Electrocatalytic activity of carbon spheres towards NADH oxidation at low overpotential and its applications in biosensors and biofuel cells†
Feng
Gao
*,
Xinying
Guo
,
Jun
Yin
,
Dan
Zhao
,
Maoguo
Li
and
Lun
Wang
Key Laboratory of Chemo/Biosensing, Anhui Key Laboratory of Functional Molecular Solids, College of Chemistry and Materials Science, Anhui Normal University, Wuhu, 241000, P. R. China. E-mail: fgao@mail.ahnu.edu.cn; Fax: +86-553-3869302
First published on 27th September 2011
Abstract
The excellent electrocatalytic activity of a micro-structured carbon material, carbon hollow spheres (CS), to the oxidation of dihydronicotinamide adenine dinucleotide (NADH) is demonstrated here. Compared to conventional bare glassy carbon electrodes, a substantial decrease by 450 mV in the overpotential of NADH electrooxidation was observed using CS coatings, with oxidation starting at ca. −0.10 V (vs.Ag/AgCl, pH 7.0). The CS-coated glassy carbon electrode (CS/GC) thus allows highly sensitive and direct amperometric detection of NADH at lower potential, ranging from 0.20 to 100 μM with a high sensitivity of 7.3 ± 0.2 nA μM−1 (i.e., 103.3 ± 2.8 nA μM−1 cm−2), low detection limit of 0.08 ± 0.03 μM, and minimization of surface fouling. With lactate dehydrogenase (LDH) as a model, a lactate biosensor with the LDH-CS/GC electrode was constructed and the biosensor shows rapid and highly sensitive amperometric response to lactate ranging from 0.5 to 12 μM with a detection limit of 3.7 ± 0.2 μM, a sensitivity of 4.1 ± 0.2 nA μM−1 (i.e., 57.9 ± 2.8 nA μM−1 cm−2), good reproducibility and excellent stability. Furthermore, the promoted direct electron transfer (DET) of bilirubin oxidase (BOD) on CS/GC electrode was investigated, and thus a membrane-less lactate/oxygen biofuel cell was assembled by using LDH-CS/GC as bioanode for lactate oxidation and BOD-CS/GC electrode for oxygen reduction, with a high open-circuit potential of 0.60 V. Such ability of CS to decrease the NADH oxidation overpotential and promote DET of blue-copper oxidases suggests great promise for dehydrogenase-based amperometric biosensors and biofuel cells.
Introduction
The electrochemical oxidation of dihydronicotinamide adenine dinucleotide (NADH) to its corresponding oxidized form, nicotinamide adenine dinucleotide (NAD+), is of particular interest in developing correlative bioelectronic devices including biosensors and biofuel cells, owing to its participation in the enzymatic catalysis reactions of more than 300 dehydrogenases known today as cofactors.1–3 Unfortunately, the direct electrooxidation of NADH at conventional bare electrodes is slow and requires significant overpotential, e.g., 1.1 V at carbon4 or 1.3 V at platinum5electrode although the thermodynamic potential of the NADH/NAD+ couple is as low as −0.54 V (vs.Ag/AgCl at pH 7.0, 25 °C).6 More seriously, the oxidation of NADH at high overpotentials leads to fouling of the electrode surface associated with the accumulation of reaction products, and consequently diminishes the analytical sensitivity, stability, and operational lifetime. To circumvent these issues, initial attempts have been concentrated on the immobilization of redox species such as monomeric or polymeric phenazine, phenothiazine, or phenoxazine dyes,7–13 compounds containing quinone functionalities,14–18 metal complexes,19–21 conducting salts,22,23redox polymers,24–28 and some special compounds29–31 on the electrode surface as electron transfer mediators to catalyze the electrooxidation of NADH. However, most such mediator-modified electrodes still suffer from intrinsic limitations such as limited stability of the mediators and their leaching from the electrodes, which reduce the sensitivity of the prepared electrodes, and therefore limit their applications. Recently, considerable efforts have been devoted to identifying new electrode materials that will reduce the overpotential and minimize surface passivation effects for the direct electrooxidation and unmediated detection of NADH. To the best of our knowledge, only a few new materials, including peptide nanotubes,32 poly(1,2-diaminobenzene) nanotubes,33 and carbon-based materials34–46 have been exploited to promote the direct oxidation of NADH at low overpotential. Among these employed electrode materials, carbon-based materials have attracted more attention. Comparing with metal electrode materials, carbon-based electrode materials are more readily available, cheaper, biocompatible and can be produced in a variety of structures. In most electrolyte solutions, carbon materials are relatively chemically inert and can keep fairly high surface activity in a wide potential window (from −1.0 to +1.0 V vs.SCE) in an aqueous solution. These distinct characteristics essentially endow carbon materials with wide applications in electrochemistry and electroanalytical chemistry.48 Up to now, different forms of carbon materials such as highly boron-doped diamond,34 preanodized screen printed electrodes,35carbon nanotubes (CNT),36–40 pyrolytic graphite,41,42carbon nanofibers,43carbon fibers,44,45 and carbon cloth46 have been exploited to facilitate the direct oxidation of NADH at lower overpotential. For these carbon-based materials, the edge-plane sites/defects and oxygen-rich groups present on the carbon surface could be responsible for NADH oxidation at lower potential.34–48The present work demonstrates the electrocatalytic activity of one new carbon structure, carbon double-shelled hollow spheres (CS), which further decreases the oxidation overpotential of NADH and minimizes the passivation effects, towards the direct oxidation of NADH. This excellent electrocatalytic behavior allows us to fabricate a highly sensitive sensor for direct and unmediated detection of NADH. Combining this electrocatalytic activity with the enzymatic activity of lactate dehydrogenase (LDH) immobilized on CS by a simple casting process, a biosensor for lactate was also proposed (as shown in Scheme 1). Furthermore, using LDH-coated CS film electrode as a bioanode for lactate oxidation, and bilirubin oxidase (BOD)-coated CS film electrode as a biocathode for oxygen reduction, which is based on facilitated direct electron transfer (DET) of BOD on CS film, a direct bioelectrocatalysis-type membrane-less lactate/O2 biofuel cell was assembled (as shown in Scheme 1).
 | ||
| Scheme 1 Illustration of dehydrogenase based mediator-free biosensor and direct bioelectrocatalysis-type biofuel cell based on CS modified electrodes. | ||
Experimental section
Reagents
L-Lactate dehydrogenase (LDH, 1.1.1.27, 891 Umg−1, from rabbit muscle), bilirubin oxidase (BOD, E.C.1.3.3.5, from Myrothecium verrucaria), and sodium L-lactate (98%) were all obtained from Sigma-Aldrich and used as received. NAD+ (disodium hydrate), NADH, bovine serum albumin (BSA) and glutaraldehyde (50%) were purchased from Fluka. All other chemicals were of analytical reagent grade. The phosphate buffer (pH 7.0, 0.1 M) was prepared with KH2PO4 and Na2HPO4, and the desired pH was modulated with NaOH or H3PO4 by a pH meter. All aqueous solutions were prepared with triple distilled and deionized water.Synthesis of carbon double-shelled hollow spheres
The synthesis of carbon double-shelled hollow spheres was synthesized according to previous lreports.49,50 In brief, the synthesis process includes three steps. Firstly, synthesis of sulfonated polystyrene hollow sphere templates: freeze-dried polystyrene hollow spheres were immersed in concentrated sulfuric acid at varied temperature and for varied time controlling the sulfonation extent. Sulfonation at 40 °C for 1 h results in the sulfonated hollow sphere templates denoted as S1. The sulfonated spheres were thoroughly rinsed with water and ethanol. Secondly, synthesis of polymer phenolic formaldehyde (PF) composite hollow spheres: the solution of PF resole resin with solid content 60 wt.% was prepared by alkaline Mg(OH)2 catalytic reaction of phenol/formaldehyde (molar ratio = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.3). Freeze-dried sulfonated polystyrene hollow sphere template S1 (0.1 g) was dispersed in 5 ml ethanol at ambient temperature. A designed amount of PF resin solution was then dropped into the dispersion under stirring within 10 min following by a further reaction for 4 h. The weight ratio of phenolic resin and sulfonated polystyrene hollow spheres is chosen as 4:1. PF composite hollow spheres were prepared after centrifugation followed by further cross-linking at 150 °C for 2 h. Finally, synthesis of carbon double-shelled hollow spheres: carbon double-shelled hollow spheres were synthesized by calcination of the phenolic composite hollow spheres at 800 °C for 2 h under a nitrogen atmosphere.
:1.3). Freeze-dried sulfonated polystyrene hollow sphere template S1 (0.1 g) was dispersed in 5 ml ethanol at ambient temperature. A designed amount of PF resin solution was then dropped into the dispersion under stirring within 10 min following by a further reaction for 4 h. The weight ratio of phenolic resin and sulfonated polystyrene hollow spheres is chosen as 4:1. PF composite hollow spheres were prepared after centrifugation followed by further cross-linking at 150 °C for 2 h. Finally, synthesis of carbon double-shelled hollow spheres: carbon double-shelled hollow spheres were synthesized by calcination of the phenolic composite hollow spheres at 800 °C for 2 h under a nitrogen atmosphere.
Preparation of CS film electrodes and enzyme-immobilized electrodes
Prior to surface modification, the glassy carbon disk electrodes (GC, 3 mm diameter, Bioanalytical System, Inc.) were first polished with 0.3 and 0.05 μm alumina slurry on a polishing cloth, and then sonicated in acetone and distilled water each for 5 min. The as-synthesized CS was dispersed into N,N-dimethylformamide (DMF) to give a homogeneous suspension (10 mg mL−1) under sonication. A 4 μL of homogeneous suspension was cast onto the surface of the GC electrode and allowed to dry under a lamp to evaporate the solvent, thus a CS-modified GC electrode (referred to as a CS/GC electrode) was obtained. To immobilize enzyme on the CS-modified film electrode, an 8 μL of a mixed aqueous solution which contained LDH (30 mg mL−1) and BSA (1%) with a volume ratio of 2:1 was coated on the CS/GC electrode. This enzyme coated electrode was then cross-linked with 2 μL of glutaraldehyde (40 mM), and dried in the fridge (4 °C). The resulting electrode (LDH-CS/GC) was rinsed with distilled water before use. The BOD modified electrode (BOD-CS/GC) was prepared in the same way as described above.
Apparatus for the characterization of CS
A transmission electron microscope (TEM, JEOL 100CX operating at 100 kV) and a scanning electron microscope (SEM, HITACHI S-4800 operating at 5 kV) were used to characterize the morphology of the spheres. A very dilute dispersion of the spheres in ethanol was dispersed onto carbon-coated copper grids for TEM observation. The ambient dried spheres were vacuum sputtered for SEM observation. FT-IR spectra were recorded on an IRPrestige-21(Shimadzu, Japan) using KBr pellet samples. Raman spectra were performed on a PE 2000 FT-Ramam spectrometer. XPS spectra were carried out using an EscaLab 220-IXL spectrometer. The survey spectra for C1s and O1s were collected and the software provided with the instrument was used to deconvolute the constituent peaks under C1s and O1s peaks and to integrate peak areas.Electrochemical measurements
A CHI 660C electrochemical potentiostat (CHI, China) was used to collect electrochemical data. A conventional three-electrode system was used with a GC, CS-GC, LDH-CS/GC, or BOD-CS/GC electrode as the working electrode, a platinum spiral wire as the counter electrode, and a KCl-saturated Ag/AgCl electrode as the reference electrode. All the experiments were performed at room temperature with 0.1 M pH 7.0 phosphate buffer as background electrolyte. The experiments were repeated a minimum of three times and the means of measurements are presented with relative standard deviations.Results and discussion
Physicochemical properties of carbon double-shelled hollow spheres
The synthesis of carbon double-shelled hollow spheres was synthesized according to previous reports and the details are shown in ESI.† The SEM and TEM images of the synthesized carbon hollow spheres are shown in Fig. 1 A and B, respectively. The carbon hollow spheres with porous shells are about 480 nm in diameter, with the hollow cavity and gap retained between the two shells about 30 nm. BET specific surface area was 194 m2 g−1, and the total pore volume was 0.36 cm3 g−1. The pores possibly originated from the release of small molecules and decomposition of the sulfonated polystyrene gel during carbonation. Elemental analysis shows that the carbon spheres contain 94.0 wt.% C, 1.0 wt.% H, 2.7 wt.% O and 1.0 wt.% S, suggesting that oxygen-containing functional groups are present on the CS surface. XRD results and selected area electron diffraction (data not shown) indicated that the carbon was glassy, which had an appreciable electron conductivity of 1.5×10−2 S cm−1. Glassy carbon of the carbon spheres was confirmed by two distinct bands at 1285 cm−1 (referred to as the D-band) and 1593 cm−1 (referred to as the G-band) in the Raman spectra (see ESI Fig. S1).The spectra are composed of two characteristic bands at 1285 cm−1 and 1593 cm−1. The origin of the D-band is attributable to the presence of surface defects such as the distributed edge planes, finite-size effects which break the translational symmetry,48,51–53 while the G-band comes from the first-order scattering of E2g symmetry.48,51–53 | ||
| Fig. 1 Typical SEM (A) and TEM (B) images of carbon double-shelled hollow spheres (CS). Inset (upper) is a single carbon sphere. | ||
Fig. S2 (see ESI) presents the sequence of transmission IR spectra of carbon sphere dispersed in KBr. The band at 3432 cm−1 is attributed to the hydroxyl (O–H) stretching vibration of the C–OH group,54 the band at 1625 cm−1 is assigned to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibrations of the carbonyl and carboxyl group, while the band at 1172 cm−1 is assigned to C–O stretching vibrations.55
O stretching vibrations of the carbonyl and carboxyl group, while the band at 1172 cm−1 is assigned to C–O stretching vibrations.55
XPS spectra of the carbon hollow sphere samples are shown in Fig. S3 (see ESI). The deconvolution of the C1s peak in the XPS spectrum shows the presence of three types of carbon bond: C–C (283.6 eV), C–O (284.5 eV) and CO (287.8 eV). The O1s spectrum shows two components at 531.5 and 533.8 eV, corresponding to CO and C–O in the OC–O functional group, respectively.55–57
Based on the surface characterization of CS, we could conclude that different oxygen-containing functional groups such as phenolic, carbonyl, and carboxyl are present on the CS surface. By deconvolving the constituent peaks under C1s and O1s peaks and integrating the peak areas, the O/C ratio was calculated to be 1.20%.
Electrochemical characterization of carbon hollow spheres
Stable voltammetric background response at CS/GC electrode was observed from −0.2 to 0.6 V in aqueous media, as shown in curve a (black line) of Fig. S4 (ESI). The cyclic voltammograms of bare GC electrode and CS-coated GC electrodes in phosphate buffer of pH 7.0 containing 0.1 M KCl show that the double-layer capacitance is higher in the case of CS-modified GC electrode (2.1 mF cm−2) than that of uncoated GC electrode (12.7 μF cm−2). The background charging current is of the order of 420 μA cm−2 for CS/GC electrode and 2.31 μA cm−2 for the uncoated GC electrode. Capacitances were calculated by summing the charge current in the positive and negative scan directions and then dividing the sum by twice the scan rate.58 Geometric area rather than the electrochemical active area is used for the determination of double layer capacitance and charging current density. The increased capacitance of CS-coated electrode is due to the high specific electrochemical active area of CS film. The charging currents remain constant during potential cycling and the film is found to be stable on the surface after continuous scanning. Furthermore, the voltammetric response in 0.1 M KCl is featureless in the working potential window (from −0.2 to 0.6 V), indicating that the CS film–electrolyte interface is almost ideally polarizable. The CS-coated GC surface is also characterized using the benchmark redox probe of Fe(CN)63−. Fig. S5 (see ESI) shows the cyclic voltammograms obtained at CS-coated GC electrode in 0.1 M phosphate buffer solution (pH 7.0) containing 5 mM Fe(CN)63− at different scan rates from 10 to 500 mV s−1. As shown in Fig. S5, well-defined redox waves are seen with ΔEp values of 61, and Ipox/Ipred values of 1.0. This indicates that the CS films exhibit reversible electron transfer kinetics for inorganic redox analytes of Fe(CN)63−/4−. In other words, CS thin films show good electrochemical activity without any pretreatment. From the linear dependence of peak current against the square root of scan rate (inset of Fig. S5), we can see that the Ipox and Ipred values vary linearly with the square root of scan rate in the range of 10–500 mV s−1. These results indicate that the current is limited by semi-infinite linear diffusion of the reactant to the interfacial reaction zone and that the reaction does not involve any surface-confined species. In addition, the same results were obtained after the CS film was exposed to laboratory air for several hours prior to voltammetric measurements. This strong tendency to resist deactivation is another attribute of the CS film.Electrocatalytic activity of CS/GC electrode to NADH oxidation and unmediated detection of NADH
The fast, reliable, and unmediated detection of NADH at low potential is of great importance in fabricating amperometric biosensors and biofuel cells based on dehydrogenases such as glucose dehydrogenase, alcohol dehydrogenase, and lactate dehydrogenase. The cyclic voltammetric responses at bare and CS/GC electrodes obtained in pH 7.0 phosphate buffer in the absence and presence of NADH at a scan rate of 20 mV s−1 are presented in Fig. 2. As shown in Fig. 2A, in the absence of NADH, no visible voltammetric response was observed (dotted line a). Upon the addition of NADH of 0.5 mM, an oxidation peak at 0.15 V could be observed (solid line b). This behavior is consistent with a strong electrocatalytic effect of carbon spheres to NADH oxidation. The CVs obtained at CS-coated GC electrode in 0.1 M phosphate buffer (pH 7.0) containing 0.5 mM NADH at different scan rates are shown in Fig. 2B. The plot of peak current vs. square root of the scan rate is given as an inset in Fig. 2B. As shown in Fig. 2B, the peak currents increase linearly with the square roots of the scan rates, indicating a diffusion-controlled redox process. However, the peak potential for NADH oxidation at the bare GC electrode was observed at 0.60 V (Fig. 2C, solid line b), which has a good agreement with the value reported previously.34,36,37,43 These comparative results indicate that the presence of CS resulted in a substantial negative shift of the potential to about 450 mV for NADH oxidation. The peak potential for NADH electrooxidation is much lower than those of other electrode materials such as peptide nanotubes,32carbon nanotubes,36–38,40carbon fibers,44boron-doped diamond,34 and pyrolytic graphite electrodes41,42 employed for direct (unmediated) oxidation of NADH in literature, as shown in Table 1. It also could be seen from Fig. 2 that the onset potential for NADH oxidation at CS/GC electrode is at ca. −0.10 V, which is 500 mV more negative than that at bare GC electrode (ca. 0.40 V). Such catalytic activity to NADH oxidation with considerably lower potential at CS/GC electrode endows the CS with potential applications in the fabrication of dehydrogenase-based biosensors and bioanode for biofuel cells. The electrocatalytic behavior of CS film to the oxidation of NADH could partially result from the oxygen-rich groups such as phenolic hydroxides, quinones, and edge plane defects present on the CS surface.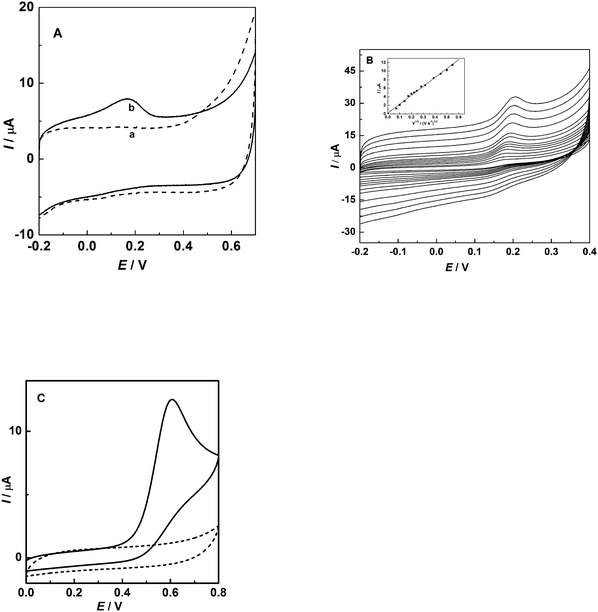 | ||
| Fig. 2 CVs of NADH at CS-coated (A) and bare (C) GC electrode in 0.1 M phosphate buffer (pH 7.0) in the presence (solid line) or absence (dotted line) of 0.5 mM of NADH with a scan rate of 20 mV s−1, and (B) CVs obtained at CS-coated GC electrode in 0.1 M phosphate buffer (pH 7.0) containing 0.5 mM NADH at different scan rates (from inside to outside: 5, 10, 20, 30, 40, 50, 60, 80, 100, 150, 200, 250, 300 mV s−1). The linear dependence of peak current on the square root of scan rate is shown in the inset. | ||
| Electrode materials | Oxidation potential (V) | Linear range (μM) | Detection limit (μM) | Sensitivity (nA μM−1) | Response time (s) | Ref. |
|---|---|---|---|---|---|---|
| MWNT, Multi-wall carbon nanotube; SWNT, single-wall carbon nanotube. The potentials refer to Ag/AgCl reference electrode. | ||||||
| Peptide nanotube/Au | 0.54 | 50–500 | — | — | — | 32 |
| Poly(1,2-diaminobenzene) nanotubule/GC electrode | 0.63 | 50–1000 | 50 | 0.099 | 15 | 33 |
| Highly boron-doped diamond electrode | 0.60 | 0.010–0.5 | 0.01 | — | — | 34 |
| Preanodized screen printed carbon electrode | 0.40 | 10–50 | 0.157 | 91.5 | — | 35 |
| MWNT/GC electrode | 0.33 | 200–5000 | — | — | 8 | 36 |
| SWNT/GC electrode | 0.36 | — | — | — | 36 | |
| MWNT-chitoson/GC | 0.34 | 5–104 | 3 | 9.19 ± 0.42 | <5 | 37 |
| Boron-doped carbon nanotube | 0.32 | 0–1405 | 0.05 | 91.5 | <5 | 38 |
| Treated CNT electrode | −0.04 ± 0.01 | 10–100 | 2.0 | 0.848 | 50 | 39 |
| MWNT-Teflon composite | 0.40 | 200–1000 | 4.375 | — | 8–10 | 40 |
| Basal plane pyrolytic graphite electrode | 0.36 | — | — | — | — | 41 |
| Edge plane pyrolytic graphite electrode | 0.42 | 2.25–36 | 0.3 | — | — | 42 |
| Carbon nanofiber | 0.08 | 0.2–686 | 0.11 | 5.6 | — | 43 |
| Carbon fiber | — | — | 7 | — | — | 44 |
| Active carbon cloth | 0.12 | 20–400 | 2 | 18 | — | 46 |
| Carbon spheres | 0.15 | 0.2–100 | 0.08 ± 0.03 | 7.3 ± 0.2 | — | This work |
Fig. 3 records the amperometric traces of the CS/GC electrode at +0.15 V to the successive addition of NADH into stirring pH 7.0 phosphate buffer. The electrode modified with CS displays a distinct NADH signal in the form of current steps after each addition of NADH to the solution and the anodic current increases rapidly and reaches a steady state within 8 s. At lower concentration of NADH, the amplified amperometric response is shown in the upper inset of Fig. 3. The relationship between current and concentration of NADH is illustrated in the lower inset of Fig. 3. The response displays a linear range from 0.20 to 100 μM with a correlation coefficient of 0.991 and a slope of 7.3 ± 0.2 nA μM−1 (i.e., 103.3 ± 2.8 nA μM−1 cm−2, using calculated geometric area as reported in literature33,37,39). The linear response range was wider than that of 10–100 μM reported recently with a electrode modified with treated carbon nanotubes at −0.05 V,39 10–50 μM with preanodized screen printed carbon electrode at 0.40 V,35 and 2.25–36 μM with edge plane pyrolytic graphite electrode at 0.40 V.42 The sensitivity is also higher than that at most of the electrode materials employed for direct detection of NADH, including poly(1,2-DAB) nanotubes,33 MWNT-Teflon composite,40boron-doped CNTs,38 and carbon nanofibers,44 as seen in Table 1.
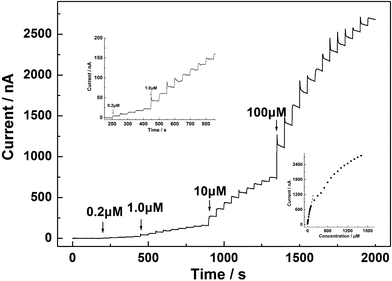 | ||
| Fig. 3 Amperometric response of CS/GC electrode to successive addition of different concentrations of NADH in 0.1 M phosphate buffer (pH 7.0) saturated with N2, at an applied potential of +0.15 V. Upper inset, amplified response curve for lower NADH concentration. Lower inset, plot of chronoamperometric current vs.NADH concentration. | ||
The operational stability of the CS/GC electrode was examined by successive assays of 50 μM NADH using the same CS/GC electrode poised at 0.15 V. The electrode was regenerated by immersing in pH 7.0 phosphate buffer after each of the measurements. Reproducible currents with a relative standard deviation (R.S.D.) of 2.1% were obtained for 11 successive assays. The reproducibility between five CS/GC electrodes fabricated independently in the same way was also investigated. The five electrodes were used for the measurements of 50 μM NADH and show a satisfactory R.S.D. of 3.8%. Therefore, the stability and reproducibility are both acceptable, suggesting the CS/GC electrode is suitable for direct determination of NADH without any mediator. The limit of detection (LOD) was also calculated based on the equation of 3Sb/k, where Sb is standard deviation of the blank measurements and k is the sensitivity of the present method (i.e., 7.3 nA μM−1). The LOD was calculated to be 0.08 ± 0.03 μM−1, which is lower than that with carbon nanofibers,43carbon fibers,44 MWNT-chitosan,37 active carbon cloth,46and treated CNT39-modified GC electrodes, as listed in Table 1.
The direct electrooxidation of NADH at high overpotential, accompanied with the generation of radical intermediates and dimerization, leads to the fouling of the electrode surface. As a consequence, the electrode deactivates rapidly and the oxidation potential shifts to more positive values. To evaluating the antifouling ability of the CS/GC electrode, the short-term stability of the electrode was examined. Fig. 4 shows the amperometric responses recorded at CS/GC electrode (curve a) with an operating potential of 0.15 V and bare GC electrode (curve b) poised at 0.6 V in 0.1 M phosphate buffer containing 100 μM NADH over a continuous period of 4000 s. It can be seen clearly that the bare GC electrode displays a rapid decay of the current signal with a loss of ca. 52.6% of the original current, whereas only 16.0% loss for CS/GC electrode is found after 4000 s. This demonstrates that the CS/GC electrode can perfectly resist the surface contamination compared with the bare GC electrode.
 | ||
| Fig. 4 Amperometric responses recorded over a continuous 4000 s of 0.1 mM NADH in 0.1 M pH 7.0 phosphate buffer at CS/GC electrode poised at 0.15 V (lower, line a) and bare GC electrode poised at 0.60 V (upper, line b). | ||
Lactate sensor
The low-potential detection of NADH makes CS extremely attractive for amperometric biosensing using various NAD+-dependent dehydrogenases. In the present study, lactate dehydrogenase was chosen as a model enzyme to fabricate a lactate sensor. Fig. 5 displays the steady-state amperometric response of LDH-CS-modified electrode at an applied potential of +0.15 V for different additions of lactate under magnetic stirring in 0.1 M pH 7.0 phosphate buffer. The LDH-CS/GC electrode responded significantly and rapidly (less than 10 s) to the changes of the concentration of lactate. The oxidation current increased linearly with lactate concentration ranging from 0.5 to 12 μM (inset in Fig. 5) with a sensitivity of 4.1 ± 0.2 nA μM−1 (i.e., 57.9 ± 2.8 nA μM−1 cm−2). The limit of detection was estimated at a signal-to-noise ratio of 3 to be 3.7 ± 0.2 μM. These analytical parameters of the present lactate biosensor are quite comparable to those of the biosensors reported in the literature, as shown in Table 2, indicating that the new structural carbon materials provide a facile platform for constructing dehydrogenase-based biosensors. | ||
| Fig. 5 Amperometric response of LDH-CS/GC electrode at an applied potential of +0.15 V to successive addition of different concentrations of L-lactate in 0.1 M phosphate buffer (pH 7.0) saturated with N2. Inset: plot of chronoamperometric current vs.L-lactate concentration. | ||
| Biosensorsa | Linear range (μM) | Detection limit (μM) | Sensitivity (nA μM−1) | Ref. |
|---|---|---|---|---|
| a LOx, lactate oxidase; LDH, lactate dehydrogenase; MWNT, multiwall carbon nanotube; PTTCA, poly-5,2′;5′,2′′-terthiophene-3′-carboxylic acid; GC, glassy carbon electrode; MB, meldola's blue; An, aniline; FAn, fluoroaniline; ITO, indium tin oxide electrode; HMF, hydroxymethylferrocene. | ||||
| LOx-mucin/albumin hydrogel | 2–1000 | 0.8 | 0.537 ± 0.007 | 59 |
| LDH-MWNT-chitoson | 5–120 | 0.76 | 0.587 | 60 |
| LDH-PTTCA-MWNT/GC | 5–90 | 1.0 | 10.6 | 61 |
| LDH-MB-MWNT/GC | 100–104 | 7.5 | 0.42 | 62 |
| LOx-poly(An-co-FAn)/ITO | 100–5500 | 100 | 1.18 | 63 |
| LDH-thick-film carbon electrode | 0–9100 | 110 | — | 64 |
| LOx-HMF-Au electrode | 0–300 | 10 | 0.77 | 65 |
| LDH-CS/GC | 0.5–12 | 3.7 ± 0.2 | 4.1 ± 0.2 | This work |
The examination of the operational stability of LDH-CS/GC enzyme electrode was carried out by successive assays of 5 μM lactate in pH 7.0 phosphate buffer using the same LDH-CS/GC electrode and the reproducible current with a R.S.D. value of 4.0% was obtained for 11 successive assays. The storage stability of the enzyme electrode was also investigated by monitoring its response to 5 μM lactate. A ca. 9.3% decrease of initial current was observed after storage for two weeks. The batch-to-batch reproducibility between five LDH-CS/GC electrodes fabricated independently in the same way was investigated through the measurements of 5 μM lactate and shows a satisfactory R.S.D. of 3.1%. The good stability and reproducibility may be attributed to the porous and three-dimensional structure of the LDH-CS film which is very efficient for retaining the bioactivity of LDH.
Direct bioelectrocatalysis-based lactate/oxygen biofuel cell
Biofuel cells (BFCs), a type of energy conversion device which employ biocatalysts instead of metal catalysts in conventional fuel cells, represent a new kind of environmentally friendly green power sources and have attracted much attention in recent years owing to their potential applications in biomedical electronic devices as an implantable miniature power source.66–71 Much research and a large number of review articles on the design and characterization of BFCs based on mediated electron transfer (MET) have been reported.66–71 In contrast, few BFCs based on direct bioelectrocatalysis have been developed.69,72,73 In the present study, a direct bioelectrocatalysis-type BFC was constructed based on CS-coated film electrode.As demonstrated above, direct bioelectrocatalysis oxidation of lactate on LDH-CS/GC electrode makes it more suitable for acting as a bioanode. Furthermore, the oxidation of lactate at low potential starting at −0.1 V will be favorable for improving the open circuit voltage of the BFC. Herein, LDH-CS/GC enzyme electrode was employed to be the bioanode.
To prepare a biocathode, bilirubin oxidase (BOD), which belongs to the family of multi-copper oxidases, was used as biocatalyst of biocathode to catalyze oxygen reduction to water. The advantage of BOD is that the enzyme works as an electrode catalyst even under neutral conditions, while most multi-copper oxidases such as laccase have an optimum pH in the region of slightly acidic conditions and do not work well under neutral conditions. The direct electron transfer of BOD has been recently achieved at some kinds of the carbon-based electrode, including spectroscopic graphite, HOPG, plastic formed carbon, CNT, and porous carbonaceous electrodes.8,72,74–79 Herein, the direct electron transfer behavior of BOD on CS-coated film electrode was also investigated. Cyclic voltammograms recorded at CS/GC electrode cross-linked with BOD in N2 (black line a), air (red line b), and oxygen (blue line c)-saturated 0.1 M phosphate buffer (pH 7.0) are shown in Fig. 6. It can be seen that a reduction current dependent on the oxygen concentration was recorded in the presence of the enzyme substrate (molecular oxygen). However, the control experiments (Fig. S6) demonstrate that no reduction currents are observed in the potential windows employed at bare GC, BOD-GC, and CS/GC electrodes in the presence of oxygen, indicating that the reduction current observed at BOD-CS/GC electrode can be attributed to oxygen reduction catalyzed by BOD. These results indicate that the direct electron transfer between BOD and GC electrode was facilitated by CS film. It also can be seen from Fig. 6 that the electrocatalytic current at BOD-CS/GC electrode starts at a potential of about +500 mV (vs.Ag/AgCl), which is slightly more positive than the formal potential of the type I Cu site of bilirubin oxidase (0.460 V)80,81 and close to the redox potential of BOD reported in literature.8,72,74–79 This demonstration further confirms that the electron transfer of the BOD could be conducted at the CS film electrodes, revealing that a low overpotential is involved in O2reduction at the prepared BOD-CS/GC biocathode. This result essentially suggests the potential application of the BOD-CS/GC electrodes for the development of new kinds of oxygen cathode of biofuel cells in neutral media.
 | ||
| Fig. 6 Cyclic voltammograms obtained at the BOD-CS/GC electrodes in 0.10 M phosphate buffer solution (pH 7.0) saturated with N2 (curve a, black line), atmosphere (curve b, red line), and O2 (curve c, blue line) with a scan rate of 10 mV s−1. | ||
By using LDH as the bioelectrocatalyst for the catalytic oxidation of lactate at the bioanode and BOD as the bioelectrocatalyst for oxygen reduction at the biocathode, we have successfully developed a direct bioelectrocatalysis-based compartment-less lactate/O2 biofuel cell. Fig. 7 displays the polarization curve and the relationship between the power density (P) and the current density (j) of the assembled carbon sphere-based lactate/O2 BFC in phosphate buffer of pH 7.0. The open circuit voltage (OCV) of the BFC is ca. 0.60 V and the power density reaches 3.13 μW cm−2 at 0.40 V. This result is quite comparable to that of direct electron transfer-based glucose/oxygen BFC reported recently.72 When the cell operated continuously with an external loading of 1 MΩ resistance in a quiescent phosphate buffer (0.10 M, pH 7.0) containing 20 mM NAD+ and 40 mM lactate under ambient air, it lost ca. 6% of its original power in the first 24 h and the power output dropped by ca. 41% after 7 days of continuous work. The performance of the assembled CS-based lactate/O2BFC is restricted to the current density of the BOD-CS/GC biocathode. We are currently working on a new strategy to improve the performance of the biocathode for O2reduction.
 | ||
| Fig. 7 Polarization curve (○) of the assembled CS-based compartment-less lactate/O2 biofuel cell and the dependence of the power output (●) on the current density in the quiescent phosphate buffer (0.1 M, pH 7.0) containing 20 mM NAD+ and 40 mM lactate under O2-saturated atmosphere. | ||
Conclusions
We have synthesized and characterized a novel carbon-based material, carbon hollow spheres. Taking advantage of its excellent catalytic activity to NADH oxidation at lower overpotential, a direct amperometric sensor for NADH detection is proposed. The resulting CS-based NADH sensor limits the electrode surface fouling and improves the operational stability, fabrication reproducibility, and sensitivity. Such ability of CS to promote the electron transfer between NADH and the electrode suggests a new, promising biocompatible platform for development of dehydrogenase-based amperometric biosensors. Thus, a lactate biosensor is constructed by a simple method and the biosensor exhibits very good analytical performance with convenient preparation, sensitive, rapid, and reproducible detection. The direct electron transfer behavior of bilirubin oxidase at CS is also investigated and the results indicate that CS could efficiently facilitate the DET between BOD and electrode. Based on these distinguished properties of CS, a direct bioelectrocatalysis-type membrane-less lactate/oxygen biofuel cell is assembled by using LDH-CS/GC as bioanode for lactate oxidation, and BOD-CS/GC as biocathode for oxygen reduction. In summary, the present results indicate that carbon spheres are a useful electrode material for the analytical detection of NADH, making it attractive for use as electrochemical transducers in biosensors and biofuel cells based on dehydrogenase-catalyzed reactions involving NAD+ as a cofactor.Acknowledgements
F. Gao is grateful for the financial support from the Natural Science Foundation of China (Grant Nos. 20705001, 21055001, 21175002), Anhui Provincial Natural Science Foundation for Distinguished Youth (Grant No. 1108085J09), the Key Project of Educational Committee of Anhui Province (Grant No. KJ2007A008), the Advanced Program for Academic and Technical Leader Candidate of Anhui Province, and the Starting Foundation for PhD of Anhui Normal University. M. Li thanks the Natural Science Foundation of China (Grant No. 21075001) for the financial support. We also give our deep thanks to Mu Yang, Zhenzhong Yang, and Lanqun Mao of Institute of Chemistry, Chinese Academy of Sciences for their helpful discussions and some assistance in characterization experiments.References
- A. Radoi and D. Compagnone, Bioelectrochemistry, 2009, 179, 126–134 CrossRef.
- I. Katakis and E. Domínguez, Microchim. Acta, 1997, 126, 11–32 CrossRef CAS.
- L. Gorton and E. Domínguez, Rev. Mol. Biotechnol., 2002, 82, 371–392 CrossRef CAS.
- J. Moiroux and P. J.Elving, Anal. Chem., 1978, 50, 1056–1062 CrossRef CAS.
- H. Jaegfeld, J. Electroanal. Chem., 1980, 110, 295–302 CrossRef.
- L. Gorton, J. Chem. Soc., Faraday Trans. 1, 1986, 82, 1245–1248 RSC.
- Y. M.Yan, W. Zheng, L. Su and L. Mao, Adv. Mater., 2006, 18, 2639–2643 CrossRef.
- F. Gao, Y. M. Yan, L. Su, L. Wang and L. Mao, Electrochem. Commun., 2007, 9, 989–996 CrossRef CAS.
- M. Zhang and W. Gorski, J. Am. Chem. Soc., 2005, 127, 2058–2059 CrossRef CAS.
- M. Zhang and W. Gorski, Anal. Chem., 2005, 77, 3960–3965 CrossRef CAS.
- P. Ramesh and S. Sampath, Anal. Chem., 2000, 72, 3369–3373 CrossRef CAS.
- F. D. Munteanu, D. Dicu, I. C. Popescu and L. Gorton, Electroanalysis, 2003, 15, 383–391 CrossRef CAS.
- K. Tanaka, K. Tokuda and T. Ohsaka, J. Chem. Soc., Chem. Commun., 1993, 1770–1772 RSC.
- N. Mano, A. Thienpont and A. Kuhn, Electrochem. Commun., 2001, 3, 585–589 CrossRef CAS.
- R. Antiochia, A. Gallina, I. Lavagnini and F. Magno, Electroanalysis, 2002, 14, 1256–1261 CrossRef CAS.
- B. Ge, Y. Tan, Q. Xie, M. Ma and S. Yao, Sens. Actuators, B, 2009, 137, 547–554 CrossRef.
- D. C. S. Tse and T. Kuwana, Anal. Chem., 1978, 50, 1315–1318 CrossRef CAS.
- B. W. Carlson and L. L. Miller, J. Am. Chem. Soc., 1985, 107, 479–485 CrossRef CAS.
- F. Y. Bernadette and R. L. Christopher, Anal. Chem., 1987, 59, 2111–2115 CrossRef.
- Q. Wu, M. Maskus, F. Pariente, F. Tobalina, V. M. Fernandez, E. Lorenzo and H. D. Abruna, Anal. Chem., 1996, 68, 3688–3696 CrossRef CAS.
- I. C. Popescu, E. Domínguez, A. Narváez, V. Pavlov and I. Katakis, J. Electroanal. Chem., 1999, 464, 208–214 CrossRef.
- B. F. Y. Yon Hin and C. R. Lowe, Anal. Chem., 1987, 59, 2111–2115 CrossRef CAS.
- C. X. Cai, K. H. Xue, Y. M. Zhou and H. Yang, Talanta, 1997, 44, 339–347 CrossRef CAS.
- P. N. Bartlett, P. R. Birkin and E. N. K. Wallace, J. Chem. Soc., Faraday Trans., 1997, 93, 1951–1960 RSC.
- A. Ciszewski and G. Milczarek, Anal. Chem., 2000, 72, 3203–3209 CrossRef CAS.
- F. Valentini, A. Salis, A. Curulli and G. Palleschi, Anal. Chem., 2004, 76, 3244–3248 CrossRef CAS.
- F. Pariente, E. Lorenzo, F. Tobalina and H. D. Abruña, Anal. Chem., 1995, 67, 3936–3944 CrossRef CAS.
- F. Pariente, F. Tobalina, M. Darder, E. Lorenzo and H. D. Abruña, Anal. Chem., 1996, 68, 3135–3142 CrossRef CAS.
- F. Pariente, E. Lorenzo and H. D. Abruna, Anal. Chem., 1994, 66, 4337–4344 CrossRef CAS.
- H. Jaegfeldt, T. Kuwana and G. Johansson, J. Am. Chem. Soc., 1983, 105, 1805–1814 CrossRef CAS.
- N. de-los-Santos-Álvarez, M. J. Lobo-Castañón, A. J. Miranda-Ordieres, P. Tuñón-Blanco and H. D. Abruña, Anal. Chem., 2005, 77, 2624–2631 CrossRef.
- M. Yemini, M. Reches, E. Gazit and J. Rishpon, Anal. Chem., 2005, 77, 5155–5159 CrossRef CAS.
- F. Valentini, A. Salis, A. Curulli and G. Palleschi, Anal. Chem., 2004, 76, 3244–3248 CrossRef CAS.
- T. N. Rao, Y. T. Miwa, D. A. Tryk and A. Fujishima, Anal. Chem., 1999, 71, 2506–2511 CrossRef CAS.
- K. S. Prasad, J. C. Chen, C. Ay and J. M. Zen, Sens. Actuators, B, 2007, 123, 715–719 CrossRef.
- M. Musameh, J. Wang, A. Merkoci and Y. H. Lin, Electrochem. Commun., 2002, 4, 743–746 CrossRef CAS.
- M. Zhang, A. Smith and W. Gorski, Anal. Chem., 2004, 76, 5045–5050 CrossRef CAS.
- C. Deng, J. Chen, X. Chen, C. Xiao, Z. Nie and S. Yao, Electrochem. Commun., 2008, 10, 907–909 CrossRef CAS.
- M. Wooten and W. Gorski, Anal. Chem., 2010, 82, 1299–1304 CrossRef CAS.
- J. Wang and M. Musameh, Anal. Chem., 2003, 75, 2075–2079 CrossRef CAS.
- R. R. Moore, C. E. Banks and R. G. Compton, Anal. Chem., 2004, 76, 2677–2682 CrossRef CAS.
- C. E. Banks and R. G. Compton, Analyst, 2005, 130, 1232–1239 RSC.
- L. Wu, X. Zhang and H. Ju, Anal. Chem., 2007, 79, 453–458 CrossRef CAS.
- W. G. Kuhr, V. L. Barrett, M. R. Gagnon, J. P. Hopper and P. Pantano, Anal. Chem., 1993, 65, 617–22 CrossRef CAS.
- M. A. Hayes and W. G. Kuhr, Anal. Chem., 1999, 71, 1720–1727 CrossRef CAS.
- C. E. Campbell and J. Rishpon, Electroanalysis, 2001, 13, 17–20 CrossRef CAS.
- C. E. Banks and R. G. Compton, Anal. Sci., 2005, 21, 1263–1268 CrossRef CAS.
- R. L. McCreery, Chem. Rev., 2008, 108, 2646–2687 CrossRef CAS.
- M. Yang, J. Ma, C. Zhang, Z. Yang and Y. Lu, Angew. Chem., Int. Ed., 2005, 44, 6727–6730 CrossRef CAS.
- M. Yang, J. Ma, S. Ding, Z. Meng, J. Liu, T. Zhao, L. Mao, Y. Shi, X. Jin, Y. Lu and Z. Yang, Macromol. Chem. Phys., 2006, 207, 1633–1639 CrossRef CAS.
- Y. Wang, D. C. Alsmeyer and R. L. McCreery, Chem. Mater., 1990, 2, 357–563 Search PubMed.
- K. Ray and R. L. McCreery, Anal. Chem., 1997, 69, 4680–4687 CrossRef CAS.
- G. Katagiri, H. Ishida and A. Ishitani, Carbon, 1988, 26, 565–571 CrossRef CAS.
- P. Ramesh and S. Sampath, Anal. Chem., 2003, 75, 6949–6957 CrossRef CAS.
- J. Rikhie and S. Sampath, Electroanalysis, 2005, 17, 762–768 CrossRef CAS.
- R. Bradley, H. X. Ling and I. Sutherland, Carbon, 1993, 31, 1115–1993 CrossRef CAS.
- S. Biniak, G. Szymanski, J. Siedlewski and A. Swaitowski, Carbon, 1997, 35, 1799–1810 CrossRef CAS.
- L. Su, F. Gao and L. Mao, Anal. Chem., 2006, 78, 2651–2657 CrossRef CAS.
- M. R. Romero, F. Ahumada, F. Fernando Garay and A. M. Baruzzi, Anal. Chem., 2010, 82, 5568–5572 CrossRef CAS.
- Y. C. Tsai, S. Y. Chen and H. W. Liaw, Sens. Actuators B, 2007, 125, 474–481 CrossRef.
- M. M.Rahman, M. J. A. Shiddiky, M. A. Rahman and Y. B. Shim, Anal. Biochem., 2009, 384, 159–165 CrossRef.
- A. C. Pereira, M. R. Aguiar, A. Kisner, D. V. Macedoa and L. T. Kubota, Sens. Actuators, B, 2007, 124, 269–276 CrossRef.
- S. Sumana, R. Singhal, A. L. Sharma, B. D. Malthotra and C. S. Pundir, Sens. Actuators, B, 2005, 107, 768–772 CrossRef.
- H. C. Yoon and H. S. Kim, Anal. Chim. Acta, 1996, 336, 57–65 CrossRef CAS.
- A. Parra, E. Casero, L. Vázquez, F. Pariente and E. Lorenzo, Anal. Chim. Acta, 2006, 555, 308–315 CrossRef CAS.
- N. Mano, F. Mao and A. Heller, J. Am. Chem. Soc., 2002, 124, 12962–12964 CrossRef CAS.
- S. C. Barton, J. Gallaway and P. Atanassov, Chem. Rev., 2004, 104, 4867–4886 CrossRef CAS.
- J. A. Cracknell, K. A. Vincent and F. A. Armstrong, Chem. Rev., 2008, 108, 2439–2461 CrossRef CAS.
- M. J. Cooney, V. Svoboda, C. Lau, G. Martin and S. D. Minteer, Energy Environ. Sci., 2008, 1, 320–337 CAS.
- F. Gao, L. Viry, M. Maugey, P. Poulin and N. Mano, Nat. Commun., 2010, 1 DOI:10.1038/ncomms1000.
- M. Zayats, B. Willner and I. Willner, Electroanalysis, 2008, 20, 583–601 CrossRef CAS.
- V. Coman, R. Ludwig, W. Harreither, D. Haltrich, L. Gorton1, T. Ruzgas and S. Shleev, Fuel Cells, 2010, 10, 9–16 CAS.
- A. Ramanavicius, A. Kausaite and A. Ramanaviciene, Biosens. Bioelectron., 2005, 20, 1962–1967 CrossRef CAS.
- S. Shleev, A. E. Kasmi, T. Ruzgas and L. Gorton, Electrochem. Commun., 2004, 6, 934–939 CrossRef CAS.
- S. Tsujimura, K. Kano and T. Ikeda, J. Electroanal. Chem., 2005, 576, 113–120 CrossRef CAS.
- S. Tsujimura, T. Nakagawa, K. Kano and T. Ikeda, Electrochemistry, 2004, 72, 437–439 CAS.
- S. Tsujimura, Y. Kamitaka and K. Kano, Fuel Cells, 2007, 7, 463–469 CrossRef CAS.
- P. Ramírez, N. Mano, R. Andreu, T. Ruzgas, A. Heller, L. Gorton and S. Shleev, Biochim. Biophys. Acta, Bioenerg., 2008, 1777, 1364–1369 CrossRef.
- V. Flexer, N. Brun, O. Courjean, R. Backov and N. Mano, Energy Environ. Sci., 2011, 4, 2097–2106 CAS.
- S. Tsujimura, A. Kuriyama, N. Fujieda, K. Kano and T. Ikeda, Anal. Biochem., 2005, 337, 325–331 CrossRef CAS.
- P. Ramírez, N. Mano, R. Andreu, T. Ruzgas, A. Heller, L. Gorton and S. Shleev, Biochim. Biophys. Acta, Bioenerg., 2006, 1757, 1634–1641 CrossRef.
Footnote |
| † Electronic Supplementary Information (ESI) available. See DOI: 10.1039/c1ra00444a/ |
| This journal is © The Royal Society of Chemistry 2011 |