for the N-alkylation of amines with alcohols under N2 without any co-catalysts. First, we show optimization of various catalyst parameters. Then, we show scope of the most efficient catalyst. Next, to provide fundamental information in this system, we show mechanistic and structural features of this new catalytic system.
Results and discussion
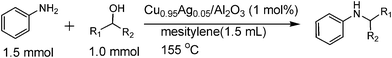
First, the influence of various catalyst parameters on the catalytic activity for N-alkylation of aniline 1 with benzylalcohol 2 was studied under the same reaction condition. We prepared a series of Al2O3-supported transition metal catalysts (M/Al2O3, M = Cu, Ag, Fe, Co, Ni, Ru, Rh, Pd, Ir, Pt, Au) with the same metal loading (1 wt%). Note that the supported catalysts in Table 1 were reduced in H2 at 600 °C and then exposed to air at room temperature. Table 1 shows catalytic results with 1 mol% of the metal catalysts after 0.5 h. Pd and Cu catalysts showed higher yield of the mono-alkylated N-benzylaniline 3 than other M/Al2O3 catalysts. A moderate activity of Pd/Al2O3 catalyst and a relatively high selectivity to the unreduced byproduct, imine 4, are consistent with the previous report by Kwon et al.7a Another disadvantage of Pd/Al2O3 is that toluene 5, produced via a cleavage of the C–O bond, is produced in 36% yield under the present reaction condition. The most effective catalyst was Cu/Al2O3, which gave a 27% yield of the product 3 and a relatively low yield of imine 4. Al2O3 did not show any activity, which excludes a contribution of the support itself as a catalyst. In our previous study,9a we reported that Ag/Al2O3 (4 mol% Ag) showed a good yield for N-alkylation of anilines with benzylalcohols in the presence of the acidic additives (5 mol% FeCl3·6H2O) in the reaction mixture (t = 24 h). However, in the additive-free condition, Ag/Al2O3 (entry 2) was ineffective in the present condition (additive free, 1 mol% Ag, t = 0.5 h). Yus and co-workers reported that a simple Cu salt, copper(II) acetate,11 catalyzed the reaction of 1 and 2 to 3 in the presence of 1 eq of base (t-BuOK). In the base-free condition, copper compounds such as Cu(OAc)2, Cu powder, and Cu2O were totally ineffective (entries 14–16). A common feature of these copper catalysts is that they do not produce 3 but 4. Apparently, the hydrogenation of imine seems to be problematic.
Table 1 Reaction of 1 with 2 by various catalystsa
|

|
| Entry |
Catalyst
|
Conv.(%) |
Yield (%) |
|
|
|
Conversion of 2 and yields of 3, 4 and 5 were determined by GC based on 2.
Al2O3 = 0.1 g
|
| |
|
|
3
|
4
|
5
|
| 1 |
Cu/Al2O3 |
36 |
27 |
6 |
0 |
| 2 |
Ag/Al2O3 |
21 |
1 |
3 |
0 |
| 3 |
Cu0.95Ag0.05/Al2O3 |
100 |
85 |
15 |
0 |
| 4 |
Fe/Al2O3 |
5 |
0 |
2 |
0 |
| 5 |
Co/Al2O3 |
7 |
0 |
2 |
0 |
| 6 |
Ni/Al2O3 |
6 |
0 |
2 |
0 |
| 7 |
Ru/Al2O3 |
7 |
0 |
1 |
0 |
| 8 |
Rh/Al2O3 |
43 |
13 |
12 |
10 |
| 9 |
Pd/Al2O3 |
100 |
28 |
36 |
36 |
| 10 |
Ir/Al2O3 |
46 |
0 |
1 |
0 |
| 11 |
Pt/Al2O3 |
34 |
2 |
10 |
9 |
| 12 |
Au/Al2O3 |
21 |
3 |
5 |
0 |
| 13b |
Al2O3 |
3 |
<1 |
<1 |
0 |
| 14 |
Cu(OAc)2 |
12 |
0 |
11 |
0 |
| 15 |
Cu powder |
18 |
0 |
15 |
0 |
| 16 |
Cu2O
|
27 |
0 |
5 |
0 |
Despite the fact that Cu/Al2O3 and Ag/Al2O3 were nearly inactive for the formation of 3, a complete conversion of aniline and a good yield of the product 3 (85% yield) were achieved, when a small amount of Ag (Cu/Ag molar ratio of 95/5) was added to Cu/Al2O3 (entry 3). To optimize the metal content in Cu0.95Ag0.05/Al2O3, the catalysts with different total metal (Cu + Ag) content (0.1, 0.5, 1, 3, 5, 10, 15, 25 wt%) were tested under the condition described in Table 1. The reaction rate (V0), measured under the condition where the conversion of 1 was below 40%, and the yield after 0.5 h are plotted as a function of the total metal content in Fig. 1. It is found that the metal content of 1 wt% gives the highest activity. A series of the CuxAg100−x/Al2O3 catalysts with different compositions (x = 10, 50, 90, 95, 99) were prepared by the co-impregnation method. It was found that the Cu/Ag molar ratio of 95/5 gave the highest 3 yield for the reaction of 1 and 2 (results not shown). Under the optimized composition (Cu/Ag = 95/5) and loading (1 wt%), the effect of the support oxides (MOx) was tested as shown in Table 2. Basic supports (CeO2, MgO) and acidic supports (SiO2–Al2O3, SiO2) gave a lower conversion of 2 and a lower yield of 3 than Al2O3 as an acid–base bi-functional support.16 This trend is consistent with that observed for the supported Ag cluster catalysts for the same reaction in the presence of co-catalyst (FeCl3·6H2O) reported in our previous study.9a To find the most effective secondary component, a series of Cu0.95M0.05/Al2O3 (M = Fe, Co, Ni, Zn, Ag, Pt, Sn) catalysts with the optimized metal loading (Cu + M = 1 wt%) and Cu/M molar ratio (95/5) were prepared. The catalytic results after 24 h are shown in Table 3. Among various bimetallic catalysts, Cu0.95Ag0.05/Al2O3 (entry 5) gave the highest yield of 3 (94%).
 |
| | Fig. 1 (○) Initial formation rate of 3 and (△) yield of 3 after 0.5 h vs. total metal content in Cu0.95Ag0.05/Al2O3. Conditions are the same as in Table 1. | |
Table 2 Reaction of 1 with 2 by Cu0.95Ag0.05/MOxa
| Entry |
Support (MOx) |
Conv. (%) |
Yield (%) |
|
3
|
4
|
|
t = 0.5 h, Cu + Ag = 1 mol% with respect to 2, 1 = 2 mmol , 2 = 1 mmol, under o-xylene (1.5 mL) reflux.
|
| 1 |
CeO2 |
65 |
23 |
32 |
| 2 |
MgO
|
42 |
0.2 |
27 |
| 3 |
Al2O3 |
100 |
85 |
15 |
| 4 |
SiO2–Al2O3 |
92 |
29 |
56 |
| 5 |
SiO2 |
21 |
0.4 |
17 |
Table 3 Reaction of 1 with 2 by Cu0.95M0.05/Al2O3a
| Entry |
M |
Conv. (%) |
Yield (%) |
|
3
|
4
|
|
t = 24 h, Cu + M = 1 mol% with respect to 2, 1 = 2 mmol , 2 = 1 mmol, under o-xylene (1.5 mL) reflux.
1 = 15 mmol , 2 = 10 mmol, catalyst = 0.1 mol%, T = 155 °C, no solvent.
|
| 1 |
Fe
|
100 |
82 |
18 |
| 2 |
Co
|
100 |
79 |
21 |
| 3 |
Ni
|
100 |
86 |
14 |
| 4 |
Zn
|
100 |
84 |
16 |
| 5 |
Ag
|
100 |
94 |
6 |
| 6b |
Ag
|
68 |
56 |
12 |
| 7 |
Pt
|
100 |
68 |
32 |
| 8 |
Sn
|
10 |
4 |
5 |
Synthetic scope of Cu0.95Ag0.05/Al2O3
From the above screening tests, the bimetallic catalyst, Cu0.95Ag0.05/Al2O3 with a metal loading of 1 wt%, was found to be the best catalyst. The catalyst showed a 94% yield of the N-alkylated amine 3, corresponding to a turnover number (TON) of 94. The yield of byproduct 4 was 6%. No formation of di-alkylated N,N-dibenzylaniline was observed. The reaction was completely terminated by removal of Cu0.95Ag0.05/Al2O3 from the reaction mixture after 44% conversion of 2; further heating of the filtrate under the reflux conditions did not give any increase in the product yield. Moreover, ICP analysis of the filtrate confirmed that the contents of Cu and Ag in the solution were below the detection limit (<0.1 ppm). These results confirm that the reaction is attributed to the heterogeneous catalysis of Cu0.95Ag0.05/Al2O3. In a solvent-free condition with a small amount of the catalyst (0.1 mol%), a moderate 3 yield (56%) was achieved after 24 h, corresponding to a TON of 560 (entry 6 in Table 3).
On the basis of the catalyst optimization results, the scope and limitation of the Cu0.95Ag0.05/Al2O3 catalyzed system with regard to various combination of amines and alcohols were examined. First, the reactions of aniline with various alcohols were conducted (Table 4). In the literature, the use of anilines in alkylation reactions with alcohols has been unsuccessful in some catalytic systems, possibly because anilines are less nucleophilic than aliphatic amines. In this catalytic system, the reactions of aliphatic alcohols (n-octanol and 2-phenylethanol) with aniline resulted in the selective mono-alkylation in high yields (entries 1 and 2). After the reaction of aniline with n-octanol, the catalyst was easily separated from the reaction mixture by centrifugation. The filtered catalyst was washed with water, followed by calcination in air at 600 °C for 10 min, and by reducing in H2 at 600 °C for 10 min. As shown in Table 4 (entry 1), the recovered catalyst was reused at least two times without any indication of catalyst deactivation. The total TON for the three successive reactions reaches 258, which is 5–15 times larger than the previously reported excellent catalytic systems using homogeneous and heterogeneous platinum-group-metal catalysts for the same reaction: TON = 49 for Cp*Ir with basic co-catalyst,6a TON = 18 for Ru(OH)x/Al2O3.8 To the best of our knowledge, our result represents the first successful example of N-alkylation of aniline with aliphatic alcohols with Cu-based catalyst without any co-catalysts in the solution. The reactions of aniline with electron-rich benzylalcohols proceeded in good yield (entries 3–5). The reactions of benzylalcohol with an electron-deficient substituent (entries 6 and 7) were unsuccessful. The secondary alcohols (entries 8–10) were tolerated with moderate yields. The reaction using tertiary alcohol (t-butyl alcohol) was also examined, but it resulted in no reaction.
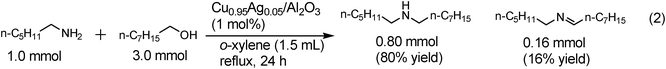
We also examined the N-alkylation of various amines with benzylalcohol. The results are summarized in Table 5. At first, the reactions of substituted anilines, including a sterically hindered one (entry 1), proceeded in good to excellent yields (entries 1–5). Benzylamine and n-octylamine were also tolerated (entries 6 and 7). For the reaction of n-octylamine with benzylalcohol the TON is 88, which is larger than that of the Cu(OH)x/Al2O3 catalyst (TON = 11).15Secondary amines, N-methylaniline (entry 8) and an aliphatic secondary amine (entry 9), were also benzylated in good to excellent yields to give tertiary amines. When the reactions were performed with a large excess of alcohol (alcohol/amine ratio = 3/1), more challenging reactions employing aliphatic amines and aliphatic alcohols as the reactants proceeded using small amounts of Cu0.95Ag0.05/Al2O3 (1 mol%). The reaction of n-hexylamine with 2-octanol gave the mono-alkylated amine in good yield (80%) based on n-hexylamineeqn (1). The reaction of n-hexylamine with n-octanol gave an 80% yield of the mono-alkylated amineeqn (2). These transformations by using heterogeneous catalysts were scarcely described in the literature except for a recent report by Heet al.10 in which Au/TiO2 showed a moderate yield (50%) for the reaction of n-hexylamine with n-decanol at the alcohol/amine ratio of 1/1.
| |  | (1) |
| |  | (2) |
Reaction mechanism and catalyst structure
As previously postulated by the group of Yamaguchi,6a Beller5a–c and Williams,4b,5d,e it is most probable that the present reaction proceeds through the hydrogen-borrowing pathway (dehydrogenation/hydrogenation cycles). As shown in Table 1, Cu/Al2O3 shows a 27 times higher 3 yield than Ag/Al2O3, indicating that Cu has an intrinsically higher activity for the present reaction. Taking into account the high Cu/Ag ratio (95/5) in Cu0.95Ag0.05/Al2O3, it is reasonable to assume that the Cu species act as the primary catalytic sites in our bimetal catalyst, Cu0.95Ag0.05/Al2O3. Zaccheria et al.17 previously reported the hydrogen transfer (Oppenauer-type) oxidation of primary and secondary alcohols to aldehydes or ketones catalyzed by the metallic Cu nanoparticle-loaded Al2O3 catalyst. Thus, the initial stage of the reaction would be the oxidation of alcohols to a carbonyl intermediate accompanied by the transitory generation of a copper hydride. To obtain information concerning the reaction mechanism, we studied kinetic experiments for the N-alkylation of aniline with benzylalcohol by Cu0.95Ag0.05/Al2O3 under the conditions described in Table 1. Fig. 2 shows a time course of the reaction. The reaction was carried out at lower temperature (140 °C) than the typical conditions (144–155 °C). In this condition, a time–conversion profile characteristic to a consecutive reaction was observed. The imine intermediate 4, formed at an initial induction period, was consumed after 3 h. After an induction period (3 h), the yield of the hydrogenated product 3 steeply increased with time. This indicates that 3 is formed through a consecutive pathway via the hydrogenation of 4. We examined the relationship between log(kX/kH) and the Hammett (σ) parameter for the reactions of 2 with benzylalcohols with an electron-donating or an electron-withdrawing substituent (Fig. 3). The order of reactivity for benzylalcohols was p-CH3O > p-CH3 > p-H > p-Cl > p-CF3. There is a fairly good linearity between log(kX/kH) and σ giving a negative slope (ρ = −4.8). This suggests that a transition state of the rate-determining step involves a positive charge at the α-carbon atom adjacent to the phenyl ring. This result suggests that β-hydride elimination of the alcohol is the rate-determining step. In Fig. 4, the reaction rates are plotted as a function of the initial concentration of aniline and benzylalcohol. The reaction rate increased with the aniline concentration up to 0.73 M, which corresponds to the reaction order (n) of 0.74 (Fig. 4A). At higher concentration, a significant negative-order dependence (n = −5.1) was observed. The rate dependence on the benzylalcohol concentration showed a large positive-order dependence (n = 2.2) in a range of 0.09–0.40 M and a negative-order dependence (n = −1.2) at higher concentration (Fig. 4B). These results suggest that benzylalcohol is involved in a rate-limiting step and aniline is not involved in the rate-limiting step. The results also rule out the hydrogenation of imine 4 as the rate determining step. From the above kinetic results we conclude that β-hydride elimination of the alcohol is the rate-determining step. The observed negative-order dependencies at the high concentration region suggest that competitive adsorption of aniline and benzylalcohol on the surface active site inhibits the catalytic reaction.
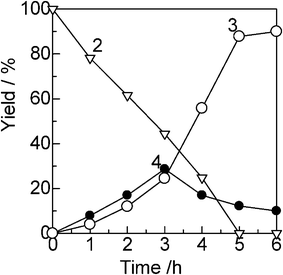 |
| | Fig. 2 Yields of unreacted alcohol 2 (▽), amine 3 (○) and imine 4 (●) by Cu0.95Ag0.05/Al2O3 (1 mol%) at 140 °C in o-xylene (1.5 mL): 1 = 1.5 mmol, 2 = 1.0 mmol. | |
 |
| | Fig. 3 Hammett plot for the reaction of aniline with p-substituted benzyl alcohols by Cu0.95Ag0.05/Al2O3. Reaction conditions are shown in Table 4. | |
 |
| | Fig. 4 Formation rate of 3 (○) as a function of the concentration of (A) aniline (Caniline = 0.09 to 2.0 M) and (B) benzyl alcohol (CBnOH = 0.09 to 2.0 M). | |
In the absence of catalyst, the reaction of benzaldehyde with aniline yielded quantitative amounts of 4 at room temperature (not shown), indicating that the imine formation is very fast. In the presence of Cu0.95Ag0.05/Al2O3 (1 mol%), the transfer hydrogenation of the imine 4 by 3 equivalents of 2-propanol gave 3 in 80% yield at 130 °C eqn (3). Under the same reaction condition, Cu/Al2O3 showed a lower yield of 3 (42%). Combined with the result that Cu0.95Ag0.05/Al2O3 shows a higher conversion of alcohol than Cu/Al2O3 in the reaction of 1 and 2 (Table 1), it is suggested that a small amount of Ag promotes the hydride transfer to the imines as well as the dehydrogenation of alcohols to carbonyl compounds. Based on the above results, a plausible mechanism is given in Fig. 5. The reaction begins with the rate-limiting dehydrogenation of alcohol by metal sites to a carbonyl compound (step 1), which reacts with amine to give an imine (step 2). Finally, hydrogen transfer from the metal-hydride species to the imine gives N-alkylated amine (step 3). In the literature of the transfer hydrogenation of imines with homogeneous catalysts, a cooperation mechanism of coordinatively unsaturated metal centers and adjacent acid/base centers is widely accepted.18 Support-specific catalysis observed in this study (Table 2) may be explained by assuming that the acid–base pair site of Al2O316 located at the metal-support interface plays a role in important steps such as deprotonation of alcohol and hydrogen transfer from metal hydride to the imines.
| |  | (3) |
To investigate the structure of metal species in Cu0.95Ag0.05/Al2O3, spectroscopic characterization experiments were carried out. X-Ray diffraction (XRD) of Cu0.95Ag0.05/Al2O3 showed a very weak Cu (111) diffraction line assignable to metallic copper. The d111 value of Cu0.95Ag0.05/Al2O3 (0.209 nm) was identical to that of Cu/Al2O3 (0.209 nm), which exclude the formation of a Cu–Ag alloy. Average particle size of Cu estimated from the half-width of the Cu (111) line was 43 nm. The XRD results indicate the presence of large Cu metal particles in Cu0.95Ag0.05/Al2O3. To study the structure of the Ag species, X-ray absorption fine structure (XAFS) was used. A high loading catalyst (Cu + Ag = 10 wt%) was adopted as a model sample, because Ag K-edge XAFS analysis of the standard Cu0.95Ag0.05/Al2O3 catalyst with metal loading of 1 wt% was unsuccessful due to a low Ag content. The X-ray absorption near-edge structures (XANES) spectrum of Cu0.95Ag0.05/Al2O3 was nearly identical to that of Ag foil, indicating a metallic state of Ag (see Fig. S1 in the Electronic Supplementary Information†). Fig. 6 shows the Fourier transforms of the extended X-ray absorption fine structure (EXAFS). Structural parameters derived from curve-fitting analysis (Fig. S2†) are listed in Table 6. The EXAFS of Cu0.95Ag0.05/Al2O3 consists of three shells: a weak Ag–O shell at bond distance (R) of 2.39 Å with coordination number (N) of 0.7, a Ag–Ag shell (N = 8.5 at R = 2.83 Å) and a Ag–Cu shell (N = 1.3 at R = 3.13 Å). The smaller Ag–Ag coordination number (8.5) than bulk silver (12) and the shorter Ag–Ag distance (2.83 Å) than bulk silver (2.89 Å) have been typically observed for metallic silver clusters with sizes below a few nanometres in our previous studies.9a,19 The observation of the Ag–Cu shell indicates that the Ag cluster has a boundary with Cu. The Ag–Cu distance (3.13 Å) is longer than that expected for a Cu–Ag alloy. The Ag–O bond distance (2.39 Å) is longer than that of Ag2O (2.04 Å). XPS spectra of Cu0.95Ag0.05/Al2O3 (Cu + Ag = 10 wt%) before and after the reaction are shown in Fig. S3.† The spectral features before and after the reaction are similar to each other. The Ag 3d5/2 peak at 368.3 eV indicates a metallic state of Ag,20 which is consistent with the EXAFS and XANES results. The Cu 2p1/2 peak at 953.1 eV is between those of Cu0 or Cu+ (952.5 eV) and Cu2+ (953.8 eV),20 suggesting that the surface of the Cu metal particle is partially oxidized. A possible structural model which accounts for the XRD and XAFS results is that small Ag nanoclusters are present on the surface of a relatively large Cu metal particle, and there are Ag–O–Cu bonds at the boundary. A strong chemical interaction between Cu and Ag has also been observed by Zhou et al.20 for CuAg/Al2O3 catalyst with Cu/Ag molar ratio of 7/3, which is effective for the selective hydrogenolysis of glycerol. Combined with the result of mechanistic studies, we propose that the unique bimetallic structure can be crucial to an effective promotion of the alcohol dehydrogenation (step 1 in Fig. 5) and the hydride transfer to the imines (step 3).
 |
| | Fig. 6 Fourier transforms of Ag K-edge EXAFS for (a) Ag foil and (b) Cu0.95Ag0.05/Al2O3 (Cu + Ag = 10 wt%). | |
Table 6 Curve-fitting analysis of Ag K-edge EXAFS of Cu0.95Ag0.05/Al2O3(Cu + Ag =10 wt%)
| Sample |
Shell |
N
a
|
R/Åb |
σ/Åc |
R
f (%)d |
|
Coordination number.
Bond distance.
Debye–Waller factor.
Residual factor.
Crystallographic data of Ag metal.
|
| Cu0.95Ag0.05/Al2O3 |
O |
0.7 |
2.39 |
0.024 |
0.9 |
| |
Ag
|
8.5 |
2.83 |
0.071 |
|
| |
Cu
|
1.3 |
3.13 |
0.046 |
|
|
Ag foil |
Ag
|
12e |
2.89e |
— |
— |
Conclusion
We have demonstrated that Al2O3-supported copper–silver bimetallic catalysts with a Cu/Ag molar ratio of 95/5 (Cu0.95Ag0.05/Al2O3) acts as an effective heterogeneous catalyst for the alkylation of anilines and aliphatic amines with various alcohols, including benzyl and aliphatic alcohols. The reaction proceeds through the hydrogen-borrowing mechanism and the dehydrogenation of alcohol is the rate-limiting step. Small Ag nanoclusters are supported on Cu nanoparticles possibly through Ag–O–Cu bonds at the silver–copper boundary. This bimetallic structure can be crucial to an effective promotion of the alcohol dehydrogenation and hydride transfer to the imines.
Experimental
General
The GC (Shimadzu GC-14B) and GCMS (Shimadzu GCMS-QP5000) analyses were carried out with a Rtx-65 capillary column (Shimadzu) using nitrogen as the carrier gas. Commercially available organic and inorganic compounds were used without further purification.
γ-Al2O3 (with surface area of 224 m2 g−1) was prepared by calcination of γ-AlOOH (Catapal B Alumina purchased from Sasol) at 600 °C for 3 h. CeO2 (140 m2 g−1) was purchased from Rhodia Electronics Catalysis. MgO (JRC-MGO-3, 19 m2 g−1) and SiO2–Al2O3 (JRC-SAL-2, Al2O3 = 13.75 wt%, 560 m2 g−1) were supplied from Catalysis Society of Japan. SiO2 (Q-10, 300 m2 g−1) was supplied from Fuji Silysia Chemical Ltd.
CuxAg1−x/Al2O3 (x = 0 to 1) catalysts were prepared by a co-impregnation method. A mixture of γ-Al2O3 and an aqueous solution of Ag(I) and Cu(II) nitrates were evaporated at 80 °C, followed by drying at 120 °C for 12 h, calcination in air at 600 °C for 2 h, and reduction in H2 at 600 °C for 10 min. The catalyst was then exposed to air at room temperature. Cu0.95Ag0.05/Al2O3 (x = 0.95) with total metal (Cu + Ag) loading of 1 wt% was used as a standard catalyst. Cu0.95Ag0.05/MOxcatalysts (Cu + Ag = 1 wt%, MOx = CeO2, MgO, SiO2–Al2O3, SiO2) were prepared by the same procedure. Cu0.95M0.05/Al2O3 (M = Fe, Co, Ni, Zn, Sn, Pt) catalysts with metal (Cu + M) loading of 1 wt% were prepared by the co-impregnation method using Cu(II) nitrate and metal nitrates of Fe(III), Co(II), Ni(II), and Zn(II) or SnCl4·5H2O or Pt(NH3)2(NO3)2. Al2O3-supported metal catalysts, M/Al2O3 (M = Fe, Co, Ni, Ru, Pd, Rh, Pt), with metal loadings of 1 wt% were prepared by the impregnation method using aqueous solution of metal nitrates (for Fe, Co, Ni), RuCl3, or aqueous HNO3 solution of Rh(NO3)3, Pd(NO3)2, or Pt(NH3)2(NO3)2. These catalyst precursors were dried at 120 °C for 12 h, followed by calcination in air at 600 °C for 2 h, and reduction in H2 at 600 °C for 10 min. Au/Al2O3 (Au = 1 wt%) was prepared by a deposition–precipitation method with HAuCl4.9a
Typical procedures of catalytic reactions
Cu0.95Ag0.05/Al2O3 (1 mol% with respect to benzylalcohol) was added to the mixture of o-xylene (1.5 mL), benzylalcohol (1.0 mmol), and aniline (1.5 or 2.0 mmol) in a reaction vessel equipped with a condenser, and filled with N2. The resulting mixture was vigorously stirred under reflux conditions (heating temperature was 155 °C). Conversion and yields of products were determined by GC using n-dodecane as an internal standard. The products were identified by H1NMR as well as by GC-MS equipped with the same column as GC and by comparison with commercially pure products.
Characterization
XRD patterns of the powdered catalysts were recorded with a Rigaku MiniFlex II/AP diffractometer with Cu-Kα radiation. The average particle size of Cu was calculated from the half-width of the Cu (111) peak at 43.20° from the XRD pattern using the Scherrer equation.
Ag K-edge XAFS measurements were performed in transmission mode at the BL01B1 in the SPring-8. The storage ring was operated at 8 GeV. An Si (111) single crystal was used to obtain a monochromatic X-ray beam. Samples were sealed in cells made of polyethylene under ambient atmosphere and XAFS spectra were taken at room temperature. The analysis of EXAFS was performed using the REX version 2.5 program (RIGAKU). The Fourier transformation of the k3-weighted EXAFS oscillation from k space to r space was performed over the range 30–140 nm−1 to obtain a radial distribution function. The inversely Fourier filtered data were analyzed with a usual curve fitting method in the k range of 30–140 nm−1. For the curve-fitting analysis, the empirical phase shift and amplitude functions for the Ag–Ag and Ag–O shells were extracted from the data for Ag foil and Ag2O, respectively. The parameters for the Ag–Cu shell have been provided by the FEFF6.
The X-ray photoelectron spectroscopy (XPS) measurements were carried out using a JEOL JPS-900MC with Al Kα anode operated at 10 mA and 10 kV. The oxygen 1s core electron levels in the support oxides were recorded. Binding energies were calibrated with respect to C 1s at 285.0 eV.
Acknowledgements
This work was supported by the Japanese Ministry of Education, Culture, Sports, Science and Technology via Grant-in-Aids for Scientific Research B (20360361) and for Young Scientists A (22686075). The X-ray absorption experiment was performed with the approval of the Japan Synchrotron Radiation Research Institute (Proposal No. 2010B1447).
References
-
(a) J. F. Hartwig, Synlett, 2006, 1283–1294 CrossRef CAS;
(b) S. L. Buchwald, C. Mauger, G. Mignani and U. Scholz, Adv. Synth. Catal., 2006, 348, 23–39 CrossRef CAS.
-
K. Weisermel and H. J. Arpe, Industrial Organic Chemistry, Wiley Interscience, New York, 2002 Search PubMed.
-
(a) R. Grigg, T. R. B. Mitchell, S. Sutthivaiyakit and N. Tongpenyai, J. Chem. Soc., Chem. Commun., 1981, 611–612 RSC;
(b) Y. Watanabe, Y. Tsuji and Y. Ohsugi, Tetrahedron Lett., 1981, 22, 2667–2670 CrossRef CAS;
(c) Y. Watanabe, Y. Morisaki, T. Kondo and T. Mitsudo, J. Org. Chem., 1996, 61, 4214–4218 CrossRef CAS;
(d) R. A. T. M. Abbenhuis, J. Boersma and G. van Koten, J. Org. Chem., 1998, 63, 4282–4290 CrossRef CAS;
(e) T. Naota, H. Takaya and S.-I. Murahashi, Chem. Rev., 1998, 98, 2599–2660 CrossRef CAS.
-
(a) G. Guillena, D. J. Ramon and M. Yus, Chem. Rev., 2010, 110, 1611–1641 CrossRef CAS;
(b) T. D. Nixon, M. K. Whittlesey and J. M. J. Williams, Dalton Trans., 2009, 753–762 RSC;
(c) M. H. S. A. Hamid, P. A. Slatford and J. M. J. Williams, Adv. Synth. Catal., 2007, 349, 1555–1575 CrossRef CAS;
(d) G. Guillena, D. J. Ramon and M. Yus, Angew. Chem., Int. Ed., 2007, 46, 2358–2364 CrossRef CAS.
-
(a) D. Hollmann, A. Tillack, D. Michalik, R. Jackstell and M. Beller, Chem.–Asian J., 2007, 2, 403–410 CrossRef CAS;
(b) A. Tillack, D. Hollmann, K. Mevius, D. Michalik, S. Bähn and M. Beller, Eur. J. Org. Chem., 2008, 4745–4750 CrossRef CAS;
(c) S. Bähn, A. Tillack, S. Imm, K. Mevius, D. Michalik, D. Hollmann, L. Neubert and M. Beller, ChemSusChem, 2009, 2, 551–557 CrossRef;
(d) M. H. S. A. Hamid, C. L. Allen, G. W. Lamb, A. C. Maxwell, H. C. Maytum, A. J. A. Watson and J. M. J. Williams, J. Am. Chem. Soc., 2009, 131, 1766–1774 CrossRef CAS;
(e) A. J. A. Watson, A. C. Maxwell and J. M. J. Williams, J. Org. Chem., 2011, 76, 2328–2331 CrossRef CAS.
-
(a) K. Fujita, Z. Li, N. Ozeki and R. Yamaguchi, Tetrahedron Lett., 2003, 44, 2687 CrossRef CAS;
(b) K.-I. Fujita, Y. Enoki and R. Yamaguchi, Tetrahedron, 2008, 64, 1943–1954 CrossRef CAS;
(c) A. Prades, R. Corberan, M. Poyatos and E. Peris, Chem.–Eur. J., 2008, 14, 11474–11479 CrossRef CAS;
(d) B. Blank, S. Michlik and R. Kempe, Chem.–Eur. J., 2009, 15, 3790–3799 CrossRef CAS;
(e) N. Andrushko, V. Andrushko, P. Roose, K. Moonen and A. Bcrner, ChemCatChem, 2010, 2, 640–643 Search PubMed;
(f) S. Michlik and R. Kempe, Chem.–Eur. J., 2010, 16, 13193–13198 CrossRef CAS.
-
(a) M. S. Kwon, S. Kim, S. Park, W. Bosco, R. K. Chidrala and J. Park, J. Org. Chem., 2009, 74, 2877–2879 CrossRef CAS;
(b) A. Corma, T. Rόdenas and M. J. Sabater, Chem.–Eur. J., 2010, 16, 254–260 CrossRef CAS;
(c) Y. Zhang, X. Qi, X. Cui, F. Shi and Y. Deng, Tetrahedron Lett., 2011, 52, 1334–1338 CrossRef CAS.
-
(a) J. W. Kim, K. Yamaguchi and N. Mizuno, J. Catal., 2009, 263, 205–208 CrossRef CAS;
(b) K. Yamaguchi, J. He, T. Oishi and N. Mizuno, Chem.–Eur. J., 2010, 16, 7199–7207 CAS.
-
(a) K. Shimizu, M. Nishimura and A. Satsuma, ChemCatChem, 2009, 1, 497–503 Search PubMed;
(b) X. Cui, Y. Zhang, F. Shi and Y. Deng, Chem.–Eur. J., 2011, 17, 1021–1028 CrossRef CAS.
- L. He, X.-B. Lou, J. Ni, Y.-M. Liu, Y. Cao, H.-Y. He and K.-N. Fan, Chem.–Eur. J., 2010, 16, 13965–13969 CrossRef CAS.
- A. Martinez-Asencio, D. J. Ramon and M. Yus, Tetrahedron Lett., 2010, 51, 325–327 CrossRef CAS.
- Y. Zhao, S. W. Foo and S. Saito, Angew. Chem., Int. Ed., 2011, 50, 3006–3009 CrossRef CAS.
-
(a) R. Martinez, D. J. Ramόn and M. Yus, Org. Biomol. Chem., 2009, 7, 2176–2181 RSC;
(b) C. Gonzalez-Arellano, K. Yoshida, R. Luque and P. L. Gai, Green Chem., 2010, 12, 1281–1287 RSC.
-
(a) E. J. Schwoegler and H. J. Adkins, J. Am. Chem. Soc., 1939, 61, 3499–3502 CrossRef CAS;
(b) A. Baiker and W. Richarz, Tetrahedron Lett., 1977, 18, 1937–1938 CrossRef;
(c) J. Runeberg, A. Baiker and J. Kijenski, Appl. Catal., 1985, 17, 309–319 Search PubMed;
(d) P. R. Likhar, R. Arundhathi, M. L. Kantam and P. S. Prathima, Eur. J. Org. Chem., 2009, 5383–5389 CrossRef CAS;
(e) T. Yamakawa, I. Tsuchiya, D. Mitsuzuka and T. Ogawa, Catal. Commun., 2004, 5, 291–295 CrossRef CAS;
(f) H. Kimura and H. Taniguchi, Appl. Catal., A, 2005, 287, 191–196 CrossRef CAS.
- J. He, K. Yamaguchi and N. Mizuno, Chem. Lett., 2010, 39, 1182–1183 CrossRef.
-
(a) C. Morterra and G. Magnacca, Catal. Today, 1996, 27, 497–532 CrossRef CAS;
(b) M. Digne, P. Sautet, P. Raybaud, P. Euzen and H. Toulhoat, J. Catal., 2004, 226, 54–68 CrossRef CAS.
- F. Zaccheria, N. Ravasio, R. Psaro and A. Fusi, Chem.–Eur. J., 2006, 12, 6426–6431 CrossRef CAS.
-
(a) R. Noyori and T. Ohkuma, Angew. Chem., Int. Ed., 2001, 40, 40–73 CrossRef CAS;
(b) R. M. Bullock, Chem.–Eur. J., 2004, 10, 2366–2374 CrossRef CAS.
- K. Shimizu, K. Ohshima and A. Satsuma, Chem.–Eur. J., 2009, 15, 9977–9980 CrossRef CAS.
- J. Zhou, L. Guo, X. Guo, J. Mao and S. Zhang, Green Chem., 2010, 12, 1835–1843 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1ra00560j |
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here.