DOI:
10.1039/C1RA00413A
(Paper)
RSC Adv., 2011,
1, 602-606
Efficient microwave-assisted synthesis of aminoxy acid conjugates†
Received
6th July 2011
, Accepted 6th July 2011
First published on 31st August 2011
Abstract
A representative set of Cbz-protected (α-aminoxyacyl)benzotriazoles was utilized as versatile building blocks for the efficient and convenient synthesis of novel α-aminoxy acid conjugates. Convenient protocols were developed for their regioselective preparation with sterically hindered nucleophiles such as sugars, terpenes, steroids and nucleosides.
Introduction
The clinical development of biologically active compounds is often hindered by undesirable biopharmaceutical properties such as low water solubility, stability, and permeability through biological membranes. Many of these drawbacks can be attenuated by derivatization of the biomolecules, e.g. the preparation of bioconjugates. Thus, amino acid esters of naturally occurring alcohols such as steroids show increased hydrophilicity.1–2 Linking amino acids with hydroxylic terpenes afforded effective medicinal agents, e.g. in the therapy of atherosclerosis.3Sugar-amino acid conjugates offer improved physiochemical and biological properties and allow access to significant diversification.4–8 Some N4 amino acid substituted derivatives of the antitumor active nucleoside analogue 1-β-D-arabinofuranosylcytosine (Ara-C, Cytarabine) showed superior biological activity and bioavailability in comparison with the parent drug.9Valaciclovir, the valine ester of aciclovir, has increased solubility in water and oral bioavailability relative to aciclovir.10
α-Aminoxy acids [RCH(ONH2)CO2H] resist enzymatic degradation and are thus of interest as bioisosteric α-amino acids and as analogs of β-amino acids.11–12 Yang et al. found that peptides including α-aminoxy acid units can adopt rigid secondary foldamer structures of considerable interest as novel analogs of β-peptides (Fig. 1).13
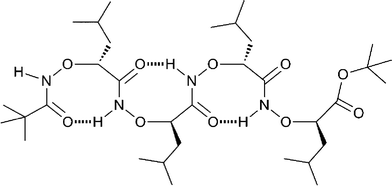 |
| | Fig. 1 Typical helical structure (1.88 helix) of an α-aminoxy peptide. | |
Repulsion between lone pairs electrons on the N and O atoms give rise to torsional characteristics that are distinctive for the N–O bond in aminoxy acids; in particular the backbone of aminoxy peptides is more rigid than that of natural peptides. For D-α-aminoxy peptides, 1.88 helices independent of side chains were observed in peptides that contain as few as two residues.14α-Aminoxy peptides have also facilitated the construction of anion receptors and channels.15
Recently, we utilized (α-aminoxyacyl)benzotriazoles in a mild general method for the preparation of aminoxyacyl amides and aminoxy hybrid peptides,16 showing advantages over the existing literature methods which used (i) coupling reagents including BOP-HOBt-NEM, HBTU-HOBt-NEM, DIC-HOBt,17EDCI-HOBt/HOAt,18TBTU/HOBt/DIEA,19DIC/HOBt;20 (ii) active esters21 or (iii) α-aminoxy diazoketones.22
N-Acylbenzotriazoles are stable solids, easy to handle, advantageous for N-, O-, C- and S-acylation, especially where the corresponding acid chlorides are unstable, toxic or difficult to prepare.23 The synthesis of chiral di-, tri- and tetrapeptides from amino acids using N-protected-(α-aminoacyl)benzotriazoles in solution phase occurs with complete retention of chirality.23
We now describe the convenient and efficient synthesis of novel aminoxy acid conjugates with sterically hindered nucleophiles such as steroids, terpenes, sugars and nucleosides using benzotriazole methodology.
Results and discussion
Synthesis of N-Cbz-protected α-aminoxy acids 3a–d, (3c + 3c′)
The N-Cbz-protected α-aminoxy acids 3a–d, (3c + 3c′) were prepared by first transforming the amino acids 1a–d, (1c + 1c′) into the corresponding α-bromocarboxylic acids 2a–d, (2c + 2c′) (40–80%, Scheme 1). Then, without further purification, 2a–d, (2c + 2c′) were reacted with benzyl hydroxycarbamate in the presence of sodium hydride to afford the desired N-protected α-aminoxy acids 3a–d, (3c + 3c′) (47–61%, Scheme 1).
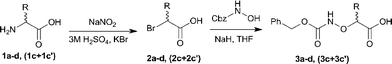 |
| | Scheme 1 Synthesis of N-Cbz-protected (α-aminoxy)carboxylic acids 3a–d, (3c + 3c′). | |
Synthesis of N-Cbz-protected (α-aminoxyacyl)benzotriazoles 4a–d, (4c + 4c′)
The N-Cbz-protected (α-aminoxyacyl)benzotriazoles 4b–d, (4c + 4c′) were prepared in good yields (76–88%) from the corresponding N-Cbz-protected α-aminoxy acids 3a–d, (3c + 3c′) according to our previously published method (Scheme 2, Table 1).16 Key intermediates 4a–d, (4c + 4c′) were characterized by 1H and 13C NMR spectroscopy as well as elemental analysis.
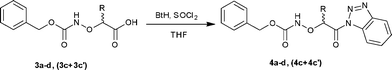 |
| | Scheme 2 Synthesis of N-Cbz-protected (α-aminoxyacyl)benzotriazoles 4a–d, (4c + 4c′). | |
Table 1
N-Cbz-protected (α-aminoxyacyl)benzotriazoles 4a–d, (4c + 4c′) prepared
| Entry |
R |
yield (%) |
mp (°C) |
|
Novel compound.
|
|
4a
|
H- |
76 |
88.0–90.0 |
|
4b
|
D-CH3- |
78 |
88.0–90.0 |
|
4c
|
D-Ph-CH2- |
88a |
90.0–91.0 |
|
(4c + 4c′) |
DL-(CH3)2CHCH2- |
82a |
114.0–116.0 |
|
4d
|
D-(CH3)2CHCH2- |
86a |
oil |
Synthesis of O-(protected-α-aminoxyacyl)sugars 6a–f, (6e + 6e′)
We first studied the preparation of the O-(protected-α-aminoxyacyl)sugars 6a–f, (6e + 6e′). In initial experiments the reaction of the N-Cbz-protected (α-aminoxyacyl)benzotriazoles 4b with 1,2:5,6-di-O-isopropylidene-D-glucose 5a was not complete after stirring for 24 h at 20 °C. However, under microwave irradiation, syntheses of the sugar conjugates 6a–e, (6e + 6e′) were accomplished within 15–30 min. Thus, coupling of the N-Cbz-protected (α-aminoxyacyl)benzotriazoles 4b–d, (4c + 4c′) with 1,2:5,6-di-O-isopropylidene-D-glucose 5a and 1,2:3,4-di-O-isopropylidene-D-galactopyranose 5b in the presence of 4-(N,N-dimethylamino)pyridine (DMAP, 0.1 equiv) at 70 °C and 100 W followed by purificationviacolumn chromatography afforded the desired O-(protected-α-aminoxyacyl)sugars 6a–e, (6e + 6e′) in 60–84% yields (Scheme 3, Table 2). In all cases completion of the reaction was monitored by TLC analysis.
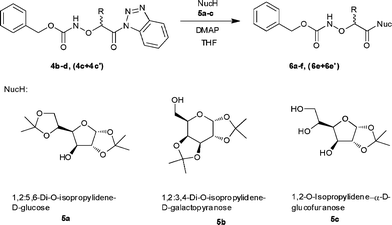 |
| | Scheme 3 Synthesis of O-(protected-α-aminoxyacyl)sugars 6a–f, (6e + 6e′). | |
Table 2
O-(Protected-α-aminoxyacyl)sugars 6a–f, (6e + 6e′) prepared
| Entry |
N-(Cbz-Protected aminoxyacyl)-benzotriazole, 4 |
NucH |
Yield (%) |
|
MW, 70 °C, 100 W, 15 min.
MW, 70 °C, 100 W, 30 min.
MW, 60 °C, 50 W, 30 min.
|
|
6a
|
Cbz-D-AOAla-Bt, 4b |
5a
|
75a |
|
6b
|
Cbz-D-AOAla-Bt, 4b |
5b
|
71a |
|
6c
|
Cbz-D-AOLeu-Bt, 4d |
5a
|
60a |
|
6d
|
Cbz-D-AOPhe-Bt, 4c |
5a
|
68a |
|
6e
|
Cbz-D-AOPhe-Bt,4c |
5b
|
84b |
| (6e + 6e′) |
Cbz-DL-AOPhe-Bt, (4c + 4c′) |
5b
|
65b |
|
6f
|
Cbz-D-AOAla-Bt, 4b |
5c
|
62c |
NMR analysis of 6a–e revealed no detectable racemization. The 1H NMR of 6a–e showed a prominent singlet for the NH proton in the range of 8.18–8.86 ppm. All methyl protons from the diisopropylidene group appeared also as singlets, supporting the absence of any undesired isomers of 6a–e. Similarly, no splitting of peaks were observed in the 13C NMR of 6a–e. In contrast, the 1H and 13C NMR of diastereomeric mixture (6e + 6e′) derived from the racemic mixture (4c + 4c′) showed clearly the presence of two isomers. For example, in 6e the NH proton gave a singlet at 8.18 ppm and the ester moiety a carbon peak at 170.66 ppm whereas the 1H-NMR of the corresponding diastereomeric mixture (6e + 6e′) showed two singlets for the NH proton at 8.14 ppm and 8.21 ppm and the 13C NMR two carbonyl carbon peaks at 170.66 and 170.74 ppm.
In addition, we investigated the O-acylation of the partially protected sugar 1,2-O-isopropylidene-α-D-glucofuranose 5c. In the presence of 0.1 equiv of DMAP the acylating agent 4b was completely consumed after heating the reaction mixture for 30 min at 60 °C under microwave irradiation (50 W) and the product 6f was obtained after purification by column chromatography in 62% yield (Table 2). Compound 5c was acylated exclusively at the primary hydroxyl group and the structure of the acylated product 6f was fully assigned using several 2D NMR techniques (see Figs. S1–S3 in ESI†).
The proton connectivity for the sugar fragment was obtained by 1H–1H gDQCOSY experiment (Fig. S2–S3†), the anomeric proton at δ = 5.79 ppm shows correlation with 4.39 ppm, the other proton chemical shifts on the sugar fragment were assigned based on their cross peaks. The most important correlations that confirm the assignment of the acylation position were seen in the 1H–13C gHMBC experiment (Fig. S1†); the diastereomeric protons at 4.00 and 4.30 ppm show three bond correlations with the carbonyl carbon at 171.8 ppm (important correlations are indicated with a single headed arrow in Fig. 2).
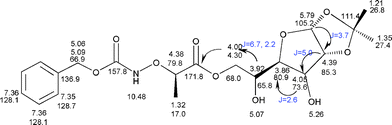 |
| | Fig. 2
1H and 13C Chemical shift assignments of 6f. | |
Synthesis of O-(protected-α-aminoxyacyl)terpenes 8a–c, (8b + 8b′)
O-(protected-α-aminoxyacyl)terpenes 8a–c, (8b + 8b′) were prepared by coupling of the N-Cbz-protected (α-aminoxyacyl)benzotriazoles 4b–d, (4c + 4c′) with hydroxysubstituted terpenes 7a,b in the presence of 4-(N,N-dimethylamino)pyridine (0.1 equiv) under microwave irradiation at 50 °C and 45 W. The crude products were purified by a simple aqueous work-up procedure to afford the desired terpene conjugates 8a–c, (8b + 8b′) as colorless oils in 50–60% yield (Scheme 4, Table 3).
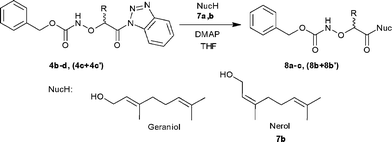 |
| | Scheme 4 Synthesis of O-(protected-α-aminoxyacyl)terpenes 8a–c, (8b + 8b′). | |
Table 3
O-(Protected-α-aminoxyacyl)terpenes 8a–c, (8b + 8b′) prepared
| Entry |
N-(Cbz-Protected aminoxyacyl)-benzotriazole, 4 |
NucH |
Yield (%)a |
|
Optimized reaction conditions: MW, 50 °C, 45 W, 30 min.
|
|
8a
|
Cbz-D-AOAla-Bt, 4b |
7b
|
60 |
|
8b
|
Cbz- D -AOPhe-Bt, 4c |
7a
|
55 |
| (8b + 8b′) |
Cbz- DL-AOPhe-Bt, (4c + 4c′) |
7a
|
54 |
|
8c
|
Cbz-D-AOLeu-Bt, 4d |
7b
|
50 |
Synthesis of O-(protected-α-aminoxyacyl)steroids 10a–d, (10b + 10b′)
The attempted preparation of steroid conjugates 10a–d, (10b + 10b′) at room temperature failed. Thus, we carried out the coupling of 4b–d, (4c + 4c′) with the steroid derivatives 9a–c under microwave irradiation (50 °C, 45 W, 50 min). TLC analysis of the reaction mixture indicated formation of the product, but the reaction did not go to completion and the desired O-(protected-α-aminoxyacyl)steroids 10a–d, (10b + 10b′) were isolated by flash column chromatography in 20–26% yield (Scheme 5, Table 4). All attempts to increase the yield of the coupling reaction by variation of temperature, power and reaction time were unsuccessful.
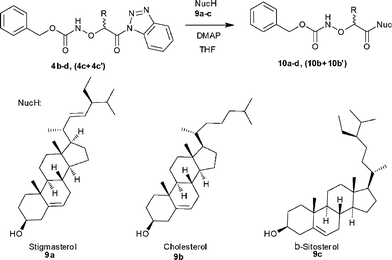 |
| | Scheme 5 Synthesis of O-(protected-α-aminoxyacyl)steroids 10a–d, (10b + 10b′). | |
Table 4
O-(Protected-α-aminoxyacyl)steroids 10a–d, (10b + 10b′) prepared
| Entry |
N-(Cbz-Protected aminoxyacyl)-benzotriazole, 4 |
NucH |
Yield (%)a |
|
Optimized reaction conditions: MW, 50 °C, 45 W, 50 min.
|
|
10a
|
Cbz-D-AOAla-Bt, 4b |
9c
|
22 |
|
10b
|
Cbz-D-AOPhe-Bt, 4c |
9b
|
25 |
| (10b + 10b′) |
Cbz-DL-AOPhe-Bt, (4c + 4c′) |
9b
|
25 |
|
10c
|
Cbz-D-AOPhe-Bt, 4c |
9c
|
26 |
|
10d
|
Cbz-D-AOLeu-Bt, 4d |
9a
|
20 |
Synthesis of N-(protected-α-aminoxyacyl)nucleosides 12a,b
Finally, we utilized the Cbz-protected (α-aminoxyacyl)benzotriazoles 4c,d for the synthesis N-(protected-α-aminoxyacyl)nucleosides 12a,b. The attempted synthesis of 12a,b under microwave irradiation provided diacylated products. Consequently, we repeated these experiments at room temperature and the reaction of 4d with cytidine and 4c with adenosine in DMF provided the corresponding N-acylated products 12a,b regioselectively in 21% and 54% yield, respectively (Scheme 6, Table 5). All products were purified by column chromatography to furnish the corresponding N-(protected-α-aminoxyacyl)nucleosides 12a,b.
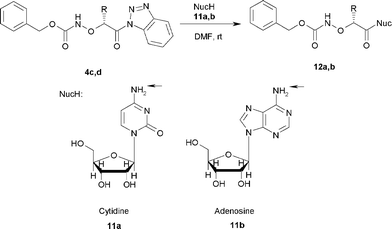 |
| | Scheme 6 Synthesis of N-(protected-α-aminoxyacyl)nucleosides 12a,b. | |
Table 5
N-(Protected-α-aminoxyacyl)nucleosides 12a,b prepared
| Entry |
N-(Cbz-Protected aminoxyacyl)-benzotriazole, 4 |
NucH |
Yield (%) |
|
12a
|
Cbz-D-AOLeu-Bt, 4d |
11a
|
21 |
|
12b
|
Cbz-D-AOPhe-Bt, 4c |
11b
|
54 |
Conclusions
Cbz-protected (α-aminoxyacyl)benzotriazoles 4a–d, (4c + 4c′) are stable, usually crystalline and readily available reagents, which were utilized as building blocks for the convenient preparation of aminoxy acid conjugates with sugars, terpenes, steroids and nucleosides. Our microwave-assisted preparation provided various Cbz-protected aminoxy acid conjugates in moderate to good yields and short reaction times without detectable racemization.
Experimental
General methods
Melting points were determined on a capillary point apparatus equipped with a digital thermometer and are uncorrected. NMR spectra were recorded in CDCl3, DMSO-d6 or CD3OD-d4 on Gemini or Mercury NMR operating at 300 MHz for 1H and 75 MHz for 13C with TMS as an internal standard. Elemental analyses were performed on a Carlo Erba-1106 instrument. All microwave assisted reactions were carried out with a single mode cavity Discover Microwave Synthesizer (CEM Corporation, NC). The reaction mixtures were transferred into a 10 mL glass pressure microwave tube equipped with a magnetic stirrer bar. The tube was closed with a silicon septum and the reaction mixture was subjected to microwave irradiation (Discover mode; run time: 60 s.; PowerMax-cooling mode). The 1H–1H gDQCOSY, 1H–1H NOESY, 1H–13C gHMQC and 1H–13C gHMBC of 6f were recorded on a Varian Inova instrument operating at 500 MHz for 1H, 125 MHz for 13C, equipped with three channels, 5 mm indirect detection probe, with z-axis gradients. The solvent was DMSO-d6 and the temperature was 25 °C. The chemical shifts for 1H and 13C were referenced to DMSO, 2.50 ppm for 1H and 39.5 ppm for 13C.
General procedure for preparation of O-(protected-α-aminoxyacyl)sugars 6a–f, (6e + 6e′)
A solution of the N-Cbz-protected (α-aminoxyacyl)benzotriazoles 4b–d, (4c + 4c′) (0.5 mmol), sugar 5a–c (0.75 mmol) and DMAP (0.05 mmol) in THF (1 mL) was subjected to microwave irradiation (power, temperature and hold time as indicated in Table 2). Each reaction mixture was allowed to cool down to room temperature and transferred to a round bottomed flask. The solvent was evaporated, and the crude product was purified by column chromatography using hexanes:ethyl acetate (gradient) as eluent to afford the O-(protected-α-aminoxyacyl)sugars 6a–f, (6e + 6e′) as analytically pure compounds.
(R)-((3aR,5R,6S,6aR)-5-((R)-2,2-Dimethyl-1,3-dioxolan-4-yl)-2,2-dimethyltetra-hydrofuro[3,2-d][1,3]dioxol-6-yl) 2-(benzyloxycarbonylaminooxy)propanoate (6a)
White microcrystals, mp 94.0–96.0 °C, 75% yield, [α]D21 = +33.2 (c 0.17 in CH2Cl2). Anal. Calcd. for C23H31NO10: C 57.37; H 6.49; N 2.91. Found C 57.23; H 6.64; N 2.76. 1H-NMR (300 MHz, CDCl3) δ, 8.77 (s, 1H), 7.41-7.28 (m, 5H), 5.89 (d, J = 3.7 Hz, 1H), 5.23-5.03 (m, 3H), 4.64-4.53 (m, 2H), 4.35-4.26 (m, 1H), 4.22 (dd, J = 8.9, 3.4 Hz, 1H), 4.13 (dd, J = 8.9, 5.7 Hz, 1H), 4.06 (dd, J = 8.9, 3.4 Hz, 1H), 1.53 (s, 3H), 1.47 (d, J = 7.0 Hz, 3H), 1.34 (s, 3H), 1.32 (s, 3H), 1.22 (s, 3H). 13C-NMR (75 MHz, CDCl3) δ, 171.3, 156.8, 135.6, 128.7, 128.7, 112.6, 110.2, 105.4, 83.6, 81.0, 80.3, 73.0, 68.0, 67.8, 27.0, 26.4, 24.8, 16.2.
General procedure for synthesis of O-(protected-α-aminoxyacyl)terpenes 8a–c, (8b + 8b′) and O-(protected-α-aminoxyacyl)steroids 10a–d, (10b + 10b′)
The N-Cbz-protected (α-aminoxyacyl)benzotriazoles 4b–d, (4c + 4c′) (1 equiv) were each added portionwise to a stirring solution of terpene 7a,b or steroid 9a–c (1 equiv) in THF (1 mL) in the presence of DMAP (0.1 equiv). The reaction mixture was subjected to microwave irradiation (power, temperature and hold time as indicated in Table 3–4) and subsequently allowed to cool to room temperature, transferred to a round bottomed flask and the solvent was evaporated. The crude terpene conjugates 8a–c, (8b + 8b′) were redissolved in ethyl acetate (10 mL), and the solutions were washed with Na2CO3 (10% w/w, 2 × 10 mL) and dried over MgSO4 to give 8a–c, (8b + 8b′). The crude steroid conjugates 10a–d, (10b + 10b′) were purified by column chromatography using hexanes:ethyl acetate (gradient) as eluent to afford the O-(protected-α-aminoxyacyl)steroids 10a–d, (10b + 10b′) as analytically pure products.
(R,E)-3,7-Dimethylocta-2,6-dien-1-yl 2-((((benzyloxy)-carbonyl)amino)oxy)-3-phenylpropanoate (8b)
Colorless oil, 55% yield, [α]D21 = +42.5 (c 0.23 in CH2Cl2). Anal. Calcd for C27H33NO5: C 71.82; H 7.37; N 3.10. Found: C 71.70; H 7.51; N 3.37. 1H-NMR (300 MHz, CDCl3) δ, 7.87 (s, 1H), 7.38-7.30 (m, 5H), 7.30-7.25 (m, 5H), 5.24 (t, J = 7.0 Hz, 1H), 5.14 (d, J = 12.3 Hz, 1H), 5.09 (d, J = 12.3 Hz, 1H), 5.07 (t, J = 7.9 Hz, 1H), 4.67 (dd, J = 6.7, 5.3 Hz, 1H), 4.61 (d, J = 7.3 Hz, 1H), 2.07-1.98 (m, 4H), 1.68 (s, 3H), 1.66 (s, 3H), 1.60 (s, 3H). 13C-NMR (75 MHz, CDCl3) δ, 170.9, 157.0, 143.3, 135.8, 135.5, 132.0, 129.5, 128.7, 128.5, 128.4, 128.4, 127.0, 123.7, 117.6, 84.5, 67.7, 62.2, 39.6, 37.2, 26.3, 25.8, 17.8, 16.6.
(2R)-(10R,13R)-17-((2R,5R)-5-Ethyl-6-methylheptan-2-yl)-10,13-dimethyl-2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradeca-hydro-1H-cyclopenta[a]phenanthren-3-yl 2-((((benzyloxy)-carbonyl)amino)oxy)-3-phenylpropanoate (10c)
White microcrystals, mp 112.0–114.0 °C, 26% yield, [α]D21 = +12.9 (c 0.22 in CH2Cl2). Anal. Calcd for C46H65NO5: C 77.60; H 9.20; N 1.97. Found: C 77.73; H 9.29; N 1.91. 1H-NMR (300 MHz, CDCl3) δ, 7.78 (s, 1H), 7.36-7.29 (m, 5H), 7.29-7.24 (m, 5H), 5.36 (d, J = 4.8 Hz, 1H), 5.20-5.16 (m, 1H), 5.16 (d, J = 12.4 Hz, 1H), 5.11 (d, J = 12.3 Hz, 1H), 5.01 (dd, J = 15.3, 8.7 Hz, 1H), 4.65 (dd, J = 6.7, 5.8 Hz, 1H), 4.62-4.58 (m, 1 H), 3.13 (s, 1H), 3.11 (d, J = 1.8 Hz, 1H), 2.25 (d, J = 7.8 Hz, 2H), 2.23-1.85 (m, 3H), 1.85-1.62 (m, 3H), 1.59-1.38 (m, 8H), 1.38-1.13 (m, 7H), 1.03-0.99 (m, 8H), 0.98-0.90 (m, 2H), 0.85-0.79 (m, 9H), 0.69 (s, 3H). 13C-NMR (75 MHz, CDCl3) δ, 170.4, 157.1, 139.4, 138.5, 135.9, 135.6, 129.6, 129.5, 128.8, 128.6, 128.5, 127.1, 123.2, 84.5, 75.4, 67.8, 57.0, 56.1, 51.5, 50.2, 42.4, 40.7, 39.8, 38.2, 37.3, 37.1, 36.8, 32.1, 29.1, 27.8, 25.6, 24.6, 21.5, 21.3, 21.2, 19.5, 19.2, 12.5, 12.3.
General procedure for synthesis of N-(protected-α-aminoxyacyl)nucleosides 12a,b
The N-Cbz-protected (α-aminoxyacyl)benzotriazoles 4c,d (1 equiv) were each added portionwise to a stirring solution of nucleosides 11a,b (1 equiv) in DMF (2 mL). The reactions were carried out at room temperature under argon atmosphere and were completed in 24 h. The solvent was evaporated under reduced pressure and the crude compounds were purified by flash column chromatography using methanol:dichloromethane (gradient) as eluent to afford the N-(protected-α-aminoxyacyl) nucleosides 12a,b.
Benzyl ((2R)-1-((1-(3,4-dihydroxy-5-(hydroxymethyl)tetra-hydrofuran-2-yl)-2-oxo-1,2-dihydropyrimidin-4-yl)amino)-4-methyl-1-oxopentan-2-yl)oxycarbamate (12a)
White microcrystals, mp 107.0–108.0 °C, 21% yield, [α]D21 = +25.9 (c 0.10 in CH2Cl2). Anal. Calcd for C23H30N4O9: C 54.54; H 5.97; N 11.06. Found: C 54.23; H 6.13; N 10.77. 1H-NMR (300 MHz, CD3OD-d4) δ, 8.59 (d, J = 7.5 Hz, 1H), 7.40 (d, J = 7.7 Hz, 1H), 7.32-7.28 (m, 5H), 5.88 (s, 1H), 5.15 (s, 2H), 4.42 (dd, J = 9.3, 4.2 Hz, 1H), 4.18-4.15 (m, 2H), 4.11-4.09 (m, 1H), 3.98 (dd, J = 12.6, 2.1 Hz, 1H), 3.80 (dd, J = 12.3, 2.7 Hz, 1H), 2.90-1.85 (m, 1H), 1.80-1.67 (m, 1H), 1.57-1.50 (m, 1H), 0.98 (d, J = 4.8 Hz, 3H), 0.94 (d, J = 6.3 Hz, 3H). 13C-NMR (75 MHz, CD3OD-d4) δ, 174.4, 164.0, 160.0, 158.0, 147.1, 137.4, 129.6, 129.5, 129.4, 98.0, 93.3, 86.7, 86.0, 76.7, 70.1, 68.6, 61.4, 41.5, 25.8, 23.7, 22.3.
Acknowledgements
We thank the University of Florida, The Kenan Foundation and King Abdulaziz University, Jeddah, Saudi Arabia for financial support. We thank Dr C. D. Hall and Dr I. Ghiviriga for helpful discussions.
References
- A. Sochanik, I. Kaida, I. Mitrus, A. Rajca and S. Szala, Cancer Gene Ther., 2000, 7, 513 CAS.
- X. Gao and L. Huang, Gene Ther., 1995, 2, 710 CAS.
- C. Laruelle and M. Lepant, FR patent 2654338, Chem. Abstr., 1991, 89, 14833 Search PubMed.
- P. W. Glunz, S. Hintermann, L. J. Williams, J. B. Schwarz, S. D. Kuduk, V. Kudryashov, K. O. Lloyd and S. Danishefsky, J. Am. Chem. Soc., 2000, 122, 7273 CrossRef CAS.
- H.-K. Han, R. L. A. de Vrueh, J. K. Rhie, K.-M. Y. Covitz, P. L. Smith, C.-P. Lee, D.-M. Oh, W. Sadée and G. L. Amidon, Pharm. Res., 1998, 15, 1154 CrossRef CAS.
- J. Kihlberg, J. Aahman, B. Walse, T. Drakenberg, A. Nilsson, C. Söderberg-Ahlm, B. Bengtsson and H. Olsson, J. Med. Chem., 1995, 38, 161 CrossRef CAS.
- H. Paulsen, R. Busch, V. Sinnwell and A. Pollex-Krüger, Carbohydr. Res., 1991, 214, 227 CrossRef CAS.
- E. G. von Roedern, E. Lohof, G. Hessler, M. Hoffmann and H. Kessler, J. Am. Chem. Soc., 1996, 118, 10156 CrossRef CAS.
- B. Liu, C. Cui, W. Duan, M. Zhao, S. Peng, L. Wang, H. Liu and G. Cui, Eur. J. Med. Chem., 2009, 44, 3596 CrossRef CAS.
- E. De Clercq, Med. Res. Rev., 2008, 28, 929 CrossRef CAS.
- M. T. Briggs and J. S. Morley, J. Chem. Soc., Perkin Trans. 1, 1979, 1, 2138 RSC.
- A. M. Brückner, P. Chakraborty, S. H. Gellman and U. Diederichsen, Angew. Chem., Int. Ed., 2003, 42, 4395 CrossRef.
- Y.-H. Zhang, K. Song, N.-Y. Zhu and D. Yang, Chem.–Eur. J., 2010, 16, 577 CrossRef CAS.
- D. Yang, F.-F. Ng, Z.-J. Li, Y.-D. Wu, K. W. K. Chan and D.-P. Wang, J. Am. Chem. Soc., 1996, 118, 9794 CrossRef CAS.
- D. Yang, J. Qu, W. Li, Y.-H. Zhang, Y. Ren, D.-P. Wang and Y.-D. Wu, J. Am. Chem. Soc., 2002, 124, 12410 CrossRef CAS.
- A. R. Katritzky, I. Avan and S. R. Tala, J. Org. Chem., 2009, 74, 8690 CrossRef CAS.
- M.-R. Lee, J. Lee, B.-H. Baek and I. Shin, Synlett, 2003, 325 CAS.
- S. Chandrasekhar, C. L. Rao, M. S. Reddy, G. D. Sharma, M. U. Kiran, P. Naresh, G. K. Chaitanya, K. Bhanuprakash and B. Jagadeesh, J. Org. Chem., 2008, 73, 9443 CrossRef CAS.
- L. Thevenet, R. Vanderesse, M. Marraud, C. Didierjean and A. Aubry, Tetrahedron Lett., 2000, 41, 2361 CrossRef CAS.
- I. Shin and K. Park, Org. Lett., 2002, 4, 869 CrossRef CAS.
- V. Dulery, O. Renaudet and P. Dumy, Tetrahedron, 2007, 63, 11952 CrossRef CAS.
- D. Yang, Y.-H. Zhang, B. Li and D.-W. Zhang, J. Org. Chem., 2004, 69, 7577 CrossRef CAS.
- A. R. Katritzky, P. Angrish and E. Todadze, Synlett, 2009, 2392 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: General procedures and characterization data for compounds 3a–d, (3c + 3c′) and 4a–d, (4c + 4c′), characterization data for compounds 6b–f, (6e + 6e′), 8a,c, (8b + 8b′), 10a,b,d, (10b + 10b′) and 12b, 2D NMR spectra for compound 6f and 1H NMR, 13C NMR spectra of all products. See DOI: 10.1039/c1ra00413a |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here.