Research progress and materials selection guidelines on mixed conducting perovskite-type ceramic membranes for oxygen production
Kun
Zhang
a,
Jaka
Sunarso
b,
Zongping
Shao
*c,
Wei
Zhou
c,
Chenghua
Sun
d,
Shaobin
Wang
a and
Shaomin
Liu
*a
aDepartment of Chemical Engineering, Curtin University, Perth WA 6845, Australia
bAustralian Research Council (ARC) Centre of Excellence for Electromaterials Science, Institute for Technology Research and Innovation, Deakin University, Burwood, Victoria 3125, Australia
cState Key Laboratory of Materials-Oriented Chemical Engineering, College of Chemistry & Chemical Engineering, Nanjing University of Technology, No. 5 Xin Mofan Road, Nanjing, 210009, P. R. China. E-mail: shaozp@njut.edu.cn (Z.Shao), Shaomin.Liu@curtin.edu.au (S. Liu); Tel: +61 8 92669056
dAustralian Institute for Bioengineering and Nanotechnology, The University of Queensland, Brisbane Qld, 4072, Australia
First published on 28th October 2011
Abstract
Oxygen production by air separation is of great importance in both environmental and industrial processes as most large scale clean energy technologies require oxygen as feed gas. Currently the conventional cryogenic air separation unit is a major economic impediment to the deployment of these clean energy technologies with carbon capture (i.e. oxy-fuel combustion). Dense ceramic perovskite membranes are envisaged to replace the cryogenics and reduce O2 production costs by 35% or more; which can significantly cut the energy penalty by 50% when integrated in oxy-fuel power plant for CO2 capture. This paper reviews the current progress in the development of dense ceramic membranes for oxygen production. The principles, advantages or disadvantages, and the crucial problems of all kinds of membranes are discussed. Materials development, optimisation guidelines and suggestions for future research direction are also included. Some areas already previously reviewed are treated with less attention.
1 Introduction
The demand for oxygen generation and removal for different applications has increased substantially in concurrent trend with the modern society development.1 Obtaining pure oxygen with low cost is a very important issue in industry. Nowadays, commercial oxygen is still produced by cryogenic distillation and pressure swing adsorption which are energy and cost-intensive technologies. Since the first report by Teraoka et al.2 on favorable oxygen permeation through dense disk ceramic membranes based on mixed ionic and electronic conducting (MIEC) perovskite oxides of La1−xSrxCo1−yFeyO3−δ composition in the 1980s, these membranes which possess oxygen ionic in conjunction with electronic conductivity at high temperatures have attracted considerable attention during the past decades. Many related studies and research progress on mixed conducting oxide materials and membranes have been published since then.3–29Numerous structures have been reported which exhibit these MIEC properties, such as perovskite, brownmillerite, orthoferrite, K2NiF4-type phase and Ruddlesden-Popper series. Among these structures, the perovskite-structured oxides have been studied intensively due to their diverse and controllable properties. The general formula of the perovskite is ABO3; the properties of which are determined by cations occupying its A- and B-site lattice. The A-site cations are mainly composed of alkaline earth, alkaline and lanthanide ions, while B-site cations are mainly composed of transition metal ions.30 When the original A-site cation of perovskite is partially substituted by another cation with a lower oxidation state than the original cations, the electrical neutrality is normally sustained by the formation of oxygen vacancies or increased oxidation state of the B-site cations. Accordingly, partial substitution of original cations with another cation having a higher oxidation state tends to inhibit the formation of oxygen vacancies or increased oxidation state of the B-site cations. The presence of oxygen vacancies here facilitates the oxygen ion movement which closely affects the oxygen ionic conductivity of the oxide while the electron hopping between the valence-variable metal ions at the B-site make electronic conduction possible. The resulting MIEC properties in turn, have granted these materials a unique separation mechanism, e.g.oxygen permeating through the membrane by dissociating and/or associating in the interface and ionic transport through the bulk instead by way of the conventional molecular diffusion (through micro-pores).
There has been a substantial number of articles published on the title subject in the last decade, which highlights the remarkable potential and increasing interest in the field. Several reviews on mixed conducting membranes for oxygen separation are available which provide the main understanding on the material composition, structure, preparation, as well as the transport mechanism of oxygen permeable membranes.31–36 For example, Sunarso et al.34 reviewed the development of mixed conducting dense ceramic membranes during the past three decades. Yang et al.35 summarised the development and challenges of perovskite type materials for oxygen transport membrane reactor applications. Liu et al.36 wrote a review focused on the transport theory of several classes of perovskite compounds, including their potential application in different areas and oxygen permeability improvement efforts by doping (e.g. partial substitution of original cations) with various metal oxide elements.
While many perovskite oxide materials have been explored over the past two decades; there are hardly any materials with sufficient practical economic value and performance for large scale applications which justify the continuing search for new materials. Furthermore, there is a lack of systematic studies to assist in materials selection. To this end, the objective of this review is to provide the recent advances of ceramic membranes for oxygen production, with its main focus on the strategy to systematically select perovskite-type MIEC membrane material composition featuring high oxygen permeability and structural stability. While the overlapping with other available reviews is avoided as much as possible; the basic fundamental aspects are retained and briefly explained to set it as a stand-alone paper which will also facilitate understanding for newcomers in the field. In addition, some perspectives are given in the last section to guide the future research in materials development.
2 Fundamentals of ceramic membranes for oxygen production
2.1 General characteristics of mixed conducting dense ceramic membranes
Among dense inorganic membranes, oxygen-ionic conducting membranes have received considerable attention in the past three decades. There are two types of these membranes; the first is the so-called electrolyte membrane, which is a pure oxygen ionic conductor blocking the migration of electrons through it.37,38 Therefore, electrodes on both sides of the membrane are required to separate electrons and oxygen ion transport; both of which are also attached to an outside electrical circuit to facilitate electron transport. Here, the driving force for the oxygen permeation is the electrical potential. Another type of membrane is the mixed conducting (MIEC) membrane allowing the transport of both oxygen ions and electrons through it. The oxygen permeation is driven by the difference of oxygen partial pressures between opposing sides of the membrane. This chemical potential gradient of molecular oxygen creates the opposing direction and inter-correlated flows of oxygen ions (O2−) and electronic charge carriers (electrons and/or holes) through the membrane.39–41 The difference in transport configuration between an electrolyte membrane and an MIEC membrane is illustrated in Fig. 1.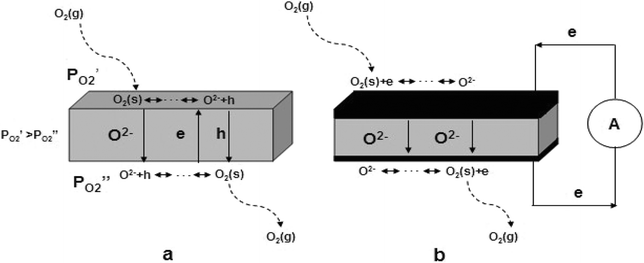 | ||
| Fig. 1 Schematic diagram of two different types of oxygen ionic conducting membranes: (a) mixed conducting membrane; (b) electrolyte membrane. | ||
Due to its membrane configuration simplicity, high oxygen permeability, absolute selectivity for oxygen (e.g. only allowing oxygen ion permeation) and catalytic activity; dense MIEC ceramic membranes can also be utilised as catalytic membrane reactors in different petro-chemistry product related processes such as coupling oxidation of methane to C2 (ethylene and/or ethane) (COM), partial oxidation of methane to syngas (POM), partial oxidation of heptanes to hydrogen (POH), selective oxidation of ethane to ethylene (SOE) and selective oxidation of propane to propylene (SOP).36,42–48 In particular, gas conversion to liquids has become more and more important currently for economic and environmental reasons. In contrast with the conventional catalyst operation mode such as packed bed reactor, the direct contact between methane and oxygen is avoided in a membrane reactor. As a result, a membrane reactor using a dense oxygen permeable ceramic membrane offers a higher selectivity and yield on the desired products. Moreover, safer operation, simultaneous oxygen separation and catalytic reaction can be realised in a single device. Thus, MIEC membranes for oxygen separation from air and membrane reactors for catalytic oxidation of light hydrocarbons to value-added products (e.g. syngas, ethylene and propylene, etc.) have become the hot topic until now.49–55
2.2 Oxygen transport
Several mechanisms such as the vacancy mechanism (i.e., via the jump of atom or ion between oxygen vacancies) interstitial mechanism (i.e., via the movement of smaller atoms from an interstitial site to one of neighboring interstitial site) and/or their combined mechanisms are available which can explain oxygen ion transport in oxides; depending on the type of existing defects.56 For perovskite type MIEC materials, it is generally known that the oxygen ion conduction is made possible by the existence of oxygen vacancies. Therefore, the following discussion of oxygen transport properties is centralised around the oxygen vacancy mechanism. At elevated temperature, oxygen vacancies are formed in oxides due to the introduction of multivalent cations with redox properties. In the presence of gaseous oxygen, oxygen vacancies react with oxygen atoms resulting in the formation of two electron holes to satisfy charge neutrality criterion.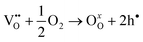 | (1) |
Oxygen permeation through MIEC membranes involves surface exchange kinetics, bulk oxygen ion and electron diffusion. Firstly, gaseous oxygen in the high oxygen partial pressure side transfers to and adsorbs on the membrane surface. The adsorbed oxygen (at the oxygen vacancy sites of the surface layer) is then reduced to lattice oxygen and migrates through the bulk layer to another side of the membrane (due to the oxygen partial pressure gradient across the membrane). Subsequently, at the lower oxygen partial pressure side of the membrane, the lattice oxygen reacts with the electron, desorbs from the membrane surface and oxidises to its gaseous form. At the same time, the multivalent metal ions on the B-site experience reduction or oxidation to compensate for the resultant charge from the oxygen ions reaction. This leads to electronic conductivity via B lattice cations through strongly overlapping B–O–B bonds by Zener double exchange mechanism process57 which is, typically, 100–1000 times higher than the oxygen ionic conductivity. Normally, the process of adsorption or desorption between the gaseous oxygen and membrane surface and the electronic conduction is very fast. Consequently, only bulk diffusion and surface reaction steps need be taken into consideration in major transport resistance evaluation.
2.3. Oxygen permeation rate determining factors
Oxygen permeation through a MIEC membrane involves oxygen surface exchange reactions and oxygen bulk diffusion, with either one, or a combination of the two, potentially being the rate-determining step(s). A number of reports have investigated the contributions of surface exchange and bulk diffusion at different membrane systems.58–62 The oxygen permeation rate (fluxes) is not only limited by intrinsic material properties, but also closely influenced by membrane thickness and practical operation conditions. Qiu et al.58 studied the influence of thickness on oxygen fluxes of a SrCo0.8Fe0.2O3−δmembrane under an Air/He atmosphere. They found that the oxygen permeation is controlled by the slow surface exchange process at the measured operation condition. Chen et al.59 investigated the oxygen permeability of La0.4Sr0.6Co0.8Fe0.2O3−δ on which they showed that the permeation process is totally limited by the bulk diffusion when the membrane thickness is between 1.25 and 2.46 mm. However, when the thickness is reduced to 0.62 mm, the permeation is controlled by both bulk diffusion and surface exchange. It is worthy to note that, even for the same material with the same membrane thickness and at similar operating conditions (i.e., oxygen partial pressure gradient and temperature), different oxygen fluxes have been reported by different researchers, which reflects the complexity of influencing variables during high temperature experimental permeation measurements. The most likely affecting factors are:1. The occurrence of non-axial oxygen diffusion due to fringe effects from sealants.
2. Interfacial reaction between the membrane surface and sealing materials.
3. Diffusion and reaction of sealing materials onto/with the oxides' surface.
4. Difficulty to reproduce exactly similar surface morphology for different membrane samples.
5. Inability to determine the real value of oxygen partial pressure on both membrane surfaces.
Understanding the oxygen permeation mechanism through the membrane is important to obtain optimised operation conditions and improved membrane materials. Numerous experimental techniques have been developed to extract bulk diffusion and surface exchange coefficients on MIEC membranes such as transient thermal gravimetric technique, coulometric titration method, isotope exchange combined with secondary ions mass spectroscopy (SIMS) technique and electrical relaxation method.63–68 Numerous empirical and/or theoretical equations based on various models have also been developed and utilised to model the experimental oxygen permeation results and obtain bulk diffusion and surface exchange information. When the oxygen permeation rate is limited by bulk diffusion, reducing the membrane thickness is effective at enhancing the oxygen permeation flux. Nevertheless, the flux enlargement cannot be obtained by reducing the thickness when the permeation rate is determined by the surface exchange kinetics. In this latter case, membrane surface modification providing higher surface area or viacatalyst deposition is more beneficial.
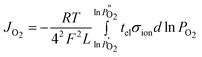 | (2) |
Where σion (S cm−1) is the ionic conductivity of the material, tel is the electronic transference number, F (C mol−1) is the Faraday constant, T/K is the absolute temperature, L (mm) is the membrane thickness, P′O2 and P′′O2 (Pa) are the high and low oxygen partial pressure on each membrane side, respectively.
For most perovskite membrane materials, σel ≫σion, Eqn (1) can be simplified into:
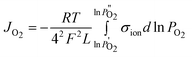 | (3) |
For the membrane material operated under the bulk diffusion limited condition, reducing the thickness can increase its oxygen fluxes.
1. The ratio of the surface exchange coefficient, Ks and the oxygen ion diffusion coefficient D*. A low Ks/D* value implies a dominant role of the surface exchange reaction.
2. Characteristic (critical) thickness, Lc = D*/Ks, e.g., the membrane thickness when the resistance from the surface reaction and bulk diffusion is equal. When L > Lc, the oxygen permeation rate is controlled by bulk diffusion. When L < Lc, the permeation rate is determined by the surface exchange reaction. The characteristic thickness is governed by the material's nature but also is influenced by the operating conditions like temperature and oxygen partial pressure.
Bouwmeester et al.70 measured the characteristic thickness of Sr-doped LaCoO3 and LaFeO3, the reported value of which is around 20–500 μm. Kilner et al.71 pointed out that the characteristic thicknesses for most perovskite oxides are around 100 μm. As a result, it is considered ineffective to reduce the membrane thickness (for improving oxygen fluxes) when the membrane thickness is already less than 100 μm. Moreover, it is worthy to note that occasionally the apparent membrane thickness does not represent the actual oxygen ion diffusion distance in the membrane bulk e.g., the distance (L) used in the calculation of the material's oxygen ionic conducting property. Ideally, the membrane should be fully densified and free of any enclosed pores. However, in most cases, it is difficult to completely avoid the presence of these enclosed pores inside the bulk of the ceramic membrane by simply adjusting the sintering temperatures. Fig. 2 compares the different oxygen bulk transport pathways through dense membranes with a fully dense structure and a porous structure without through connections. Such a porous structure containing enclosed pores (Fig. 2) exerts negative effects on the oxygen transport by increasing the transport resistance. There are two possible transport paths for oxygen to pass through these enclosed pores. One is via the more distorted pathway surpassing the isolated cavities. Another is via molecular oxygen gas diffusion which involves several cycles of surface reactions and charge transfer between oxygen ions and molecular oxygen. In both paths, large transport resistance dominates, which will definitely slow the oxygen permeation rate.
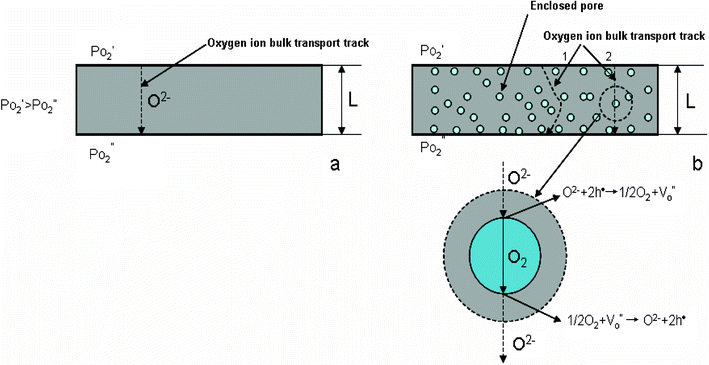 | ||
| Fig. 2 Diagram of oxygen bulk transport processes through dense membranes, (a) ideal fully dense membrane; (b) membrane with some closed non-connected pores. | ||
Accordingly, the reduction or elimination of these isolated pores is expected to improve the membrane performance. A possible reason for these isolated pore formations is the trapped air among the starting ceramic particles. High-energy ball milling procedure can modify the powder properties and to some extent reduce the amount of these pores during the sintering. The resultant membranes with a more densified structure demonstrate better oxygen permeation flux (by about 20%) relative to the original BSCF membranes.72 Another example is on LSCF hollow fibre membranes prepared using phase inversion and sintering. When water is used as the internal and external coagulants, the membrane is characterised by the presence of two arrays of isolated finger-like pores with lengths up to 30 μm sandwiched by three fully densified layers.73 Permeability improvement can be obtained by opening these isolated pores onto both membrane surfaces by acid-modification. This increases the permeation flux through LSCF hollow fiber membranes by a factor of up to 18 over the original fibers.73
Surface treatment and modification are also commonly applied to improve oxygen fluxes in membranes operating in surface reaction dominated regime. In this respect, surface modification is achieved via deposition of a porous layer or more reactive layer with favorable oxygen exchange properties. Lee et al.74 enhanced the oxygen permeation fluxes from ∼0.33 ml cm−2 min−1 to ∼1.07 ml cm−2 min−1 at 772 °C through 0.1 cm thick SrCo0.8Fe0.2O3−δ dense membrane by depositing a porous layer made from the same material. Kharton et al.75 carried out surface modification by coating Ag onto a La0.3Sr0.7CoO3−δmembrane surface; improving the permeation fluxes from ∼0.26 ml cm−2 min−1 to ∼0.34 ml cm−2 min−1 at 770 °C.
In practice, it is rare that the oxygen permeation of a certain ceramic membrane in a fixed configuration is controlled only by bulk transport or surface exchange. The combination of both steps normally contributes to the oxygen fluxes. Different operation conditions such as temperature and pressure normally translates to different operating regime. For example, in a La0.6Sr0.4Co0.2Fe0.8O3−δ disk membrane, the oxygen permeation was reported to be governed by the slow kinetics of interfacial exchange at oxygen partial pressure below 1–3 kPa and by bulk diffusion at 10–100 kPa.66 The controlling step for oxygen permeation through the same membrane is also affected by the operating temperature. For example, at 700 °C, the permeation fluxes through La0.6Sr0.4Co0.2Fe0.8O3−δ hollow fiber membranes with an Ag coating increase from 0.01 ml cm−2 min−1 to 0.1 ml cm−2 min−1 (increased by a factor of 10). However, upon increasing the temperature up to 900 °C, the increasing factor is only 3 as compared with the non-modified sample.76 This observation may imply that at a lower temperature range, the oxygen permeation was largely controlled by slow surface exchange kinetics and surface modification can effectively improve the oxygen permeation flux; nevertheless, when the temperature was gradually increased to higher temperatures (≥900 °C), the controlling step shifted to bulk diffusion so that the surface modification became less effective.
2.4 Ceramic hollow fibre membranes
Most of the currently encountered membrane-based gas separation is performed using a molecular sieving membrane, a purely physical process which relies on the relative size of the molecules and the porosity and pore size of the membrane to separate different gas molecules. In contrast, oxygen separation via dense mixed conducting ceramic membranes as discussed here involves the surface ionic reactions and bulk ionic diffusion. Currently, most of the related research work in this area uses disk-shaped or tubular membranes. Although a multiple planar stack can be designed to enlarge the membrane area to afford a plant-scale output production, numerous engineering related problems easily arise from flat designs such as sealing, connection, and pressure resistance. Therefore, tubular membranes have been developed to overcome these problems, nevertheless their small surface to volume ratios as well as their thick bulk transport layer substantially trade-off their advantages particularly towards practical applications. The manufacture of monolith structures with a chessboard arrangement might be considered further to improve the ratio of surface area to volume. Still, the problems related to the manifold complexity and required sealing for different gas streams exists in this particular system design. From another fundamental standpoint of further elucidating the relationship between the membrane structure and properties, these conventional membrane configurations are also considered unattractive. The relatively large membrane thickness (in symmetric structure) utilised in these conventional designs and the fact that they operate in the bulk diffusion regime instead of surface reaction regime means that these membranes are not being operated in possible optimum conditions. In this regard, asymmetric hollow fibre membranes seem to find their niche not only in the membrane fundamental studies but also towards scaling up to industrial applications due to their very thin transport layer, their large membrane area per unit volume and ease of sealing.The synthesis method is based on the combined phase inversion and sintering technique where the viscous poly/polyethersulfone solution in NMP (1-methyl-2-pyrrolidinone) and water can be used as the binder and coagulants, respectively.9,77–79 The resultant hollow fibre membranes with an outside diameter of 0.7–2.0 mm and thickness of 0.2–0.5 mm featuring thin dense layers supported on porous structures of the same perovskite materials are able to deliver up to 7.6 ml min−1 cm−2 at 900 °C at atmospheric air/helium gradient. The improvement in oxygen fluxes here is due to the thinner thickness of the separating layer in the membrane structure with respect to the conventional disk or tubular shaped membranes.
Depending on the material compositions, some perovskite hollow fibres can be very robust and provide sufficiently high mechanical strength and chemical stability. For example, a proof-of-concept membrane system having a maximum capacity of 3.1 L min−1 O2 with >99.5% purity using La0.6Sr0.4Co0.2Fe0.8O3 (LSCF) perovskite hollow fibre membranes has been achieved during operation of more than a 1000 h period.80 Due to the thinner membrane thickness, the perovskite hollow fibres effectively operate on the surface reaction kinetics regime evidenced by the possible improvement of oxygen fluxes by up to a factor of 20 when their membrane surfaces were etched as well as when the metal catalysts (e.g.Ag, Pt or Pd) were deposited on the membranes' surface.8,76 All of these phenomena were specific to hollow fibre membranes and have been of great substantial value towards elucidating the membrane transport theory and relationship between structure and properties. Notably, the change of binder system and internal coagulant to prepare the hollow fibres, which are often overlooked by researchers, also greatly affects the membrane performance. For example, the usage of sulfur-free polyetherimide (PEI) polymer as a new binder to replace the polysulfone or polyether avoids the contamination of the hollow fibre membranes by the reaction between sulfur oxide and metal oxides in the perovskite structure, which in turn enhances the membrane purity and translates into high performance with oxygen fluxes higher than 10 ml min−1 cm−2 possible at atmospheric air/helium gradient.81,82 Another example is the effect of internal coagulant on the membrane properties. During the phase inversion process, the amount of internal coagulant used to shape the hollow fibre geometry is very minimal, yet playing a vital role in influencing morphology of the hollow fibre. When a mixture of 10% EtOH and 90% NMP replaced DI water as the internal coagulant, a complete new honeycomb structured hollow fibre membrane with a single thin densified layer was produced.83 This single dense layer hollow fibre membrane delivers much higher O2 fluxes with an improvement factor up to 15 compared to the traditional multi-dense layer sandwiched membrane structure. More interesting is that the delicate architecture of the hollow fibre membrane is completed in one sintering step in striking contrast to the multi-step and time-consuming procedures to prepare conventional supported thin asymmetric membranes. All of these versatile hollow fibre membrane skills can be expanded directly to any other new ceramic membrane materials and ready to make the contribution towards the future commercial targets in air separation units.
2.5 Non-perovskite compounds
There are various mixed conducting materials designed for the potential application of oxygen permeation membrane. In terms of the structure, it can be simply classified into two main groups, non-perovskite and perovskite. For now, most studies put their focus on the oxygen permeation based on perovskite structure because of their excellent oxygen ion and electron conducting capacity. Therefore, in this section we firstly give a brief introduction on the non-perovskite oxides as ceramic membranes. As well known, the properties of a material are closely related to its structure. The structure of a material not only determines its performance in practical applications, but can also provide insight and understanding into why a certain material possesses excellent capability. In the past few decades, materials with fluorite, brownmillerite, orthorhombic K2NiF4-type and Sr4Fe6−xCoxO13 structures have shown promise as oxygen transport membranes due to their MIEC properties.84–87 However, the significant drawback to practical adoption is their lower ambipolar conductivity (with respect to cubic perovskite).84,88Among many structures, the fluorite-type structure is one of the most common crystal structures favorable for oxygen ion conduction which is represented by the cubic structure of ZrO2 at high temperature (in excess of 2370 °C).89,90 Doping of CaO, Y2O3 or some other trivalent metal ions into ZrO2 has been used to stabilize the cubic structure of ZrO2 to room temperature. This doping strategy also leads to the formation of oxygen vacancies as a result of the charge neutrality criterion (since Ca or Y has lower valence state than Zr) which further rationalises the oxygen ionic conductivity in this doped fluorite materials. While some transition metal ions, such as TiO2, CeO2, Tb2O3.5 and CuO can be introduced into the material to provide electronic conductivity; the oxygen ionic conductivity in fluorite type material normally accounts for a much larger portion of total conductivity (with respect to electronic conductivity).84,91–93
Partial substitution of the original A-site and B-site cation with other cations normally leads to the distortion from the cubic symmetry to other structure symmetries, most of which were reported as the brownmillerite structure.94 Further on, when the oxygen vacancy concentration is large at elevated temperature, oxygen vacancies prefer ordered distribution to minimize the Gibbs free energy with consequential structure transition into orthorhombic symmetry as observed in SrCo0.8Fe0.2O3−δ. The brownmillerite structure is represented by A2BB′O5 formula. Its crystal lattice can be pictured as an anion deficient perovskite with one-sixth of its oxygen ions empty. Oxygen vacancies are ordered in alternate BO2 planes of the cubic structure such that the alternate (110) row of oxide anions is missing.30 In comparison with the cubic perovskite, the ordering will result in the unit cell expansion of materials. Since the migration energy for oxygen ions in the ordered structure is substantially higher than that in the oxygen vacancy-disordered structure; the oxygen ionic transport in the former structure is not as mobile as in the latter structure.
A sharp change in the oxygen flux behavior with temperature change has been commonly noted mostly on the mixed conducting membrane containing brownmillerite structure at a specific temperature range, due to the effect of the order–disorder transition of oxygen vacancies on the oxygen ion transport in such oxides. The relationship between oxygen permeability and phase transition in mixed conducting membranes are of interest. The transport of oxygen ions in the perovskite-related mixed conducting oxides is also affected by the distribution state of oxygen vacancies. In parallel with a structural transition from cubic perovskite to brownmillerite, the oxygen vacancies change their packing state from disordered into ordered arrangements. In the latter state, in particular, oxygen ions are hardly mobile because of the ordering and pinning states of the oxygen vacancies; to the extent that transport is restricted in a two-dimensional ac-plane instead of a three dimensional bulk phase in the former state.95 Bouwmeester et al.96,97 observed that SrCo0.8Fe0.2O2.5 experienced a zero oxygen release process because of the formation of the vacancy-ordered brownmillerite phase. The absence of ordered vacancy in Ba0.5Sr0.5Co0.8Fe0.2O3−δ, on the other hand leads to its higher oxygen permeation fluxes. Nevertheless, some materials with the brownmillerite structure still show considerable oxygen permeability. Eltron Research98–100 reported oxygen conducting capability for some brownmillerite materials which are equivalent to or exceed the value for perovskite materials, the highest flux of which under syngas production can reach 10–12 ml min−1 cm−2. The reason for the high oxygen permeation through the brownmillerite oxides is likely to be related to their high concentration of intrinsic oxygen vacancy and low activation energy for ionic conductivity. Moreover, it is interesting that not only the order-disorder of oxygen vacancies, but also the order-disorder of A-site cation in perovskite-related oxide can influence the oxygen permeability. Recently, numerous A-site ordered perovskites have attracted a considerable attention as potential alternative mixed conducting materials.101–103 Due to the ordering of A-site cations, these materials have an ordered layered structure which reduces the strength of oxygen binding and provides disorder-free channels for oxygen transport. In spite of significantly fast oxygen exchange and diffusion kinetics, the oxygen permeation flux of A-site ordered layered membranes, i.e., LnBaCo2O5+δ, is much lower than that from the highly-permeable Ba0.5Sr0.5Co0.8Fe0.2O3−δmembranes under similar operating conditions. It is well known that the layered material distributes the oxygen vacancies in the LnOδ planes. In such a structure, oxygen ions can transport quickly but their transport is only limited to a confined two-dimensional plane of the single crystals of the oxides. When oxygen permeates through the dense polycrystalline ceramic membrane, the oxygen ion transport will not be a straight path but a detoured or round about path due to the random crystal orientation in the ceramic bulk. In this case, the actual oxygen diffusion distance will be much longer than the real membrane thickness.104 Therefore, LnBaCo2O5+δoxides should be more appropriate for usage as a thin catalyst coating layer to improve the surface exchange kinetics rather than as bulk dense membrane materials.105–106
Dual-phase membranes have also been widely investigated.107–111 The dispersion of a metallic phase electronic conductor phase into an oxygen ion conducting phase, e.g.Pd metal into stabilised zirconia, allows oxygen ion and electron transport through separate phases. Unfortunately, such membranes usually demonstrate low oxygen permeability which might be due to the non-ideal mixing of the two phases. Chen et al.110 reported the preparation of a dual-phase membrane using several dual-phase composites such as erbia stabilized bismuth silver or gold. It was observed that in order to form an electronically conducting phase network, around 40% volume of metal is required. The oxygen transport was controlled by bulk diffusion of oxygen ions in the oxide phase for membranes with a thickness equal to or more than 1.0 mm. As the membrane thickness decreases further, both bulk diffusion and surface exchange determine the oxygen permeation rate. Dual-phase membranes have also been prepared using two ceramic material phases with one providing large ionic conductivity and another offering large electronic conductivity. For example, a dual-phase membrane with the fluorite type material, Ce0.8Gd0.2O2−δ as the oxygen ion conducting phase and the perovskite type material, La0.7Sr0.3MnO3−δ as the electron conducting phase have been prepared by Kharton et al.111 The utilisation of perovskite structure in this latter system which is less stable than the fluorite structure alone especially in long term and harsh operating condition normally limits the stability of these membranes. The advantage of the dual-phase membrane concept nevertheless lies in the possibility to tune its stability by choosing proper phase component; both of which contribute to its stability. Very recently, a dual-phase membrane consisting of alkaline earth free CO2-stable spinel-type material, NiFe2O4 and Co-free fluorite type material like Ce0.9Gd0.1O2−δ have been developed which demonstrates very stable fluxes during a 100 h operation when CO2 is used as sweep gas.112
3 Perovskite compounds
In a broad definition, a perovskite is any material with the same crystal structure type as calcium titanium oxide, e.g.CaTiO3, which has a face cubic centered structure with oxygen atoms occupying the face centers. Perovskite derives their name from this compound, which was first discovered in the Ural mountains in Russia by Gustav Rose in 1839. Around 90% of the elements in the periodic table can form the perovskite structure. The ideal perovskite structure is cubic with a structure formula of ABO3 on which A is the larger cation and B is the smaller cation.30 Perovskite can also be regarded as a framework structure (ReO3–type framework), constructed from corner-sharing BO6 octahedra with A ions located in twelve-coordinated interstices, as shown in Fig. 3. Alternatively, the structure can be viewed with a B cation in the center, as shown in Fig. 3 on which each oxygen ion is coordinated with two B ions and four A ions. It was pointed out that the most important pre-requisite for a stable perovskite structure is the existence of a stable BO3 skeletal sub-lattice. From the geometric point of view, the ionic radius of the B cation should be larger than 0.051 nm to achieve a stable octahedral coordination in perovskite oxides. The BO3 skeletal sub-lattice can be further stabilised by putting a large A-site cation at the center of the eight BO6 octahedral. The presence of the A-site cation generally distorts the BO3 skeletal sub-lattice while it tries to attain optimal A metal cation–oxygen ion bond length, whereby the lowest radius of the A cation is 0.09 nm.113 When the distortion is very large, non-cubic crystal geometries such as orthorhombic or rhombohedral are preferred. The relationship between the tolerance limits and size of ions are defined as follows:114,115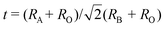 | (4) |
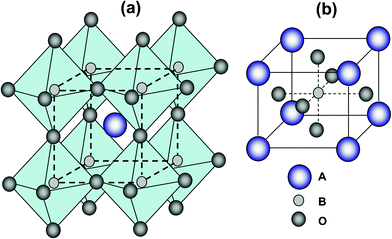 | ||
| Fig. 3 The structure of perovskite ABO3, (a) corner-sharing (BO6) octahedra with A ions located in 12-coordinated interstices, (b) B-site cation at the center of the cell. | ||
Where RA, RB and RO represent the ionic radii of A-site cation (12-coordination), B-site cation (6-coordination) and oxygen ion (1.40 Å), respectively. Ideally, t should be equal to 1 for the cubic structure to form. In practice, the cubic structure exists in between the limits of 0.75 < t < 1.0; particularly when t = 0.8 and 0.9. If the resultant t is lower than 0.9 but larger than 0.75, a cooperative buckling of the corner shared octahedron takes place, leading to orthorhombic distortion. It is obtained by tilting the BO6 octahedra so that the A atoms are displaced along the (110) pseudo-cubic directions or (010) direction. When there is no octahedral buckling, a small deformation from cubic to rhombohedral symmetry might take place. This occurs for t between 0.9 and 1.0. In perovskite oxides, the difference in the ionic radiuses of dopants and outer electronic structure can easily cause the distortion from the ideal cubic structure.
It must be noted that the total charge of A and B cations should be equal to the total charge of the oxygen ions for the electrical neutrality criterion to be maintained. This can be obtained by different means of charge distribution on the form of A1+B5+O3, A2+B4+O3 or A3+B3+O3. For that reason, partial substitution of A and B cation by other cations with different ionic radius size and oxidation state is made possible while maintaining the perovskite structure. A relatively wide range of allowed t values (between 0.75 and 1.0) also means that nonstoichiometry is normally observed in perovskite oxides by the result of either A-site or B-site cation deficiency, oxygen anion deficiency or excess. In the cation deficiency case, the A-site cation may be partially empty without the occurrence of perovskite network collapse due to the support from the BO3 skeletal lattice. In contrast, B-site cation vacancies are not favorable energetically due to the large formal charge and the small size of the B cations in perovskites. As a rule, in perovskite oxides, oxygen vacancy is more easily created and reported than cation deficiency. To some extent, the oxygen vacancy amount is particularly important and can be adjusted to control the oxygen ionic conduction. This can be accomplished for example by doping with ions of similar size but having different valence state. For example, some La3+ ions in LaBO3 can be replaced by Sr2+ to form La1−xSrxBO3−δ which results in the additional formation of oxygen vacancies.16,116,117 When B cations can adopt a mixed-valence state, charge neutrality can be achieved by simultaneous formation of oxygen vacancies and change in the valence state of the B cations. Therefore, perovskite oxides are capable of demonstrating both high oxygen ion conductivity attributed to high oxygen vacancies concentration and high electronic conductivity from mixed-valence state of B-site cations. The concentration of oxygen vacancies can also be increased by partially substituting the original B-site cation with other lower valence state cations such as Cu or Ni ions, which naturally exhibit a divalent oxidation state. In case the valence state of the B cations is fixed, charge neutrality can only be maintained by the formation of oxygen vacancies rendering the oxides being predominantly ionic conductors.
In practical applications, the oxygen permeation performance depends on several factors such as the materials' nature, the membrane thickness and operating conditions. It was firstly reported by Teraoka et al. that membranes based on La1−xSrxCo1−yFeyO3−δ having cubic perovskite structure showed oxygen permeability at elevated temperature.2 Since then, Co-based perovskite oxides have attracted considerable interest as oxygen membranes especially due to their very high oxygen fluxes, although their chemical stability to resist reducing gases and CO2 is low. Nevertheless, Co-free oxides have been pursued intensively since the last decade due to their substantially better stability in long term and harsh condition operation.118,119 At this point, it is worth mentioning that a trade-off occurs between oxygen permeability and stability. On introducing cobalt which is redox reactive, oxygen permeability is improved with stability being compromised. Therefore, in this section, the oxygen permeation performance of both compounds might be of interest for readers which will be expanded in two different subsections.
3.1 Cobalt-containing perovskites
Numerous MIEC materials with high oxygen permeability come from cobalt containing perovskites such as SrCoO3−δ based oxides.120–126 The oxygen permeation fluxes through SrCoO3−δ based membranes are one or two orders of magnitude greater than those through stabilised ZrO2. However, pure SrCoO3−δ does not exhibit cubic structure; instead displays the typical 2-H type hexagonal structure regardless of the preparation methods and operation conditions used. It will experience structure transformation from hexagonal (during which no oxygen permeation is observed) to defect cubic at about 900 °C. It has been widely known that partial substitution of A- or B-site cations in cobalt-based perovskite oxides with other metal cations can stabilize the cubic structure to room temperature; thereby improving the oxygen permeability at low temperature. Teraoka et al.2 investigated the oxygen permeation based on La1−xSrxCo1−yFeyO3−δ perovskite systems. They found that the oxygen permeation fluxes constantly increases with increased Sr and Co content in parallel with increasing oxygen vacancy concentration in the oxide. The A-site or B-site doping effect on the oxygen permeability was further studied on cobalt containing perovskite.127,128 For A-site doping on La0.6A0.4Co0.8Fe0.2O3−δ materials with fixed cations at the B-site, the oxygen permeation fluxes decreases in the sequence of Ba > Ca > Sr > Na. For B-site doping on La0.6Sr0.4B0.8Fe0.2O3−δ materials, the oxygen permeation flux decreases in the sequence of Cu > Ni > Co > Fe > Cr > Mn but again with fixed A-site cations.Following this, many investigations on the oxygen permeation through SrCo0.8Fe0.2O3−δmembrane were performed.96,129,130 The permeation results under various temperatures and oxygen partial pressures in conjunction with the modeling results devised that the oxygen permeation rate through 1–2 mm thickness SrCo0.8Fe0.2O3−δoxide is controlled by surface exchange.58 Its very high oxygen permeability mainly results from the high oxygen vacancy concentration as reflected by the oxygen content (3 − δ) between 2.33 and 2.58 under a wide temperature and pressure range from 1173 K and 0.05 kPa to 873 K and 100 kPa. However, high oxygen vacancy concentration also brings excessive free energy into the oxygen vacancy disordered system. To dissipate this extra free energy, the oxygen vacancies tend to associate with each other and the ordered form is generated, leading to the structure transition from cubic to orthorhombic. Therefore, at temperatures less than 1073 K and pressures lower than 10 kPa, oxygen vacancy ordered orthorhombic brownmillerite phase, Sr2Co1.6Fe0.4O5, was observed in the SrCo0.8Fe0.2O3−δoxide.131 The oxygen vacancy ordering not only inhibits the oxygen permeation through membranes but also induces cracks in the membrane due to the internal stress in the membrane as a result of the phase transition and the volume change. To improve the structural stability of SrCo0.8Fe0.2O3−δ and stabilize its cubic structure to lower temperature (<800 °C) and oxygen partial pressure (<10 kPa), partial substitution of Sr2+ at the A-site by other metal ions such as La3+ and Ba2+ has been performed successfully.13,127,128,132
Prado et al.132 reported that the increase of La content in LaxSr1−xCo0.8Fe0.2O3−δ results in the decrease of valence state of Fe and Co ions and the increase of oxygen ion content. At x = 0.4, e.g. represented by La0.4Sr0.6Co0.8Fe0.2O3−δ compound, no brownmillerite phase exist in the oxide; only the cubic perovskite structure was observed under the experimental condition (20 °C < T < 900 °C, 1 × 10−3 kPa < PO2 < 1×10−3 kPa). This study demonstrates that doping a certain amount of La into the A-site can enhance the stability of SrCo0.8Fe0.2O3−δoxide. Limitations however exist in here since increasing La content leads to an apparent decrease in its ionic conductivity. Shao et al.12 developed Ba0.5Sr0.5Co0.8Fe0.2O3−δ on which 50% mole of Sr at the A-site is substituted by Ba. This compound has improved stability and oxygen permeability (with respect to SrCo0.8Fe0.2O3−δ). They also investigated the Ba doping effect at the A-site (at different amounts of Ba) on the oxygen permeation performance of SrCo0.8Fe0.2O3−δ.13 By introducing a proper amount of Ba into SrCo0.8Fe0.2O3−δ, the structural stability of the material was improved while the oxygen permeability was also modified. The optimum Ba substituting content is within the range of x = 0.3–0.5. The introduction of Ba also stabilised the lower oxidation states of B-site metal ions. For example, Co and Fe ions prefer to exist in the form of Co3+ and Fe3+ instead of Co4+ and Fe4+. The increased amount of B-site cations with low valence state is expected to enlarge the tolerance factor (t) which explains why BaxSr1−xCo0.8Fe0.2O3−δ has a stable cubic structure up to room temperature. Accordingly, increasing the concentration of B-site cations with low valence state also enhances the oxygen vacancy concentration, leading to the enhanced oxygen ionic conductivity. McIntosh et al.97 compared the stability and oxygen permeability of Ba0.5Sr0.5Co0.8Fe0.2O3−δ and SrCo0.8Fe0.2O3−δoxide. In contrast with SrCo0.8Fe0.2O3−δ, no oxygen vacancy ordered distribution was observed in Ba0.5Sr0.5Co0.8Fe0.2O3−δoxide. Moreover, the oxygen content of Ba0.5Sr0.5Co0.8Fe0.2O3−δoxide is much higher than that of SrCo0.8Fe0.2O3−δoxide and even higher than that of ordered orthorhombic brownmillerite structure. In addition, Zeng et al.133 reported that the oxygen permeation through the Ba0.5Sr0.5Co0.8Fe0.2O3−δ (BSCF) membrane is controlled by the surface reaction even at the large membrane thickness of 1 mm. To this end, slow surface exchange kinetics on the membrane surface together with high oxygen ionic conductivity on the bulk material results in the large discrepancy between the oxygen partial pressure in the gaseous phase and membrane surface e.g. higher oxygen partial pressure on the membrane surface with respect to oxygen partial pressure in the atmosphere. However, the structural stability of the BSCF is questioned at intermediate temperature. It was reported that the oxygen permeability of the BSCF membrane decreased by 50% after 240 h of operation at 750 °C due to the growth of hexagonal phase.134 The structural instability for BSCF is ascribed to the oxidation of B-site cations which leads to its reduced ionic radius and increased Goldschmidt tolerance factor at intermediate temperature.135 It was further confirmed by electron energy loss spectroscopy that the change in the valence state of cobalt which accounts for 80 mole% of the B-site cation on BSCF at above 500 °C.136 This issue can be alleviated by the partial substitution of Ba in BaCoO3−δ by other cations with a smaller ionic radius such as La and Sr. Through such substitution, the ionic radius discrepancy between the A-site and B-site cations can be reduced so that a lower tolerance factor can be obtained to stabilise the cubic structure.135
Introducing metal ions with a fixed valence state into the B-site can also be resorted to improve the structural stability of membrane. This method is more favorable to preserve high oxygen vacancy concentration in cobalt-containing perovskites and therefore ensuring higher ionic conductivity. Tong et al.137 prepared BaCo0.4Fe0.6−xZrxO3−δ whereby the Fe in the B-site is partially substituted by Zr. Using this series of materials, 2200 h stable operation in the membrane reactor was achieved; highlighting the excellent structural stability of BaCo0.4Fe0.6−xZrxO3−δ. Zeng et al.138 reported that Sc3+ doping into the B-site of SrCoO3−δ can stabilise the cubic structure; making it a promising membrane material for oxygen separation. Nagai et al.125 conducted systematic research on the B-site cation doping onto SrCoO3−δ based mixed conducting oxides. They found that SrCo0.9Nb0.1O3−δ exhibited the best cubic structural stability and the highest oxygen permeation flux of 4.24 ml min cm−2 at 900 °C.
3.2 Cobalt-free perovskites
On current status, the practical application of cobalt-containing perovskite membranes is very limited due to their two main drawbacks. Firstly, they have a very low structure stability. It has been reported that their structure could start to collapse at an oxygen partial pressure as low as only 10−5kPa albeit substantial stabilization effect from dopants. The cobalt ion is redox reactive and therefore very unstable under such a reduced atmosphere; at which the reduction of cobalt ions to metal takes place resulting in the cubic to non-cubic structure transition which deteriorates the oxygen permeability. Secondly, they normally have a high thermal expansion coefficient, thus potentially introducing large stress across the membrane setup and sealing under asymmetric atmospheres and thermal cycling. Cracks might be easily formed, especially under a large oxygen gradient which would result in membrane failure during operation. Recently, a large number of cobalt-free compounds have been developed; most of which are perovskite-type oxides.118,119,139–143 Ishihara et al.139 incorporated transition metals (e.g.Fe or Ni) into LaGaO3−δ based oxides with a high oxygen ionic conductivity and low thermal expansion coefficient. The permeation results showed that the La0.8Sr0.2Ga0.6Fe0.4O3−δmembrane has higher oxygen fluxes than La0.6Sr0.4Co0.2Fe0.8O3−δ under similar operating conditions. Yaremchenko et al.140 hypothesised that B-site doping of LaGaO3−δ based oxides with bivalent cations (e.g.Mg, Ni or Cu) may improve ionic conductivity and more preferable than doping alkaline earth (e.g. Ca, Sr and Ba) into the A-site. They found that partial filling of the B-site cation by Mg leads to an increase in both electronic and oxygen ionic conductivity due to an increase in the average valence state of the transition metal ions and the oxygen vacancies concentration, respectively. Zhu et al.119 developed cobalt-free membrane based on BaCexFe1−xO3−δ, which contains two phases, a Ce-rich phase and an Fe-rich phase; with only the latter phase favorable for the oxygen transport. Wang et al.118 synthesized cobalt-free membrane material of Ba0.5Sr0.5Zn0.2Fe0.8O3−δ based on that fact that the substituted SrFeO3−δ has mixed oxygen ionic and electronic conductivity. This material exhibits high oxygen-permeation fluxes and good stability at high temperature. Efimov et al.141 developed a Ba0.5Sr0.5Fe0.8Cu0.2O3−δmembrane with a cubic structure. This material demonstrates good phase stability at high and intermediate temperatures and higher oxygen permeability in comparison to other Fe-based perovskite membranes. The oxygen fluxes of the Ba0.5Sr0.5Fe0.8Cu0.2O3−δmembrane were maintained at a stable value for 200 h at 1023 K. However, the main limitation of cobalt-free perovskite oxides lies in their substantially lower oxygen permeation fluxes (with respect to cobalt-containing perovskites). The condensed list of oxygen fluxes values through cobalt-containing and cobalt-free perovskite MIEC membranes as reported in literatures are listed in Table 1.| Materials | Temperature (°C) | J O2 × 106 (mol cm−2s−1) | Thickness/mm | Reference |
|---|---|---|---|---|
| Cobalt-containing | ||||
| SrCo0.8Fe0.2O3−δ | 850 | 0.1738 | 1 | 129 |
| La0.6Sr0.4CoO3−δ | 860 | 0.7649 | 1.5 | 127 |
| La0.6Sr0.4Co0.2Fe0.8O3−δ | 1000 | 0.0253 | 1 | 2 |
| La0.6Sr0.4Co0.8Fe0.2O3−δ | 860 | 0.4591 | 1.5 | 127 |
| La0.6Sr0.4Co0.8Mn0.8O3−δ | 860 | 0.372 | 1.5 | 127 |
| La0.6Sr0.4Co0.8Ni0.2O3−δ | 860 | 1.076 | 1.5 | 127 |
| La0.8Sr0.2Co0.3Ga0.7O3−δ | 700 | 0.233 | 0.5 | 130 |
| Ba0.5Sr0.5Co0.8Fe0.2O3−δ | 900 | 1.563 | 1.8 | 13 |
| Ba0.5Sr0.5Co0.8Fe0.2O3−δ | 950 | 3.266 | 0.22 | 9 |
| BaBi0.4Co0.2Fe0.4O3−δ | 925 | 0.599 | 1.5 | 12 |
| SrCo0.9Nb0.1O3−δ | 900 | 3.155 | 1 | 125 |
| SrCo0.8Sc0.2O3−δ | 900 | 2.306 | 1 | 123 |
| SrCo0.9Ti0.1O3−δ | 900 | 1.36 | 0.65 | 122 |
| Cobalt-free | ||||
| La0.8Sr0.2Fe0.3Ga0.7O3−δ | 700 | 0.438 | 0.5 | 139 |
| La0.8Sr0.2Ni0.3Ga0.7O3−δ | 700 | 0.261 | 0.5 | 139 |
| LaGa0.5Ni0.5O3−δ | 950 | 0.066 | 1 | 140 |
| BaCe0.15Fe0.85O3−δ | 900 | 0.311 | 1 | 119 |
| Ba0.5Sr0.5Zn0.2Fe0.8O3−δ | 950 | 0.260 | 1.45 | 118 |
| Ba0.5Sr0.5Cu0.8Fe0.2O3−δ | 850 | 0.393 | 1 | 141 |
3.3 Selection of perovskite for oxygen permeation membrane
Although many perovskite MIEC membrane materials have been investigated and reported; only very few materials meet the requirements for practical application. First thing to be considered is the oxygen permeability. Current target for economic feasibility is 10 cm3 cm−2 min−1.144 Secondly, phase and structural stability is also crucial. Particularly when the membrane is utilized for partial oxidation or coupling reaction, it is subjected to large oxygen partial pressure gradient attributed to the exposure of one membrane surface to very low oxygen partial pressure environment (∼10−19 atm) e.g. when hydrogen, carbon monoxide or methane is introduced.In this case, developing and selecting the materials with high permeability, stable structure in long term practical operation, cheap cost and sufficient mechanical strength are of interest. Unfortunately, there is no straightforward theory for this. Empirical rules and analogies have been largely used to guide research. Additionally, a particular material exerts MIEC property only at specific temperatures and oxygen partial pressures regime. Thus, a good material at one set of conditions might not be a good material at different set of conditions.
Several available empirical relationships between oxygen ionic transport and perovskite oxide crystal structure include the average metal–oxygen bond energy, the lattice opening degree, the saddle point formed by two A- and one B-site; all of which were reported as promising methods to predict potential perovskite materials having high ionic conductivity and low activation energy for oxygen transport.145–147 Additionally, several studies on the stability of perovskites under reducing atmospheres showed that the properties of long-range disordered oxygen vacancies in lattice is largely affected by the binding energy of oxygen–metal ion.
3.3.1.1 Oxygen migration. As an oxygen ion conductor, two basic conditions should be met: firstly, a certain amount of mobile ions; secondly, passage for the transport of oxygen ions. It is known that oxygen permeability of the membrane is closely related to its electrical conductivity. In perovskite MIEC materials, the electronic conductivity is usually much higher than the oxygen ionic conductivity. Therefore, ionic conductivity is the primary variable to enhance the oxygen permeation fluxes expressed as:145
| σiT = A × exp(−Ea/kT) | (5) |
Where T is the absolute T/K, Ea is the sum of activation energy of oxygen mobility and defect formation energy (J mol−1), A is an exponential factor. To a certain temperature, increasing the A value or decreasing the Ea value improves the oxygen ionic conductivity substantially. Neglecting the change of carrier concentration at various temperatures, the exponential factor A can be written as:
| A = Cγ(Z2e2/k)ao2νoexp(ΔSm/k) | (6) |
Where C is the concentration of charge carriers, γ is the geometric factor. Z·e stands for the charge carrier number, ao is the jump distance, νo is the jump odds. Generally, Ea is equal to ΔHm (ΔHm for migration enthalpy). C can be determined from the concentration of oxygen vacancies which is exchanged with oxygen ions, namely,
| C = No[V..o](1 − V..o) | (7) |
N o is the number of the oxygen ions per unit volume, [V..o] is the oxygen vacancy concentration. When [V..o] is small, it is reasonable to assume C ≈ No[V..o]. The increase in oxygen vacancy concentration results in the increase of the exponential factor A. Consequently, the oxygen ionic conductivity is enhanced. This theory is based on the assumption that the oxygen vacancies act as the free carrier, without any association between them. However, in most cases, especially at low temperatures, the relationship between the oxygen vacancy concentration and charge carriers concentration is not proportional. Moreover, the association will lead to the increase of oxygen migration energy, which causes the decline of oxygen ionic conductivity. Eqn (5) implies that the oxygen permeability improvement can be achieved by lowering the activation energy. The oxygen migration activation energy (Ea) has a very close relationship with cavity size (rc), lattice free volume (Fv) and metal–oxygen average bond energy (ABE).146,147
During oxygen ion migration, oxygen ions need to go through the narrowest space called the saddle point, which consists of one B cation and two A cations with definition as:
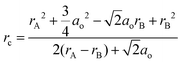 | (8) |
Where rc is the critical radius, rA and rB are the A and B cation radii, respectively. ao is the crystal parameter. rc is usually less than 1.10 Å, which is smaller than the size of the oxygen ion (1.40 Å). Therefore, the relaxation effect happens when the oxygen ions go through the cavity. A substantial amount of energy is consumed in this migration which greatly contributes to the enhanced oxygen ion migration.
The lattice free volume is the difference between the lattice volume and the total volume of ions that constitute the crystal; which is defined as:
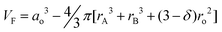 | (9) |
where ro is the oxygen ionic radius (1.40 Å). Eqn (9) shows that the lattice free volume increases with increasing average radius of A-site and B-site cations. Larger lattice free volume provides lower oxygen ion migration energy. Since rc is normally less than the size of oxygen ions, it is expected that increasing the crystal free volume is more beneficial to enhance oxygen ionic transport.
In perovskite oxides, the formation of oxygen vacancies is closely related with A–O and B–O bond strength. To obtain high oxygen ionic conductivity, elemental composition with low average binding energy (ABE) should be selected. ABE affects both oxygen surface exchange kinetics and the oxygen ionic bulk diffusion. The ABE value can be calculated as follows:
| ABE = {[ΔHf°(AmOn) − mΔH°A − D(O2)/n]/12 m} + {[ΔHf° (Bm′On′) − m′ΔH°B − D(O2)/n′]/6 m′} | (10) |
Where ΔHf°(AmOn) and ΔHf° (Bm′On′) are the standard formation enthalpy of the oxides and ΔH°A and ΔH°B are the sublimation energy of the elementary substance, D(O2) is the decomposition energy of oxygen.
3.3.1.2 Stability. Stable structure at a wide range of temperature and oxygen partial pressure is required for practical use. Structure transformation usually leads to a dramatic change in the oxygen permeability and lattice volume; causing substantial internal stress with subsequent crack of membrane. The structure stability is closely related with the properties of its element components such as ion radius, ion valence and the reactivity of the related-metal.
Perovskite oxides require different cations to be incorporated into their A- and B-sites to form stable cubic structure. A study on the influence of A-site cations shows that the structural stability of LnCoO3−δ under a reducing atmosphere increases with the increasing of ionic radius of lanthanides because of the 12-coordinated structure of the large size Ln ions.148 Furthermore, the valence state of the metal ions doped into the A-site of the perovskite affects not only the defect formation but also the structural stability. For example, increasing the amount of Sr doping in LaCoO3−δ brought about increasing oxygen vacancies and Co4+ concentrations and the respective enhanced oxygen bulk diffusion with negative effect on the structural stability. On the other hand, when a cation with a high valence state is introduced into the A-site of LaCoO3−δ, the structure stability will be improved due to the increased amount of Co2+ content. The properties of the B-site cations also have a significant impact on the structural stability. The valence of B-site cation, in particular dominantly determines the structural stability. Generally, the cubic to non-cubic structure transition is caused by the valence state transition of the B-site cations from high to low valence. To this end, suppressing the valence change of the B-site cations would suppress the structure transition. For example, several perovskite oxides with stable valence B-site cations such as Ti4+, Cr3+, Sc3+ and Nb5+ possess stable cubic structures.122,125,126, 138,140 Nevertheless, the oxygen permeability is compromised due to the high oxygen migration energy in these perovskite oxides.
From the ionic radius point of view, a large ionic radius for the B-site cation is beneficial to preserve the cubic structure. This can be rationalised in terms of the stress reduction inside the lattice structure attributable to the mismatch between the size of the A-site and B-site cations. Thus, the tolerance factor (t) has been largely used as a guideline to correlate the structure formation with the size of the A and B cations. Several studies are available that explain the stabilisation of the cubic perovskite using the tolerance factor adjustment by doping with different cations.13,42 However, it is generally difficult to pinpoint the exact effect of cation combination incorporated in the perovskite by calculating the tolerance factor since the calculated tolerance factor differs substantially depending on the assumed ionic radius of the B-site cation which in turn, is governed by the particular valence and spin state adopted by the B-site cation.
Besides cation radius, the valence state of the cations also plays an important role on the stability of the perovskite oxides, especially for the B-site cations. The distance between the B-site cations with a relatively high valence state is shorter in the hexagonal 2H-type structure than in the cubic structure since the hexagonal structure has face-shared oxygen while the cubic structure has corner-shared ones. The large electrostatic repulsion between the B-site cations from increasing the average valence state of the B-site cation can make the hexagonal structure more unstable than the cubic structure. It has been observed that Zr4+, Ti4+, Nb5+ doped perovskite oxides demonstrate excellent stability.122,125,126 Thus, B-site cations with a high valence state are preferred for cubic structure stability.
3.3.1.3 Guidelines. Previous sub-sections discuss several important parameters and approximate rules for the development of perovskite MIEC membrane materials. Important guidelines are summarised below, namely:
1. To maintain the perovskite structure, the tolerance factor of the selected metal cations should be ranged between 0.75–1.0, particularly around 1.0 for the high electronic conductivity, ionic conductivity and structural stability at the practical conditions.
2. The ionic radius of the A- and B-site cations should ideally be as large as possible. This is to maximise the lattice free volume for facilitating mobile oxygen transport. Moreover, at elevated temperature, the thermal reduction of the B-site multi-valence cation and the appearance of oxygen vacancies generally induce the expansion of structure and lattice volume, which may result in the distortion of crystal structure. Large lattice volumes resulted from the large size of A- and B-site cations would suppress the structure transition because large lattice volume gained from large size of A- and B-site cations lead to larger tolerance factor (compared with smaller lattice volume) and larger resistance to structure transition due to lattice volume change. Also, in the presence of oxygen vacancies, the electrostatic repulsion between cations in the lattice needs to be considered which tends to be unfavourable towards structure stability. With increasing lattice volume, this repulsion is reduced as a result of low mutual exposure and long distance between cations. Moreover, the elements selected in the perovskite oxides should be with the low ABE to decrease the migration activation energy of the oxygen ions for the high ionic conductivity.
3. The valence state of the A-site cations should be low, e.g. divalent (+2) to enhance the oxygen ionic conductivity by increasing the oxygen vacancy concentration and the exponential factor A. On the other hand, it should be noticed that the ionic conductivity would be decreased because of the oxygen vacancy association and ordering when the oxygen vacancy concentration increases to some extent. Therefore, A-site cations should be selected carefully.
4. Two type of cations should ideally be incorporated in B-site cation; one which is redox reactive (transition metal ion) and another one which has a fixed and high valence stable to counteract the redox effect from the first cation. It is preferred that the amount of second cation is low to avoid substantial degradation in electronic conductivity. While the valence change of the B-site cation is important for the oxygen vacancies creation at elevated temperature as well as the electronic conductivity, the valence change might be detrimental towards structural stability since the ionic radius change promoted by the valence state change bring might lead to structure distortion.
5. Considering the positive effects of A-site cation ordering on the improvement of oxygen transport, the development of new perovskite membranes with improved morphology, where the individual crystals can be oriented in one direction parallel to the membrane thickness direction to avoid the problem of the detoured and elongated transport path usually encountered in conventional randomly orientated polycrystalline ceramic membranes, will also be an interesting area. The successful fabrication of such membranes may greatly improve the oxygen permeation flux as compared to the similar membranes prepared from A-site cation ordered duoble perovskite oxides with randomly oriented crystalines. The available physical deposition method may be a nice technique to fabricate such thin membranes with special crystalline orientation
4. Application of mixed conducting membranes for clean energy delivery
Intense research and development on ceramic MIEC membranes has brought renewed interest and increasing attention especially to realise their great potential for clean energy applications.54,149–151 Today, CO2 emission due to the global coal-fire power plant is over 2 billion tons per year. Severe global climate change is challenging the traditional power generation system.152CO2 capture technology is highly required and can be achieved by retrofitting the existing energy infrastructure to sustain the fossil fuel usage without contributing emission. Among the three strategies suggested (oxyfuel, pre- or post-combustion capture), oxyfuel combustion is more competitive because of the relatively less technological requirement to achieve almost zero emission and the high flexibility of retrofitting to the existing power stations. However, current tonnage of O2 production by cryogenic process is expensive and energy intensive. Coupling a cryogenic air separation unit at the front end of a coal gasifier or oxy-fuel power plant will reduce the overall efficiency of a power plant from around 40% to 30%,153,154 which can only be used in the demonstration phase. For a long term profitable operation, new cost-effective oxygen production methods must be adopted to replace the cryogenic method because of its lower energy efficiency. MIEC properties in these membranes at high temperature particularly, allow for their use in high temperature conditions in fossil fuel combustion on power plants, fossil fuel gasification or liquefaction. For oxy-fuel technology, an O2–CO2 recycle combustion process is applied in which highly pure oxygen (95% or higher) is fed to the combustion chamber and a major part of the CO2 (∼80%) rich exhaust gas is recycled back to maintain the combustion temperature at a permissible level. Instead of using air as oxidant gas; a O2–CO2 mixture is used which makes the flue gas contain mainly CO2 and small amounts of acid such as SOx and NOx. By integrating MIEC membranes into the air separation unit, the stream of recycled CO2 (from the combustion process) can be utilized as a sweep gas to carry the permeated oxygen from the membrane back to the combustion chamber. Compared with the conventional cryogenic distillation to separate O2, this advanced technology has been predicted to reduce O2 production costs by 35% or more.155 When integrated into oxyfuel power plants, this cost-effective method to supply O2 will significantly cut the energy penalty by 50% for CO2 capture reflected by the simplicity in process design and lower operation cost and heat integration opportunities.156For fossil fuel gasification or liquefaction such as coal gasification and natural gas conversion (syngas or C2hydrocarbons), perovskite membrane reactors also offer a promising future, since by coupling the MIEC membranes as membrane and reactor; air can be directly used as the oxidant allowing oxygen separation and catalytic oxidation in one process. Furthermore, the heat generated from the reforming reaction can sustain the elevated temperature required for oxygen permeation. Such coupling thus simplifies the process and a reduced production cost can be projected. Another significant driving factor comes from the environmental perspective with the large interest to convert natural fossil fuel to high-grade and high purity fuels using membrane reactors instead of firing the fossil fuel directly and venting or flaring the resultant natural gas which would contribute to global warming. Overall, MIEC membranes are ideal for clean energy delivery. Yet, the structural and mechanical instability of perovskite oxides in the presence of CO2 or SO2 still remains the primary limiting factor for their wide scale application.
5. Summary and future development
Principles, advantages, progress and application problems of all kinds of dense ceramic membranes for air separation in the literature have been reviewed with emphasis on the discussion and exploration of perovskite material selection. The prospect of developing oxygen selective MIEC membrane materials with targeted transport properties to render this technology economically competitive seems promising. Still, performance improvement and cost reduction are required to meet the practical operation condition and commercialisation purpose. The approaches of current research works have been focused on manufacturing an asymmetric membrane with a porous thick substrate and a dense thin film layer, developing dual-phase membrane or membrane surface modification. Although major improvements have been obvious, the exploration into new perovskite materials are still under way due to the very limited number of perovskite oxide compositions showing both good structure stability and oxygen permeability. Below are our recommendations for future work in this area.i) Molecular dynamic simulation
Most past and current research has been heavily based on the empirical rules and analogy on which random introduction of different metal cations into the A- and B-sites of the perovskite oxides was performed. To this end, molecular dynamic simulations might be a potentially good alternative to explain the oxygen transport properties of perovskite MIEC membranes.157,158 To accurately describe the atomic interaction and bond breakage/formation associated with oxygen diffusion and surface reactions, first principle calculations are highly beneficial. In recent years, density functional theory (DFT) has been widely employed to study the mobility and permeation of oxygen in various oxides, like CeO2,159BiFeO3,160La2NiO4+δ,161SrMO2.5 (M = Fe, Co),162CeMxO2−x (M = La, Lu, Y),163 BaCo0.7Fe0.3−xNbxO3−δ,164etc. A primary function of such computations is to clarify the electronic structures of complex oxides, particularly the electronic states close to the Fermi level, which play the dominant role in oxygen adsorption and dissociation. In addition, computational calculations are employed to understand the effect of defects on oxygen mobility and surface permeation. One of the central topics is oxygen vacancy, which has been widely found in complex oxides and may play a critical role for surface reactions involved in O2 production. However, pure DFT has its limitations in the description of oxygen vacancies in those oxides, and thus advanced theoretical treatments, like DFT + U165 and hybrid DFT,166 are essential, which bring significant computational cost and are very expensive for complex oxides. Consequently one of the challenges for theoretical calculation and modeling is the description of oxygen vacancy and its role. In addition, it may provide some clues and even guidelines towards quantitative selection of A-site and B-site cations in perovskite oxides.ii) New perovskite membrane materials with improved structural phase stability
Several perovskite materials are available which have been reported to retain their cubic structure during hundreds of hours of oxygen permeation.144,167,168 Yet, this performance lifetime is inferior to membranes used in an oxygen ionic conductor configuration which can be operated continuously without observed degradation for months and even years. At this time, another limitation is the relatively low poisoning resistance of perovskite oxides to acidic gas such as sulfur dioxide and carbon dioxide. The understanding of the interactions of acidic gas and perovskite oxides (primarily surface) is very limited and warrants further research. An innovative concept is currently being pursued intensively by European researchers in developing a very thin dense perovskite layer on top of thick porous substrate.169 This concept is very promising as a very stable perovskite composition is chosen to ensure long lifetime operation whilst a very thin film is engineered to obtain very high flux. Additionally, the mechanical support is provided by the porous substrate. The successful development of this strategy is still in process and requires more intense investigation on the proper coating and advanced fabrication techniques such as physical or chemical vapor deposition techniques to achieve defect-free thin film layers. The thermo–mechanical aspects of these asymmetric membranes cannot be overlooked as well as it forms the crucial part of their practical operation. Therefore, the development of perovskite membrane materials featuring excellent structure stability whilst maintaining high ionic and electronic conductivity still serves as an important future challenge.iii) Ion conducting ceramic membranes with external short circuit for oxygen separation
Summarising all the results of currently developed MIEC ceramic membranes for O2 separation, we can see that well qualified ceramic membrane addressing all the application criteria is rarely encountered. There is always a balance between the chemical stability and oxygen flux with improvement in one property but lowering of the other one. In order to get out from this dilemma, very recently, Zhang et al.170 have put forward a new membrane concept consisting of robust oxygen ion conducting ceramic membrane (i.e., fluorite-based SDC) with two surface platinum coating layers together with external short circuit as schematically illustrated in Fig. 4a. In this concept, the mixed conducting function can be realised via the oxygen ion diffusion inside the fluorite bulk and electronic conduction along the external metal wire avoiding the mutual obstruction often resulted from the conventional dual-phase membrane synthesised by powder mixing. When silver paste is used to seal the ceramic membranes between the two different gas chambers, the external wire is no longer required as the electronic conduction can be completed via the silver sealing as long as silver sealing connects with the coated porous metal layer as displayed in Fig. 4b. This new membrane concept has been verified by the direct detection and measurement of electrical currents which agreed very well with the theoretical calculation based on the equation correlating Faraday constant and oxygen flux. Given that the high O2 fluxes, the intermediate operating temperature and the well-known high stability of the fluorite membrane structure, this new membrane concept is possibly making a breakthrough in the field of oxygen production for clean energy delivery.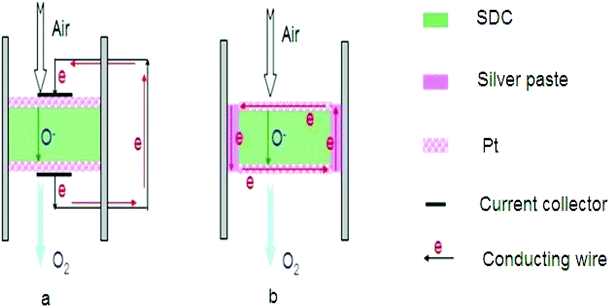 | ||
| Fig. 4 The schematic of the new membrane concept. | ||
Acknowledgements
The authors gratefully acknowledge the financial supports from the Australian Research Council (DP0878849 and DP0985578) and the "National Science Foundation for Distinguished Young Scholars of China” under contract number of 51025209.References
- R. M. Thorogood, Gas Sep. Purif., 1991, 5, 83–94 CrossRef CAS.
- Y. Teraoka, H. M. Zhang, S. Furukawa and N. Yamazoe, Chem. Lett., 1985,(11), 1743–1746 CrossRef CAS.
- Y. S. Lin, W. Wang and J. Han, AIChE J., 1994, 40, 786–798 CrossRef CAS.
- D. P. Fagg, I. P. Marozau, A. L. Shaula, V. V. Kharton and J. R. Frade, J. Solid State Chem., 2006, 179, 3347–3356 CrossRef CAS.
- D. P. Fagg, A. L. Shaula, V. V. Kharton and J. R. Frade, J. Membr. Sci., 2007, 299, 1–7 CrossRef CAS.
- H. J. M. Bouwmeester, H. Kruidhof, A. J. Burggraaf and P. J. Gellings, Solid State Ionics, 1992, 53–56, 460–468 CrossRef CAS.
- Y. Zeng and Y. S. Lin, J. Catal., 2000, 193, 58–64 CrossRef CAS.
- S. Liu, X. Tan, Z. Shao and J. C. Diniz da Costa, AIChE J., 2006, 52, 3452–3461 CrossRef CAS.
- S. Liu and G. R. Gavalas, J. Membr. Sci., 2005, 246, 103–108 CrossRef CAS.
- J. E. ten Elshof, H. J. M. Bouwmeester and H. Verweij, Solid State Ionics, 1995, 81, 97–109 CrossRef CAS.
- J. E. ten Elshof, H. J. M. Bouwmeester and H. Verweij, Appl. Catal., A, 1995, 130, 195–212 CrossRef CAS.
- Z. P. Shao, W. S. Yang, Y. Cong, H. Dong, J. H. Tong and G. X. Xiong, J. Membr. Sci., 2000, 172, 177–188 CrossRef CAS.
- Z. P. Shao, G. X. Xiong, J. H. Tong, H. Dong and W. S. Yang, Sep. Purif. Technol., 2001, 25, 419–429 CrossRef CAS.
- V. V. Kharton, A. P. Viskup, E. N. Naumovich and N. M. Lapchuk, Solid State Ionics, 1997, 104, 67–78 CrossRef CAS.
- V. V. Kharton, A. V. Kovalevsky, V. N. Tikhonovich, E. N. Naumovich and A. P. Viskup, Solid State Ionics, 1998, 110, 53–60 CrossRef CAS.
- J. W. Stevenson, T. R. Armstrong, R. D. Carmeim, L. R. Pederson and L. R. Weber, J. Electrochem. Soc., 1996, 143, 2722–2729 CrossRef CAS.
- J. F. Vente, W. G. Haije and Z. S. Rak, J. Membr. Sci., 2006, 276, 178–184 CrossRef CAS.
- S. P. S. Badwal and F. T. Ciacchi, Adv. Mater., 2001, 13, 993–996 CrossRef CAS.
- U. Balachandran and B. Ma, J. Solid State Electrochem., 2006, 10, 617–624 CrossRef CAS.
- D. Bayraktar, S. Diethelm, T. Graule, J. V. Herle and P. Holtappels, J. Electroceram., 2009, 22, 55–60 CrossRef CAS.
- X. Chen, H. Liu, Y. Wei, J. Caro and H. Wang, J. Alloys Compd., 2009, 484, 386–389 CrossRef CAS.
- J. Sunarso, J. Motuzas, S. Liu and J. C. Diniz da Costa, J. Membr. Sci., 2010, 361, 120–125 CrossRef CAS.
- C. S. Chen, S. Ran, W. Liu, P. H. Yang, D. K. Peng and H. J. M. Bouwmeester, Angew. Chem., Int. Ed., 2001, 40, 784–786 CrossRef CAS.
- Y. Cheng, H. Zhao, D. Teng, F. Li, X. Lu and W. Ding, J. Membr. Sci., 2008, 322, 484–490 CrossRef CAS.
- Q. Li, X. Zhu, Y. Hea and W. Yang, Sep. Purif. Technol., 2010, 73, 38–43 CrossRef CAS.
- X. Dong, G. Zhang, Z. Liu, Z. Zhong, W. Jin and N. Xu, J. Membr. Sci., 2009, 340, 141–147 CrossRef CAS.
- X. Zhu, S. Sun, Y. Cong and W. Yang, J. Membr. Sci., 2009, 345, 47–52 CrossRef CAS.
- J. Kniep and Y. S. Lin, Ind. Eng. Chem. Res., 2010, 49, 2768–2774 CrossRef CAS.
- K. Watenabe, M. Yuasa, T. Kida, Y. Teraoka, N. Yamazoe and K. Shimanoe, Adv. Mater., 2010, 22, 2367–2370 CrossRef CAS.
- M. A. Peña and J. L. G. Fierro, Chem. Rev., 2001, 101, 1981–2017 CrossRef.
- S. M. Hashim, A. R. Mohamed and S. Bhatia, Adv. Colloid Interface Sci., 2010, 160, 88–100 CrossRef CAS.
- H. J. M. Bouwmeester and A. J. Burggraaf, Membr. Sci. Technol., 1996, 4, 435–528 CrossRef CAS.
- A. F. Sammells and M. V. Mundschau, Nonporous inorganic membranes for chemical processing, Weinheim, Willey-VCH, 2006 Search PubMed.
- J. Sunarso, S. Baumann, J. M. Serra, W. A. Meulenberg, S. Liu, Y. S. Lin and J. C. Diniz da Costa, J. Membr. Sci., 2008, 320, 13–41 CrossRef CAS.
- W. Yang, H. Wang, X. Zhu and L. Lin, Top. Catal., 2005, 35, 155–167 CrossRef CAS.
- Y. Liu, X. Tan and K. Li, Catal. Rev. Sci. Eng., 2006, 48, 145–198 CAS.
- D. Eng and M. Stoukides, Catal. Rev. Sci. Eng., 1991, 33, 375–412 CAS.
- M. Stoukides, Catal. Rev. Sci. Eng., 2000, 42, 1–70 CAS.
- V. V. Kharton, A. A. Yaremchenko, A. V. Kovalevsky, A. P. Viskup, E. N. Naumovich and P. F. Kerko, J. Membr. Sci., 1999, 163, 307–317 CrossRef CAS.
- Y. Zeng and Y. S. Lin, J. Membr. Sci., 1998, 150, 87–98 CrossRef CAS.
- B. C. H. Steele, Curr. Opin. Solid State Mater. Sci., 1996, 1, 684–691 CrossRef CAS.
- S. J. Xu and W. J. Thomson, AIChE J., 1997, 43, 2731–2740 CrossRef CAS.
- W. Wang and Y. S. Lin, J. Membr. Sci., 1995, 103, 219–233 CrossRef CAS.
- T. Nozaki and K. Fujimoto, AIChE J., 1994, 40, 870–877 CrossRef CAS.
- F. T. Akin and Y. S. Lin, J. Membr. Sci., 2002, 209, 457–467 CrossRef CAS.
- J. E. ten Elshof, H. J. M. Bouwmeester and H. Verweij, Appl. Catal., A, 1995, 130, 195–212 CrossRef CAS.
- J. E. ten Elshof, B. A. van Hassel and H. J. M. Bouwmeester, Catal. Today, 1995, 25, 397–402 CrossRef CAS.
- Z. P. Shao, H. Dong, G. Xiong, Y. Cong and W. Yang, J. Membr. Sci., 2001, 183, 181–192 CrossRef CAS.
- U. Balachandran, J. T. Dusek, P. S. Maiya, B. Ma, R. L. Mieville, M. S. Kleefisch and C. A. Udovich, Catal. Today, 1997, 36, 265–272 CrossRef CAS.
- H. Luo, Y. Wei, H. Jiang, W. Yuan, Y. Lv, J. Caro and H. Wang, J. Membr. Sci., 2010, 350, 154–160 CrossRef CAS.
- H. J. M. Bouwmeester, Catal. Today, 2003, 82, 141–150 CrossRef CAS.
- W. Zhu, W. Han, G. Xiong and W. Yang, AIChE J., 2008, 54, 242–248 CrossRef CAS.
- H. Wang, A. Feldhoff, J. Caro, T. Schiestel and S. Werth, AIChE J., 2009, 55, 2657–2664 CrossRef CAS.
- X. Tan, K. Li, A. Thursfield and I. S. Metcalfe, Catal. Today, 2008, 131, 292–304 CrossRef CAS.
- H. Jiang, Z. Cao, S. Schirrmeister, T. Schiestel and J. Caro, Angew. Chem., Int. Ed., 2010, 49, 5656–5660 CrossRef CAS.
- D. M. Smyth, Annu. Rev. Mater. Sci., 1985, 15, 329–357 CrossRef CAS.
- C. Zener, Phys. Rev., 1951, 82, 403–405 CrossRef CAS.
- L. Qiu, T. H. Lee, L.-M. Lie, Y. L. Yang and A. J. Jacobson, Solid State Ionics, 1995, 76, 321–329 CrossRef CAS.
- C. S. Chen, Z. P. Zhang, G. S. Jiang, C. G. Fan and W. Liu, Chem. Mater., 2001, 13, 2797–2800 CrossRef CAS.
- S. J. Xu and W. J. Thomson, Chem. Eng. Sci., 1999, 54, 3839–3850 CrossRef CAS.
- L. Ge, Z. P. Shao, K. Zhang, R. Ran, J. C. Diniz da Costa and S. Liu, AIChE J., 2009, 55, 2603–2613 CrossRef CAS.
- X. Tan and K. Li, AIChE J., 2002, 48, 1469–1477 CrossRef CAS.
- R. Bredesen, F. Mertins and T. Norby, Catal. Today, 2000, 56, 315–324 CrossRef CAS.
- R. J. Chater, S. Carter, J. A. Kilner and B. C. H. Steele, Solid State Ionics, 1992, 53–56, 859–867 CrossRef CAS.
- J. E. ten Elshof, M. H. R. Lankhorst and H. J. M. Boumeester, J. Electrochem. Soc., 1997, 144, 1060–1067 CrossRef.
- J. E. ten Elshof, M. H. R. Lankhorst and J. J. M. Bouwmeester, Solid State Ionics, 1997, 99, 15–22 CrossRef CAS.
- T. H. Lee, Y. L. Yang, A. J. Jacobson, B. Abeles and M. Zhou, Solid State Ionics, 1997, 100, 77–85 CrossRef CAS.
- S. Kim, S. Wang, X. Chen, Y. L. Yang, N. Wu, A. Ignatiev, A. J. Jacobson and B. Abeles, J. Electrochem. Soc., 2000, 147, 2398–2406 CrossRef CAS.
- C. Wagner, Prog. Solid State Chem., 1975, 10, 3–16 CrossRef.
- H. J. M. Bouwmeester, H. Kruidhof and A. J. Burggraaf, Solid State Ionics, 1994, 72, 185–194 CrossRef CAS.
- J. A. Kilner, in 2nd Int. Symp. On Ionics and Mixed Conducting Ceramics, ed. Warrell W. L., Tuller H. L., Electrochem. Soc., New Jersey, 1994, pp 174–189 Search PubMed.
- D. Gao, J. Zhao, W. Zhou, R. Ran and Z. P. Shao, J. Membr. Sci., 2011, 366, 203–211 CrossRef CAS.
- Z. Wang, H. Liu, X. Tan, Y. Jin and S. Liu, J. Membr. Sci., 2009, 345, 65–73 CrossRef CAS.
- T. H. Lee, Y. L. Yang, A. J. Jacobson, B. Abeles and S. Milner, Solid State Ionics, 1997, 100, 87–94 CrossRef CAS.
- V. V. Kharton, A. V. Kovalevsky, A. A. Yaremchenko, F. M. Figueiredo, E. N. Naumovich, A. L. Shaulo and F. M. B. Marques, J. Membr. Sci., 2002, 195, 277–287 CrossRef CAS.
- X. Tan, Z. Wang, H. Liu and S. Liu, J. Membr. Sci., 2008, 324, 128–135 CrossRef CAS.
- X. Tan, Y. Lui and K. Li, Ind. Eng. Chem. Res., 2005, 44, 61–66 CrossRef CAS.
- T. Schiestel, M. Kilgus, S. Peter, K. J. Caspary, H. Wang and J. Caro, J. Membr. Sci., 2005, 258, 1–4 CrossRef CAS.
- K. Li, X. Tan and Y. Lui, J. Membr. Sci., 2006, 272, 1–5 CrossRef CAS.
- X. Tan, Z. Wang, B. Meng, X. Meng and K. Li, J. Membr. Sci., 2010, 352, 189–196 CrossRef CAS.
- A. Leo, S. Smart, S. Liu and J. C. Diniz da Costa, J. Membr. Sci., 2011, 368, 64–68 CrossRef CAS.
- J. Sunarso, S. Liu, Y. S. Lin and J. C. Diniz da Costa, Energy Environ. Sci., 2011, 4, 2516–2519 CAS.
- N. Liu, X. Tan, B. Meng and S. Liu, Sep. Purif. Technol., 2011, 80, 396–401 CrossRef CAS.
- H. Arashi and H. Naito, Solid State Ionics, 1992, 53–56, 431–435 CrossRef CAS.
- C. S. Chen, B. A. Boukamp, H. J. M. Bouwmeester, G. Z. Cao, H. Kruidhof, A. J. A. Winnubst and A. J. Burggraaf, Solid State Ionics, 1995, 76, 23–28 CrossRef CAS.
- A. A. Yaremchenko, V. V. Kharton, M. V. Patrakeev and J. R. Frade, J. Mater. Chem., 2003, 13, 1136–1144 RSC.
- M. A. Daroukh, V. V. Vashook, H. Ullmann, F. Tietz and I. A. Raj, Solid State Ionics, 2003, 158, 141–150 CrossRef.
- V. V. Kharton, A. A. Yaremchenko and E. N. Naumovich, J. Solid State Electrochem., 1999, 3, 303–326 CrossRef CAS.
- B. C. H. Steele, Mater. Sci. Eng., B, 1992, 13, 79–87 CrossRef.
- T. H. Etsell and S. N. Flengas, Chem. Rev., 1970, 70, 339–352 CrossRef CAS.
- Y. Nigara, J. Mizusaki and M. Ishigame, Solid State Ionics, 1995, 79, 208–211 CrossRef CAS.
- Y. Nigara, Y. Kosaka, K. Kawamura, J. Mizusaki and M. Ishigame, Solid State Ionics, 1996, 86–88, 739–744 CrossRef CAS.
- J. Han, Y. Zeng and Y. S. Lin, J. Membr. Sci., 1997, 132, 235–243 CrossRef CAS.
- W. T. A. Harrison, T. H. Lee, Y. L. Yang, D. P. Scarfe, L. M. Liu and A. J. Jacobson, Mater. Res. Bull., 1995, 30, 621–630 CrossRef CAS.
- Z. Q. Deng, W. S. Yang, W. Liu and C. S. Chen, J. Solid State Chem., 2006, 179, 362–369 CrossRef CAS.
- H. Kruidhof, H. J. M. Bouwmeester, R. H.E. v. Doorn and A. J. Burggraaf, Solid State Ionics, 1993, 63–65, 816–822 CrossRef CAS.
- S. McIntosh, J. F. Vente, W. G. Haije, D. H. A. Blank and J. M. Bouwmeester, Solid State Ionics, 2006, 177, 1737–1742 CrossRef CAS.
- A. F. Sammells, M. Schwartz, R. A. Mackay, T. F. Barton and D. R. Peterson, Catal. Today, 2000, 56, 325–328 CrossRef CAS.
- A. F. Sammells, R. L. Cook, J. H. White, J. J. Osborne and R. C. MacDuff, Solid State Ionics, 1992, 52, 111–123 CrossRef CAS.
- M. Schwartz, B. F. Link and A. F. Sammells, J. Electrochem. Soc., 1993, 140, L62–63 CrossRef CAS.
- A. A. Taskin, A. N. Lavrov and Y. Ando, Prog. Solid State Chem., 2007, 35, 481–490 CrossRef CAS.
- A. Tarancon, S. J. Skinner, R. J. Chater, F. H. Ramirez and J. A. Kilner, J. Mater. Chem., 2007, 17, 3175–3181 RSC.
- A. Chang, S. J. Skinner and J. A. Kilner, Solid State Ionics, 2006, 177, 2009–2011 CrossRef CAS.
- K. Zhang, L. Ge, R. Ran, Z. Shao and S. Liu, Acta Mater., 2008, 56, 4876–4889 CrossRef CAS.
- H. Gu, H. Chen, L. Gao and L. Guo, Electrochim. Acta, 2009, 54, 7094–7098 CrossRef CAS.
- Y. Lin, R. Ran, C. Zhang, R. Cai and Z. Shao, J. Phys. Chem. A, 2010, 114, 3764–3772 CrossRef CAS.
- T. J. Mazanec, T. L. Cable and J. G. Frye, Solid State Ionics, 1992, 53–56, 111–118 CrossRef CAS.
- J. Kim and Y. S. Lin, AIChE J., 2000, 46, 1521–1529 CrossRef CAS.
- T. H. Lee, Y. L. Yang and A. J. Jacobson, Solid State Ionics, 2000, 134, 331–339 CrossRef CAS.
- C. S. Chen, H. Kruidhof, H. J. M. Bouwmeester, H. Verweij and A. J. Burggraaf, Solid State Ionics, 1996, 86–88, 569–572 CrossRef CAS.
- V. V. Kharton, A. V. Kovalevsky, A. P. Viskup, F. M. Figueiredo, A. A. Yaremchenko, E. N. Naumovich and F. M. B. Marques, J. Electrochem. Soc., 2000, 147, 2814–2821 CrossRef CAS.
- H. Luo, K. Efimov, H. Jiang, A. Feldhoff, H. Wang and J. Caro, Angew. Chem., Int. Ed., 2011, 50, 759–763 CrossRef CAS.
- L. G. Tejuca, J. L. Fierro and J. M. D. Tascon, Adv. Catal., 1989, 36, 237–328 CrossRef CAS.
- V. M. Goldschmidt, Skrifer Norske Videnskaps. Akad. Oslo I. Mat. Matur. Klasse, 1926, 8, 7–156 Search PubMed.
- N. Ramadass, Mater. Sci. Eng., 1978, 36, 231–239 CrossRef CAS.
- A. V. Kovalevsky, V. V. Kharton, V. N. Tikhonovich, E. N. Naumovich, A. A. Tonoyan, O. P. Reut and L. S. Boginsky, Mater. Sci. Eng., B, 1998, 52, 105–116 CrossRef.
- V. V. Kharton, E. N. Naumovich, P. P. Shuk, A. K. Demin and A. V. Nikolaev, Electrochemistry, 1992, 28, 1693–1702 CAS.
- H. Wang, C. Tablet, A. Feldhoff and J. Caro, Adv. Mater., 2005, 17, 1785–1793 CrossRef CAS.
- X. Zhu, H. Wang and W. Yang, Chem. Commun., 2004,(9), 1130–1131 RSC.
- J. F. Vente, S. McIntosh, W. G. Haije and H. J. M. Bouwmeester, J. Solid State Electrochem., 2006, 10, 581–588 CrossRef CAS.
- S. Diethelm and J. V. Herle, J. Eur. Ceram. Soc., 2004, 24, 1319–1323 CrossRef CAS.
- X. Chen, L. Huang, Y. Wei and H. Wang, J. Membr. Sci., 2011, 368, 159–164 CrossRef CAS.
- P. Y. Zeng, Z. P. Shao, S. Liu and Z. Xu, Sep. Purif. Technol., 2009, 67, 304–311 CrossRef CAS.
- J. M. Kim, G. J. Hwang, S. H. Lee, C. S. Park, J. W. Kim and Y. H. Kim, J. Membr. Sci., 2005, 250, 11–16 CrossRef CAS.
- T. Nagai, W. Ito and T. Sakon, Solid State Ionics, 2007, 177, 3433–3444 CrossRef CAS.
- H. Lu, J. P. Kim, S. H. Son and J. H. Park, Mater. Sci. Eng., B, 2010, 166, 135–140 CrossRef CAS.
- Y. Teraoka, T. Nobunaga and N. Yamazoe, Chem. Lett., 1988,(3), 503–506 CrossRef CAS.
- Y. Teraoka, H. M. Zhang, K. Okamoto and N. Yamazoe, Mater. Res. Bull., 1988, 23, 51–62 CrossRef CAS.
- L. Tan, L. Yang, X. Gu, W. Jin, L. Zhang and N. Xu, J. Membr. Sci., 2004, 230, 21–27 CrossRef CAS.
- A. A. Yaremchenko, E. V. Tsipis, A. V. Kovalevsky, J. C. Waerenborgh and V. V. Kharton, Solid State Ionics, 2011, 192, 259–268 CrossRef CAS.
- S. McIntosh, J. F. Vente, W. G. Haije, D. H. A. Blank and H. J. M. Bouwmeester, Solid State Ionics, 2006, 177, 833–842 CrossRef CAS.
- F. Prado, N. Grunbaum, A. Caneiro and A. Manthiram, Solid State Ionics, 2004, 167, 147–154 CrossRef CAS.
- P. Y. Zeng, Z. H. Chen, W. Zhou, H. X. Gu, Z. P. Shao and S. M. Liu, J. Membr. Sci., 2007, 291, 148–156 CrossRef CAS.
- K. Efimov, Q. Xu and A. Feldhoff, Chem. Mater., 2010, 22, 5866–5875 CrossRef CAS.
- S. Švarcová, K. Wiik, J. Tolchard, H. Bouwmeester and T. Grande, Solid State Ionics, 2008, 178, 1787–1791 CrossRef.
- M. Arnold, H. Wang and A. Feldhoff, J. Membr. Sci., 2007, 293, 44–52 CrossRef CAS.
- J. Tong, W. Yang, B. Zhu and R. Cai, J. Membr. Sci., 2002, 203, 175–189 CrossRef CAS.
- P. Y. Zeng, R. Ran, Z. H. Chen, W. Zhou, H. X. Gu, Z. P. Shao and S. Liu, J. Alloys Compd., 2008, 455, 465–470 CrossRef CAS.
- T. Ishihara, T. Yamada, H. Arikawa, H. Nishiguchi and Y. Takita, Solid State Ionics, 2000, 135, 631–636 CrossRef CAS.
- A. A. Yaremchenko, V. V. Kharton, A. P. Viskup, E. N. Naumovich, V. N. Tikhonovich and N. M. Lapchuk, Solid State Ionics, 1999, 120, 65–74 CrossRef CAS.
- K. Efimov, T. Halfer, A. Kuhn, P. Heitjans, J. Caro and A. Feldhoff, Chem. Mater., 2010, 22, 1540–1544 CrossRef CAS.
- K. Watanabe, D. Takauchi, M. Yuasa, T. Kida, K. Shimanoe, Y. Teraoka and N. Yamazoe, J. Electrochem. Soc., 2009, 156, E81–E85 CrossRef CAS.
- Y. Tsuruta, T. Todaka, H. Nisiguchi, T. Ishihara and Y. Takita, Electrochem. Solid-State Lett., 2001, 4, E13–E15 CrossRef CAS.
- P. V. Hendriksen, P. H. Larsen, M. Mogensen, F. W. Poulsen and K. Wiik, Catal. Today, 2000, 56, 283–295 CrossRef CAS.
- K. A. Kilner and J. Brook, Solid State Ionics, 1982, 6, 237–252 CrossRef.
- R. L. Cook and A. F. Sammells, Solid State Ionics, 1991, 45, 311–321 CrossRef CAS.
- R. L. Cook, R. C. Macduff and A. F. Sammells, J. Electrochem. Soc., 1990, 137, 3309–3310 CrossRef CAS.
- T. Arakawa, N. Ohara and J. Shiokawa, J. Mater. Sci., 1986, 21, 1824–1827 CrossRef CAS.
- T. Shimura, M. Itoh and H. Iwahara, J. Appl. Electrochem., 1998, 28, 683–688 CrossRef CAS.
- A. Leo, S. Liu and J. C. Diniz da Costa, Int. J. Greenhouse Gas Control, 2009, 3, 357–365 CrossRef CAS.
- S. Smart, C. X. C. Lin, L. Ding, K. Thambimuthu and J. C. Diniz da Costa, Energy Environ. Sci., 2010, 3, 268–278 CAS.
- K. Brands, D. Uhlmann, S. Smart, M. Bram and J.C. Diniz da Costa, J. Membr. Sci., 2010, 359, 110–114 CrossRef CAS.
- K. Andersson and F. Jhonsson, Energy Convers. Manage., 2006, 47, 3487–3498 CrossRef CAS.
- M. Pehnt and J. Henkel, Int. J. Greenhouse Gas Control, 2009, 3, 49–66 CrossRef CAS.
- G. J. Stiegel, Separations Technology VI: New perspectives on very large scale operations, Fraser Island, Australia, 2004 Search PubMed.
- H. Stadler, F. Beggel, M. Habermehl, B. Persigehl, R. Kneer, M. Modigellaan and P. Jeschke, Int. J. Greenhouse Gas Control, 2011, 5, 7–15 CrossRef CAS.
- C. A. J. Fisher, M. Yoshiya, Y. Iwamoto, J. Ishii, M. Asanuma and K. Yabuta, Solid State Ionics, 2007, 177, 3425–3431 CrossRef CAS.
- C. Wessel, M. W. Lumey and R. Dronskowski, J. Membr. Sci., 2011, 366, 92–96 CrossRef CAS.
- C. Frayret, A. Villesuzanne, M. Pouchard, F. Mauvy, J. Bassat and J. Grenier, J. Phys. Chem. C, 2010, 114, 19062–19070 CAS.
- S. Sæterli, S. M. Selbatch, P. Ravindran, T. Grande and R. Holmestad, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 064102 CrossRef.
- C. Frayret, A. Villesuzanne and M. Pouchard, Chem. Mater., 2005, 17, 6538–6544 CrossRef CAS.
- W. Paulus, H. Schober, S. Eibl, M. Johnson, T. Berthier, O. Hernandez, M. Ceretti, M. Plazanet, K. Conder and C. Lamberti, J. Am. Chem. Soc., 2008, 130, 16080–16085 CrossRef CAS.
- C. Frayret, A. Villesuzanne, M. Pouchard, F. Mauvy, J. Bassat and J. Grenier, J. Phys. Chem. C, 2010, 114, 19062–19076 CAS.
- P. Shen, X. Liu, H. Wang and W. Ding, J. Phys. Chem. C, 2010, 114, 22338–22345 CAS.
- V. Anisimov, J. Zaanen and O. Andersen, Phys. Rev. B: Condens. Matter, 1991, 44, 943–954 CrossRef CAS.
- B. Janesko, T. Henderson and G. Scuseria, Phys. Chem. Chem. Phys., 2009, 11, 443–454 RSC.
- L. Ge, R. Ran, K. Zhang, S. Liu and Z. Shao, J. Membr. Sci., 2008, 318, 182–190 CrossRef CAS.
- W. Ito, T. Nagai and T. Sakon, Solid State Ionics, 2007, 178, 809–816 CrossRef CAS.
- M. Czyperek, P. Zapp, H. J. M. Bouwmeester, M. Modigell, K. Ebert, I. Voigt, W. A. Meulenberg, L. Singheiser and D. Stöver, J. Membr. Sci., 2010, 359, 149–159 CrossRef CAS.
- K. Zhang, Z. Shao, C. Li and S. Liu, Energy Environ. Sci., Search PubMed , in revision.
| This journal is © The Royal Society of Chemistry 2011 |