Enantioselective organocatalytic phospha-Michael reaction of α,β-unsaturated aldehydes†
Xiaoyan
Luo
,
Zhiqiang
Zhou
,
Xin
Li
,
Xinmiao
Liang
and
Jinxing
Ye
*
Engineering Research Center of Pharmaceutical Process Chemistry, Ministry of Education, School of Pharmacy, East China University of Science and Technology, 130 Meilong Road, Shanghai 200237, China. E-mail: yejx@ecust.edu.cn
First published on 25th August 2011
Abstract
The enantioselective organocatalytic phospha-Michael reaction of α,β-unsaturated aldehydes with diaryl phosphine oxides is explored for the first time using (S)-2-(diphenyl(trimethylsilyloxy)methyl)pyrrolidine as a catalyst. It afforded 1,4-addition adducts in good to excellent yields with up to 99% ee.
Introduction
Chiral organophosphorus compounds have wide and important applications in agrochemistry, medical chemistry and metal-catalyzed or organocatalytic reactions.1 They are traditionally obtained by chiral resolution or use of chiral auxiliaries.2 The asymmetric method to generate P–C bonds with chiral centers at or near phosphorus has gained much attention, and great progress has been made.3 The catalytic asymmetric reactions of trivalent phosphine compounds containing a P–H bond to electron-deficient olefins,4 such as α,β-unsaturated aldehydes5 and nitroalkenes,6 have provided direct access to chiral phosphine compounds.3 Recently, the direct addition of secondary phosphine oxides [R2P(O)H] has been achieved.7 For example, the enantioselective Michael addition of diphenyl phosphite to nitroolefin was carried out using chiral catalysts.8 Moreover, there have been many reports on the enantioselective hydrophosphonylation of imines.9 However, to the best of our knowledge, no example was reported on the asymmetric Michael reaction of pentavalent diaryl phosphine nucleophiles and α,β-unsaturated aldehydes. Moreover, there still has inherent difficulties due to the strong preference for 1,2-addition over 1,4-addition in the reaction of α,β-unsaturated aldehydes.Recently, our group has reported the enantioselective organocatalytic phospha-Michael reaction of α,β-unsaturated ketones and diaryl phosphine oxides using a multifunctional catalyst.10 It has been demonstrated that iminium activation is achieved in Michael-type β-functionalization for trivalent phosphine nucleophiles.5a,5d On the basis of these researches, we explored firstly chemo- and enantioselective organocatalytic reaction between pentavalent diaryl phosphine oxides and α,β-unsaturated aldehydes catalyzed by Jørgensen-Hayashi catalyst.11
Results and discussion
The reaction of diphenylphosphine oxide (1a) and cinnamaldehyde (2a) in the absence of catalyst gave sole 1,2-addition product 4 without any desired 1,4-addition product 3a (Table 1, entries 1–3). It is a strong background reaction for the development of the catalytic asymmetric reaction. Gratifyingly, the reaction could produce the 1,4-addition product 3a in the presence of proline or its derivative catalysts (Table 1, entries 4-6), which was due to the iminium ion formation from the condensation of the aldehyde 2a with the catalyst. Especially, 67% conversion with 71% ee was achieved when 6b was used as a catalyst (Table 1, entry 6). In order to optimize the reaction condition, different reaction solvents were screened. The reaction proceeded with nearly 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 of the ratio of 3a/4 in THF, MeCN, EtOAc or toluene (Table 1, entries 7–10). When alcohol was used as solvent, the chemoselectivity of the reaction increased. However, the enantioselectivity decreased to 51% ee (Table 1, entries 11–12). Finally, it was found that CH2Cl2 was the most suitable solvent for this procedure, and 1:0.23 ratio of 3a/4 was obtained with 82% ee and 92% of conversion (Table 1, entry 14). Then, co-catalysts were tested, and the results showed that they could slightly increase the enantioselectivity and decrease the conversion and chemoselectivity (Table 1, entries 15–17). Using PhCO2Li as a co-catalyst, lowering the reaction temperature and increasing the loading of catalyst 6b are all beneficial to the enantio- and chemoselectivity (Table 1, entries 18 and 19). But the rate of reaction was slowed by lowering the reaction temperature. Thus, the reaction was carried out at −4 °C using 20 mol% of catalyst 6b, the rate was speeded with the improvement of the chemo- and enantioselectivity (Table 1, entry 20). However, under this condition, an 88% ee value was obtained using 2-FPhCO2Li instead of PhCO2Li (Table 1, entry 21). Lowering the reaction temperature from −4 °C to −20 °C resulted in the decrease of the conversion and chemoselectivity despite improving the enantioselectivity (Table 1, entry 22). It should be noted that 77% of conversion with 92% ee and 1:0.62 of the ratio of 3a/4 was given in the presence of 10 mol% of catalyst 6b and 20% mol of PhCO2Li (Table 1, entry 23). This indicated that the co-catalyst PhCO2Li did not change the chemoselectivity and reduce the enantioselectivity at −20 °C. Therefore, in the absence of PhCO2Li, the reaction was tested at −20 °C using catalyst 6b (Table 1, entry 24). 92% ee value with 80% of conversion and 1:0.05 ratio of 3a/4 were obtained, which proved that 6b was the main cause of 1,4-addtion reaction. In order to improve the yield of 1,4-addtion adduct, the reaction was extended to 36 h, and produced the desired product with 94% of isolated yield and 94% ee (Table 1, entry 25).
:1 of the ratio of 3a/4 in THF, MeCN, EtOAc or toluene (Table 1, entries 7–10). When alcohol was used as solvent, the chemoselectivity of the reaction increased. However, the enantioselectivity decreased to 51% ee (Table 1, entries 11–12). Finally, it was found that CH2Cl2 was the most suitable solvent for this procedure, and 1:0.23 ratio of 3a/4 was obtained with 82% ee and 92% of conversion (Table 1, entry 14). Then, co-catalysts were tested, and the results showed that they could slightly increase the enantioselectivity and decrease the conversion and chemoselectivity (Table 1, entries 15–17). Using PhCO2Li as a co-catalyst, lowering the reaction temperature and increasing the loading of catalyst 6b are all beneficial to the enantio- and chemoselectivity (Table 1, entries 18 and 19). But the rate of reaction was slowed by lowering the reaction temperature. Thus, the reaction was carried out at −4 °C using 20 mol% of catalyst 6b, the rate was speeded with the improvement of the chemo- and enantioselectivity (Table 1, entry 20). However, under this condition, an 88% ee value was obtained using 2-FPhCO2Li instead of PhCO2Li (Table 1, entry 21). Lowering the reaction temperature from −4 °C to −20 °C resulted in the decrease of the conversion and chemoselectivity despite improving the enantioselectivity (Table 1, entry 22). It should be noted that 77% of conversion with 92% ee and 1:0.62 of the ratio of 3a/4 was given in the presence of 10 mol% of catalyst 6b and 20% mol of PhCO2Li (Table 1, entry 23). This indicated that the co-catalyst PhCO2Li did not change the chemoselectivity and reduce the enantioselectivity at −20 °C. Therefore, in the absence of PhCO2Li, the reaction was tested at −20 °C using catalyst 6b (Table 1, entry 24). 92% ee value with 80% of conversion and 1:0.05 ratio of 3a/4 were obtained, which proved that 6b was the main cause of 1,4-addtion reaction. In order to improve the yield of 1,4-addtion adduct, the reaction was extended to 36 h, and produced the desired product with 94% of isolated yield and 94% ee (Table 1, entry 25).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Cat. | Additive | Solvent | Conv.b(%) | 3a/4b | eec (%) |
| a Without other notes, 1a was 0.25 mmol, 2a was 0.5 mmol, the catalyst was 0.025 mmol, solvent was 0.5 ml, reaction time was 24 h at 25 °C. nd = not determined. b The values were obtained by 31P NMR analyses. The conversion values were relative to products 3a and 4. c The values of 7aa were obtained by HPLC analyses using a Chiralpak IA column with racemic product as standards after reduction of aldehydes with NaBH4. d The reaction temperature was 40 °C. e The reaction temperature was −20 °C. f The reaction time was 36 h at room temperature. g 0.05 mmol catalyst was used. h The values correspond to the conversion of product 7aa were obtained by GC analyses. i 0.05 mmol PhCO2Li was used. j The data was the isolated yield of 3a. | ||||||
| 1 | — | no | CH2Cl2 | >99 | 0/100 | |
| 2d | — | no | CH2Cl2 | 77 | 0/100 | |
| 3e | — | no | CH2Cl2 | 74 | 0/100 | |
| 4f | 5 g | no | MeOH | 38h | nd | −8 |
| 5f | 6a g | no | MeOH | 21h | nd | 77 |
| 6f | 6b g | no | MeOH | 67h | nd | 71 |
| 7 | 6b | no | THF | 77 | 1/1.11 | 82 |
| 8 | 6b | no | MeCN | 95 | 1/1 | 77 |
| 9 | 6b | no | EtOAc | 92 | 1/1.10 | 80 |
| 10 | 6b | no | toluene | 93 | 1/1.05 | 86 |
| 11 | 6b | no | MeOH | 90 | 1/0.26 | 67 |
| 12 | 6b | no | EtOH | 90 | 1/0.23 | 51 |
| 13 | 6b | no | CHCl3 | 99 | 1/0.32 | 80 |
| 14 | 6b | no | CH2Cl2 | 99 | 1/0.23 | 82 |
| 15 | 6b | PhCO2Li | CH2Cl2 | 90 | 1/0.36 | 84 |
| 16 | 6b | 2-FPhCO2H | CH2Cl2 | 84 | 1/0.32 | 86 |
| 17 | 6b | 2-FPhCO2Li | CH2Cl2 | 86 | 1/0.24 | 86 |
| 18 | 6b g | PhCO2Li | CH2Cl2 | 90 | 1/0.17 | 86 |
| 19d | 6b | PhCO2Li | CH2Cl2 | 85 | 1/0.18 | 88 |
| 20d | 6b g | PhCO2Li | CH2Cl2 | 97 | 1/0.06 | 90 |
| 21d | 6b g | 2-FPhCO2Li | CH2Cl2 | 96 | 1/0.05 | 88 |
| 22e | 6b g | PhCO2Li | CH2Cl2 | 90 | 1/0.11 | 92 |
| 23e | 6b | PhCO2Li i | CH2Cl2 | 77 | 1/0.62 | 92 |
| 24e | 6b g | no | CH2Cl2 | 80 | 1/0.05 | 92 |
| 25ef | 6b g | no | CH2Cl2 | 94j | nd | 94 |
The steric effect of the phosphine oxide was considered under the above optimized condition, and the results are shown in Table 2. It turned out that the yield was reduced sharply with the addition of the aliphatic substituted group on the aromatic ring. Moreover, the yield was decreased when the steric hindrance of the substituted group became bigger. The ortho position substitution resulted in great increase of the enantioselectivity (entry 2). However, meta position or para position substitution could not bring forth an improvement of the enantioselectivity despite of the sterically more hindered diisopropyl or tertiary butyl substituted groups (entries 3–5). When the substituted groups were at the same position, their steric hindrances had an obvious influence on the yield and enantioselectivity (entries 3 and 4).
|
|
|||
|---|---|---|---|
| Entry | R | Yieldb (%) | eec (%) |
| a Unless otherwise noted, all reactions were performed with 0.5 mmol of 2a, 0.25 mmol of 1 and 0.05 mmol of 6b in CH2Cl2 (0.5 mL) for 36 h. b Isolated yield of product after column chromatography. c Determined by chiral HPLC. | |||
| 1 | C6H5,1a | 94, 7aa | 94 |
| 2 | 2,6-(Me)2C6H3, 1b | 71, 7ba | 98 |
| 3 | 3,5-(Me)2C6H3, 1c | 78, 7ca | 86 |
| 4 | 3,5-(iPr)2C6H3, 1d | 66, 7da | 92 |
| 5 | 4-(CH3)3CC6H4, 1e | 64, 7ea | 89 |
To explore the scope of this type of Michael addition, a number of different α,β-unsaturated aldehydes2b–o were used with 1a under the optimized reaction condition, and the results are presented in Table 3. It turned out that the reaction proceeded with aromatic α,β-unsaturated aldehydes in good to excellent enantioselectivities, regardless of electron-donating or electron-withdrawing substituents on the phenyl ring (entries 1–12). The substituent position on the aromatic ring played an important role in the yield. For example, 2-chlorocinnamaldehyde (2b) reacted with 1a and gave the desired product in 82% yield while only 59% yield was obtained by 3-chlorocinnamaldehyde (2c) and 1a (entries 1 and 2). Although the reaction time of 4-chlorocinnamaldehyde (2d) with 1a was 12 h less than that of 3-chlorocinnamaldehyde (2c) with 1a, their yields were nearly the same (entries 2 and 3). In addition, among three different bromocinnamaldehydes, the yield of 3-bromocinnamaldehyde (2f) with 1a was the lowest (entries 4–6). 99% of enantioselectivity was obtained when 4-nitrocinnamaldehyde (2i) was used (entry 8). To our delight, the reaction was also suitable for aliphatic α,β-unsaturated aldehydes. (E)-hex-2-enal (2n) was tested and the adduct was produced in 77% yield with 82% ee values (entry 13). Unfortunately, when (E)-4-methylpent-2-enal (2o) was used, only a 37% yield was obtained, though the excess enantioselectivity was 99% (entry 14), which was due to the steric hindrance effect.
|
|
|||
|---|---|---|---|
| Entry | R | Yieldb (%) | eec (%) |
| a Unless otherwise noted, all reactions were performed with 0.5 mmol of 2, 0.25 mmol of 1a and 0.05 mmol of 6b in 0.5 mL CH2Cl2 for 48 h. b Isolated yield of product after column chromatography c Determined by chiral HPLC. d The reaction time was 36 h and the temperature was from -15 °C to -20 °C. | |||
| 1 | 2-ClC6H4, 2b | 82, 7ab | 95 |
| 2 | 3-ClC6H4, 2c | 59, 7ac | 91 |
| 3d | 4-ClC6H4, 2d | 56, 7ad | 92 |
| 4 | 2-BrC6H4, 2e | 98, 7ae | 90 |
| 5 | 3-BrC6H4, 2f | 64, 7af | 88 |
| 6d | 4-BrC6H4, 2g | 87, 7ag | 90 |
| 7 | 4-FC6H4, 2h | 75, 7ah | 94 |
| 8 | 4-NO2C6H4, 2i | 77, 7ai | 99 |
| 9 | 3-MeC6H4, 2j | 93, 7aj | 90 |
| 10d | 4-MeC6H4, 2k | 51, 7ak | 90 |
| 11 | 2-MeOC6H4, 2l | 87, 7al | 86 |
| 12d | 4-MeOC6H4, 2m | 41, 7am | 90 |
| 13 | Pr, 2n | 77, 7an | 82 |
| 14 | i Pr, 2o | 36, 7ao | 99 |
Recrystallization by slow volatilization of EtOH gave white crystals of 7ag that were suitable for single crystal X-ray diffraction.12 Finally, the absolute configuration of product 7ag was determined to be S product by X-ray crystallographic analysis (Fig. 1).
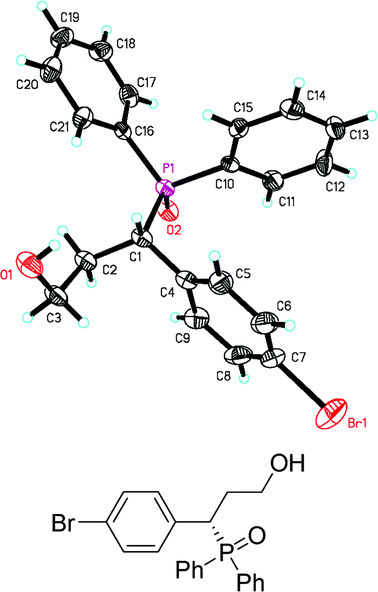 | ||
| Fig. 1 X-ray crystal structure of 7ag. | ||
According to the absolute configuration of the chiral Michael product 7ag from X-ray crystal structure analysis, a catalytic cycle was proposed (Fig. 2).
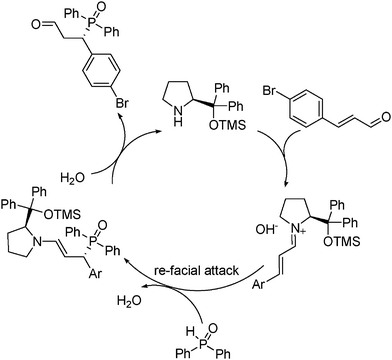 | ||
| Fig. 2 Proposed catalytic cycle. | ||
Conclusions
In conclusion, we have developed an enantioselective organocatalytic reaction between pentavalent diaryl phosphine oxides and α,β-unsaturated aldehydes using simple chiral pyrrolidine derivative Jørgensen-Hayashi catalyst. The catalytic reaction afforded 1,4-addition adducts in good to excellent enantioselectivities and yields for a broad range of enals, including aromatic and aliphatic α,β-unsaturated aldehydes. The iminium catalyst leads to the 1,4-addition reaction which can not proceed inherently. This organocatalytic approach offers a new and easy method for the direct preparation of chiral phosphine oxide derivatives from pentavalent diaryl phosphine oxides and α,β-unsaturated aldehydes.Experimental
General methods
All solvents and inorganic reagents were from commercial sources and used without purification unless otherwise noted. 1H NMR spectra, 13C NMR and 31P NMR spectra were recorded on a Bruker AV-400 spectrometer (400 MHz, 100 MHz and 162 MHz). Chemical shifts for protons are reported in parts per million downfield from tetramethylsilane and are referenced to residual protium in the NMR solvent. Chemical shifts for carbon are reported in parts per million downfield from tetramethylsilane and are referenced to the carbon resonances of the solvent . Data are represented as follows: chemical shift, integration, multiplicity (br = broad, s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet), coupling constants in Hertz (Hz). High resolution mass spectra (EI+) were measured on a Waters Micromass GCT spectrometer, electron energy 70 eV. Optical rotations were measured on an Autopol III automatic polarimeter. High performance liquid chromatography (HPLC) was performed on an Agilent 1 200 Series chromatographs using a Chiral column (IA and IC) (0.46cm × 25 cm) as noted.General procedure for asymmetric Michael addition and reduction
Diaryl phosphine oxide (0.25 mmol) in CH2Cl2 (0.5 mL) was added to (S)-2-(diphenyl-(trimethylsilyloxy)methyl)pyrrolidine (16.2 mg, 0.05 mmol) and α,β-unsaturated aldehyde (0.5 mmol). The reaction mixture was stirred for the time indicated in the tables and then 1.0 mL MeOH and NaBH4 (18.9 mg, 0.5 mmol) was added. The reaction mixture was stirred at room temperature for 1 h. The solvent was removed under vacuum. The residue was purified by silica gel chromatography to afford the appropriate product. Yields of the products are given in the tables.(S)-3-(diphenylphosphoryl)-3-phenylpropan-1-ol (Table 2, Entry 1)
The product was obtained in 94% yield, white solid. [α]D20.1 = −42.4 (c 0.67, CH3OH); mp = 188–189 °C. 1H NMR (400 MHz, CD3OD): δ (ppm) 7.98–7.94 (m, 2H), 7.86 (d, J = 8.4 Hz, 1H), 7.60–7.55 (m, 2H), 7.49–7.44 (m, 3H), 7.36–7.32 (m, 1H), 7.26–7.22 (m, 4H), 7.16–7.11 (m, 2H), 4.02–3.96 (m, 1H), 3.46–3.40 (m, 1H), 3.25–3.21 (m, 1H), 2.25–2.14 (m, 1H), 2.08–1.98 (m, 1H). 13C NMR (100 MHz, CD3OD): δ (ppm) 136.4, 133.4, 132.8, 132.5, 132.4, 132.1, 132.0, 131.2, 131.1, 130.6, 130.2, 130.1, 129.8, 129.5, 129.4, 129.3, 128.3, 60.1, 43.6, 33.1. 31P NMR (CD3OD): δ (ppm) 36.8 (s). HRMS (EI): exact mass calculated for M+ (C21H21O2P) requires m/z 336.1279, found m/z 336.1284. The enantiomeric excess was determined by HPLC. [IA-H column, 220 nm, Hexane : EtOH = 7
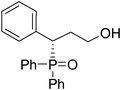 :1, 0.6 mL/min]: 14.464 min (major), 16.210 min (minor), ee = 94%.
:1, 0.6 mL/min]: 14.464 min (major), 16.210 min (minor), ee = 94%.
(S)-3-(bis(2,6-dimethylphenyl)phosphoryl)-3-phenylpropan-1-ol (Table 2, Entry 2)
The product was obtained in 71% yield, white solid. [α]D14.2 = −116.2 (c 0.53, CH3OH); mp = 140–142 °C. 1H NMR (400 MHz, CD3COCD3): δ (ppm) 7.36–7.34 (m, 2H), 7.29–7.25 (m, 1H), 7.11–7.06 (m, 5H), 7.01–6.97 (m, 1H), 6.75 (d, J = 3.6 Hz, 1H), 6.73 (d, J = 3.6 Hz, 1H), 4.23–4.18 (m, 1H), 3.57–3.50 (m, 1H), 3.42–3.35 (m, 1H), 2.61 (s, 6H), 2.59–2.53 (m, 1H), 2.49–2.41 (m, 1H), 2.33 (s, 6H). 13C NMR (100 MHz, CD3COCD3): δ (ppm) 143.5, 143.4, 143.2, 143.1, 139.5, 137.8, 132.4, 132.3, 132.2, 132.1, 132.0, 131.5, 131.4, 131.3, 129.5, 129.4, 128.6, 61.2, 48.0, 35.3, 24.4, 24.3, 23.8, 23.7. 31P NMR (CD3COCD3): δ (ppm) 42.9 (s). HRMS (EI): exact mass calculated for M+ (C25H29O2P) requires m/z 392.1905, found m/z 392.1909. The enantiomeric excess was determined by HPLC. [IA-H column, 220 nm, Hexane: EtOH = 9
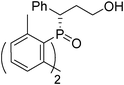 :1, 0.6 mL/min]: 27.162 min (major), 16.968 min (minor), ee = 98%.
:1, 0.6 mL/min]: 27.162 min (major), 16.968 min (minor), ee = 98%.
(S)-3-(bis(3,5-dimethylphenyl)phosphoryl)-3-phenylpropan-1-ol (Table 2, Entry 3)
The product was obtained in 78% yield, yellow solid. [α]D12.5 = −52.0 (c 0.45, CH3OH); mp = 174–176 °C. 1H NMR (400 MHz, CD3 COCD3): δ (ppm) 7.51 (d, J = 11.2 Hz, 2H), 7.32–7.30 (m, 2H), 7.16 (d, J = 10.8 Hz, 2H), 7.06–7.02 (m, 3H), 6.99–6.96 (m, 1H), 6.82 (s, 1H), 3.95–3.90 (m, 1H), 3.34–3.28 (m, 1H), 3.17–3.09 (m, 1H), 2.22 (s, 3H), 2.05 (s, 3H), 2.00–1.94 (m, 1H), 1.90–1.83 (m, 1H). 13C NMR (100 MHz, CD3COCD3): δ (ppm) 140.0, 139.9, 139.3, 139.2, 139.0, 136.1, 135.1, 134.7, 134.2, 132.1, 131.9, 130.6, 130.5, 130.4, 130.3, 129.7, 128.4, 60.8, 43.9, 34.8, 22.3, 22.1. 31P NMR (CD3COCD3): δ (ppm) 31.3 (s). HRMS (EI): exact mass calculated for M+ (C25H29O2P) requires m/z 392.1905, found m/z 392.1904. The enantiomeric excess was determined by HPLC. [IA-H column, 220 nm, Hexane:EtOH = 15
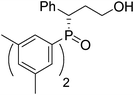 :1, 0.7 mL/min]: 14.411 min (major), 16.757 min (minor), ee = 87%.
:1, 0.7 mL/min]: 14.411 min (major), 16.757 min (minor), ee = 87%.
(S)-3-(bis(3,5-diisopropylphenyl)phosphoryl)-3-phenylpropan -1-ol (Table 2, Entry 4)
The product was obtained in 66% yield, yellow solid. [α]D14.7 = −32.0 (c 0.29, CH3OH); mp = 146–148 °C. 1H NMR (400 MHz, CD3OD): δ (ppm) 7.67 (dd, J = 11.2, 0.8 Hz, 2H), 7.39 (m, 1H), 7.26–7.25 (m, 2H), 7.21–7.15 (m, 3H), 7.12–7.09 (m, 3H), 3.96–3.90 (m, 1H), 3.51–3.46 (m, 1H), 3.28–3.25 (m, 1H), 3.01 (q, J = 6.8 Hz, 2H), 2.76 (q, J = 6.8 Hz, 2H), 2.25–2.16 (m, 1H), 2.14–2.08 (m, 1H), 1.31 (d, J = 2.4 Hz, 6H), 1.29 (d, J = 2.4 Hz, 6H), 1.12 (d, J = 6.8 Hz, 6H), 1.08 (d, J = 7.2 Hz, 6H). 13C NMR (100 MHz, CD3OD): δ (ppm) 151.2, 151.1, 150.2, 150.1, 136.7, 132.9, 131.9, 131.3, 131.2, 130.1, 129.8, 129.4, 128.3, 128.1, 128.0, 127.6, 127.5, 60.0, 44.2, 35.6, 35.4, 33.2, 24.6, 24.4, 24.3. 31P NMR (CD3OD): δ (ppm) 38.6 (s). HRMS (EI): exact mass calculated for M+ (C33H45O2P) requires m/z 504.3157, found m/z 504.3158. The enantiomeric excess was determined by HPLC. [IC-H column, 220 nm, Hexane:EtOH = 17
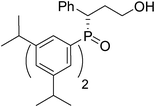 :1, 0.7 mL/min]: 12.856 min (major), 14.123 min (minor), ee = 92%.
:1, 0.7 mL/min]: 12.856 min (major), 14.123 min (minor), ee = 92%.
(S)-3-(bis(4-tert-butylphenyl)phosphoryl)-3-phenylpropan-1-ol (Table 2, Entry 5)
The product was obtained in 64% yield, white solid. [α]D13.4 = −38.2 (c 0.54, CH3OH); mp = 274–275 °C. 1H NMR (400 MHz, CD3OD): δ (ppm) 7.87–7.83 (m, 2H), 7.62–7.60 (m, 2H), 7.39–7.34 (m, 2H), 7.31–7.28 (m, 3H), 7.25–7.22 (m, 2H),7.14 (d, J = 7.2 Hz, 2H), 3.96–3.90 (m, 1H), 3.45–3.40 (m, 1H), 3.25–3.20 (m, 1H), 2.18 (brs, 1H), 2.09–1.98 (m, 2H), 1.32 (s, 9H), 1.19 (s, 9H). 13C NMR (100 MHz, CD3OD): δ (ppm) 155.6, 155.0, 130.9, 130.8, 130.6, 130.5, 129.8, 129.7, 128.4, 127.9, 127.3, 126.8, 125.8, 125.6, 125.3, 125.0, 124.8, 58.7, 42.3, 34.5, 34.3, 31.7, 30.1, 30.0. 31P NMR (CD3OD): δ (ppm) 37.4 (s). HRMS (EI): exact mass calculated for M+ (C29H37O2P) requires m/z 448.2531, found m/z 448.2527. The enantiomeric excess was determined by HPLC. [IA-H column, 220 nm, Hexane:EtOH = 15
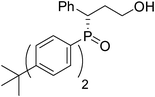 :1, 0.7 mL/min]: 23.943 min (major), 20.064 min (minor), ee = 90%.
:1, 0.7 mL/min]: 23.943 min (major), 20.064 min (minor), ee = 90%.
(S)-3-(2-chlorophenyl)-3-(diphenylphosphoryl)propan-1-ol (Table 3, Entry 1)
The product was obtained in 82% yield, yellow oil. [α]D19.3 = −44.9 (c 0.80, CH3OH); 1H NMR (400 MHz, CD3OD): δ (ppm) 8.06–8.02 (m, 2H), 7.79 (d, J = 7.6 Hz, 1H), 7.71–7.64 (m, 3H), 7.46–7.38 (m, 3H), 7.33-7.24 (m, 3H), 7.22–7.16 (m, 2H), 4.70–4.64 (m, 1H), 3.50–3.44 (m, 1H), 3.31–3.26 (m, 1H), 2.30-2.22 (m, 1H), 2.19–2.12 (m, 1H). 13C NMR (100 MHz, CD3OD): δ (ppm) 136.6, 134.8, 133.7, 133.1, 132.9,132.5, 132.4, 132.0, 131.9, 131.4, 130.5,130.4, 130.3, 129.8, 129.3, 129.2, 128.3, 60.1, 39.2, 33.8. 31P NMR (CD3OD): δ (ppm) 36.6 (s). HRMS (EI): exact mass calculated for M+ (C21H20ClO2P) requires m/z 370.0889, found m/z 370.0891. The enantiomeric excess was determined by HPLC. [IC-H column, 220 nm, Hexane:EtOH = 9
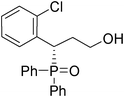 :1, 0.7 mL/min]: 27.206 min (major), 43.637 min (minor), ee = 95%.
:1, 0.7 mL/min]: 27.206 min (major), 43.637 min (minor), ee = 95%.
(S)-3-(3-chlorophenyl)-3-(diphenylphosphoryl)propan-1-ol (Table 3, Entry 2)
The product was obtained in 59% yield, white solid. [α]D19.9 = −45.0 (c 0.61, CH3OH); mp = 176–178 °C. 1H NMR (400 MHz, CD3 COCD3): δ (ppm) 8.07 (t, J = 8.8 Hz, 2H), 7.76–7.71 (m, 2H), 7.59–7.49 (m, 4H), 7.40–7.31 (m, 4H), 7.21–7.13 (m, 2H), 4.20–4.15 (m, 1H), 3.50–3.44 (m, 1H), 3.32–3.21 (m, 1H), 2.20–2.12 (m, 1H), 1.99–1.95 (m, 1H). 13C NMR (100 MHz, CD3 COCD3): δ (ppm) 140.2, 134.2, 132.5, 132.0, 131.9, 131.7, 131.6, 130.8, 130.7, 130.4, 129.7, 129.6, 129.5, 129.1, 128.9, 127.6, 59.5, 42.6, 33.5. 31P NMR (CD3 COCD3): δ (ppm) 30.8 (s). HRMS (EI): exact mass calculated for M+ (C21H20ClO2P) requires m/z 370.0889, found m/z 370.0890. The enantiomeric excess was determined by HPLC. [IC-H column, 220 nm, Hexane:EtOH = 8
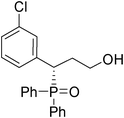 :1, 0.8 mL/min]: 13.539 min (major), 19.117 min (minor), ee = 91%.
:1, 0.8 mL/min]: 13.539 min (major), 19.117 min (minor), ee = 91%.
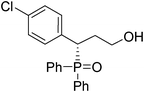
(S)-3-(4-chlorophenyl)-3-(diphenylphosphoryl)propan-1-ol (Table 3, Entry 3)
The product was obtained in 56% yield, white solid. [α]D16.4 = −60.8 (c 0.60, CH3OH); mp = 224–226 °C. 1H NMR (400 MHz, CD3OD): δ (ppm) 8.04–8.00 (m, 2H), 7.68–7.60 (m, 3H), 7.58–7.53 (m, 2H), 7.45–7.41 (m, 1H), 7.37–7.28 (m, 4H), 7.19 (d, J = 8.4 Hz, 2H), 4.11–4.06 (m, 1H), 3.52–3.46 (m, 1H), 3.31–3.24 (m, 1H), 2.26–2.16 (m, 1H), 2.13–2.04 (m, 1H). 13C NMR (100 MHz, CD3OD): δ (ppm) 133.9, 132.8, 132.1, 131.6, 131.3, 131.2, 131.0, 130.9, 130.6, 130.5, 128.8, 128.7, 128.1, 128.0, 127.9, 58.5, 41.5, 31.5. 31P NMR (CD3OD): δ (ppm) 36.4 (s). HRMS (EI): exact mass calculated for M+ (C21H20ClO2P) requires m/z 370.0889, found m/z 370.0880. The enantiomeric excess was determined by HPLC. [IA-H column, 220 nm, Hexane:EtOH = 6
 :1, 0.7 mL/min]: 11.517 min (major), 14.776 min (minor), ee = 92%.
:1, 0.7 mL/min]: 11.517 min (major), 14.776 min (minor), ee = 92%.
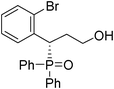
(S)-3-(2-bromophenyl)-3-(diphenylphosphoryl)propan-1-ol (Table 3, Entry 4)
The product was obtained in 98% yield, yellow oil. [α]D19.7 = −42.3 (c 0.84, CH3OH); 1H NMR (400 MHz, CD3OD): δ (ppm) 8.06–8.02 (m, 2H), 7.80 (d, J = 7.6 Hz, 1H), 7.71–7.64 (m, 3H), 7.44–7.35 (m, 5H), 7.27–7.23 (m, 2H), 7.12–7.08 (m, 1H), 4.68–4.62 (m, 1H), 3.49–3.44 (m, 1H), 3.35–3.29 (m, 1H), 2.32-2.22 (m, 1H), 2.18–2.11 (m, 1H). 13C NMR (100 MHz, CD3OD): δ (ppm) 135.2, 132.5, 132.3, 131.7, 131.4, 131.1,131.0, 130.7, 130.6, 130.4, 130.1, 129.0, 128.9, 128.7, 127.9, 127.8, 127.6, 126.4, 58.7, 40.9, 32.7. 31P NMR (CD3OD): δ (ppm) 36.7 (s). HRMS (EI): exact mass calculated for M+ (C21H20BrO2P) requires m/z 414.0384, found m/z 414.0385. The enantiomeric excess was determined by HPLC. [IC-H column, 220 nm, Hexane:EtOH = 8
 :1, 0.8 mL/min]: 22.736 min (major), 37.672 min (minor), ee = 90%.
:1, 0.8 mL/min]: 22.736 min (major), 37.672 min (minor), ee = 90%.
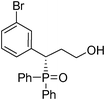
(S)-3-(3-bromophenyl)-3-(diphenylphosphoryl)propan-1-ol (Table 3, Entry 5)
The product was obtained in 64% yield, yellow solid. [α]D20 = −43.4 (c 0.84, CH3OH); mp = 171–173 °C. 1H NMR (400 MHz, CD3OD): δ (ppm) 8.04–8.00 (m, 2H), 7.66–7.62 (m, 3H), 7.58–7.49 (m, 3H), 7.44–7.41 (m, 1H), 7.37–7.29 (m, 4H), 7.14–7.10 (m, 1H), 4.10–4.05 (m, 1H), 3.53–3.47 (m, 1H), 3.31–3.26 (m, 1H), 2.27–2.17 (m, 1H), 2.12–2.04 (m, 1H). 13C NMR (100 MHz, CD3OD): δ (ppm) 137.8, 132.7, 132.1, 131.6, 131.4, 131.0, 130.9, 130.6, 130.5, 130.4, 130.0, 129.6, 128.9, 128.8, 128.5, 128.1, 128.0, 121.9, 58.5, 41.9, 31.5. 31P NMR (CD3OD): δ (ppm) 36.4 (s). HRMS (EI): exact mass calculated for M+ (C21H20BrO2P) requires m/z 414.0384, found m/z 414.0381. The enantiomeric excess was determined by HPLC. [IC-H column, 220 nm, Hexane:EtOH = 8
 :1, 0.6 mL/min]: 19.045 min (major), 27.052 min (minor), ee = 88%.
:1, 0.6 mL/min]: 19.045 min (major), 27.052 min (minor), ee = 88%.
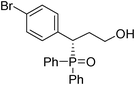
(S)-3-(4-bromophenyl)-3-(diphenylphosphoryl)propan-1-ol (Table 3, Entry 6)
The product was obtained in 87% yield, white solid. [α]D15.6 = −51.1 (c 0.49, CH3OH); mp = 226–228 °C. 1H NMR (400 MHz, CD3OD): δ (ppm) 8.04–8.00 (m, 2H), 7.66–7.60 (m, 3H), 7.58–7.53 (m, 2H), 7.45–7.42 (m, 1H), 7.37–7.34 (m, 4H), 7.24–7.21 (m, 2H), 4.10–4.05 (m, 1H), 3.52–3.46 (m, 1H), 3.31–3.24 (m, 1H), 2.26–2.18 (m, 1H), 2.12–2.02 (m, 1H). 13C NMR (100 MHz, CD3OD): δ (ppm) 134.4, 132.1, 131.6, 131.5, 131.4, 131.1, 131.0, 130.9, 130.8, 130.6, 130.5, 128.8, 128.7, 128.1, 128.0, 120.8, 58.5, 41.6, 31.5. 31P NMR (CD3OD): δ (ppm) 36.2 (s). HRMS (EI): exact mass calculated for M+ (C21H20O2PBr) requires m/z 414.0384, found m/z 414.0375. The enantiomeric excess was determined by HPLC. [IC-H column, 220 nm, Hexane:EtOH = 6
 :1, 0.6 mL/min]: 14.331 min (major), 18.652 min (minor), ee = 90%.
:1, 0.6 mL/min]: 14.331 min (major), 18.652 min (minor), ee = 90%.
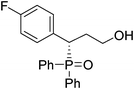
(S)-3-(4-fluorophenyl)-3-(diphenylphosphoryl)propan-1-ol (Table 3, Entry 7)
The product was obtained in 75% yield, yellow solid. [α]D19.8 = −56.6 (c 0.54, CH3OH); mp = 186–188 °C. 1H NMR (400 MHz, CD3OD): δ (ppm) 8.05–8.00 (m, 2H), 7.64–7.60 (m, 3H), 7.57–7.52 (m, 2H), 7.42–7.39 (m, 1H), 7.34–7.30 (m, 4H), 6.95–6.91 (m, 2H), 4.12–4.06 (m, 1H), 3.52–3.47 (m, 1H), 3.30–3.25 (m, 1H), 2.26–2.16 (m, 1H), 2.13–2.04 (m, 1H). 13C NMR (100 MHz, CD3OD): δ (ppm) 163.3, 132.1, 131.7, 131.5, 131.4, 131.3, 131.0, 130.9, 130.6, 130.5, 128.8, 128.7, 128.1, 128.0, 114.8, 114.6, 58.5,41.3, 31.6. 31P NMR (CD3OD): δ (ppm) 36.6 (s). HRMS (EI): exact mass calculated for M+ (C21H20FO2P) requires m/z 354.1185, found m/z 354.1183. The enantiomeric excess was determined by HPLC. [IC-H column, 220 nm, Hexane:EtOH = 9
 :1, 0.6 mL/min]: 20.573 min (major), 27.805 min (minor), ee = 94%.
:1, 0.6 mL/min]: 20.573 min (major), 27.805 min (minor), ee = 94%.
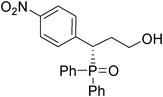
(S)-3-(4-nitrophenyl)-3-(diphenylphosphoryl)propan-1-ol (Table 3, Entry 8)
The product was obtained in 77% yield, yellow solid. [α]D20 = −42.4 (c 0.54, CH3OH); mp = 196–198 °C. 1H NMR (400 MHz, CD3OD): δ (ppm) 8.06 (d, J = 8.4 Hz, 3H), 7.87 (d, J = 8.8 Hz, 1H), 7.68–7.64 (m, 3H), 7.58–7.53 (m, 4H), 7.45–7.41 (m, 1H), 7.37–7.33 (m, 2H), 4.33–4.27 (m, 1H), 3.52–3.47 (m, 1H), 3.29–3.22 (m, 1H), 2.34–2.25 (m, 1H), 2.16–2.10 (m, 1H). 13C NMR (100 MHz, CD3OD): δ (ppm) 143.4, 132.3, 131.8, 131.0, 130.9, 130.8, 130.7, 130.5, 130.4, 129.0, 128.9, 128.8, 128.3, 128.2, 126.7, 125.5, 122.8, 58.4, 42.3, 31.5. 31P NMR (CD3OD): δ (ppm) 35.7 (s). HRMS (EI): exact mass calculated for M+ (C21H20NO4P) requires m/z 381.1130, found m/z 381.1134. The enantiomeric excess was determined by HPLC. [IC-H column, 220 nm, Hexane:EtOH = 8
 :1, 0.8 mL/min]: 31.233 min (major), ee = 99%.
:1, 0.8 mL/min]: 31.233 min (major), ee = 99%.
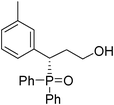
(S)-3-(3-methylphenyl)-3-(diphenylphosphoryl)propan-1-ol (Table 3, Entry 9)
The product was obtained in 93% yield, white solid. [α]D19 = −51.0 (c 0.82, CH3OH); mp = 185–187 °C. 1H NMR (400 MHz, CD3OD): δ (ppm) 8.03–7.98 (m, 2H), 7.66–7.59 (m, 3H), 7.54–7.49 (m, 2H), 7.41–7.36 (m, 1H), 7.32–7.28 (m, 2H), 7.11–7.06 (m, 3H), 6.98 (d, J = 6.8 Hz, 1H),4.03–3.97 (m, 1H), 3.52–3.46 (m, 1H), 3.31–3.27 (m, 1H), 2.28–2.19 (m, 1H), 2.12–2.02 (m, 1H). 13C NMR (100 MHz, CD3OD): δ (ppm) 137.7, 134.7, 132.0, 131.8, 131.4, 131.1, 131.0, 130.8, 130.7, 130.6, 130.5, 128. 8, 128.7, 128.0, 127.9, 127.8, 127.6, 126.8, 58.7, 42.1, 31.6, 20.0. 31P NMR (CD3OD): δ (ppm) 36.9 (s). HRMS (EI): exact mass calculated for M+ (C22H23O2P) requires m/z 350.1436, found m/z 350.1438. The enantiomeric excess was determined by HPLC. [IA-H column, 220 nm, Hexane:EtOH = 9
 :1, 0.7 mL/min]: 16.248 min (major), 17.574 min (minor), ee = 90%.
:1, 0.7 mL/min]: 16.248 min (major), 17.574 min (minor), ee = 90%.
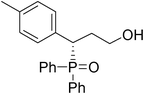
(S)-3-(4-methylphenyl)-3-(diphenylphosphoryl)propan-1-ol (Table 3, Entry 10)
The product was obtained in 51% yield, white solid. [α]D15.6 = −62.2 (c 0.58, CH3OH); mp = 232–234 °C. 1H NMR (400 MHz, CD3OD): δ (ppm) 8.03–7.98 (m, 2H), 7.66–7.59 (m, 3H), 7.59–7.51 (m, 2H), 7.42–7.38 (m, 1H), 7.33–7.29 (m, 2H), 7.20–7.17 (m, 2H), 7.02 (d, J = 8.0 Hz, 2H), 4.03–3.98 (m, 1H), 3.51–3.45 (m, 1H), 3.31–3.26 (m, 1H), 2.25 (s, 3H), 2.22–2.17 (m, 1H), 2.11–2.03 (m, 1H). 13C NMR (100 MHz, CD3OD): δ (ppm) 136.7, 131.9, 131.8, 131.6, 131.4, 131.0, 130.9, 130.8, 130.7, 130.6, 129.6, 129.6, 128.8, 128.6, 128.6, 128.6, 128.0, 127.8, 58.7, 41.6, 31.6, 19.6. 31P NMR (CD3OD): δ (ppm) 36.9 (s).HRMS (EI): exact mass calculated for M+ (C22H23O2P) requires m/z 350.1436, found m/z 350.1434. The enantiomeric excess was determined by HPLC. [IC-H column, 220 nm, Hexane:EtOH = 6
 :1, 0.7 mL/min]: 15.652 min (major), 23.167 min (minor), ee = 90%.
:1, 0.7 mL/min]: 15.652 min (major), 23.167 min (minor), ee = 90%.
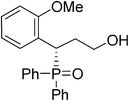
(S)-3-(2-methoxylphenyl)-3-(diphenylphosphoryl)propan-1-ol (Table 3, Entry 11)
The product was obtained in 87% yield, yellow solid. [α]D19.2 = −31.0 (c 0.90, CH3OH); mp = 152–157 °C. 1H NMR (400 MHz, CD3OD): δ (ppm) 7.96–7.91 (m, 2H), 7.62–7.50 (m, 4H), 7.38–7.29 (m, 3H), 7.21–7.17 (m, 2H), 7.10 (t, J = 8.0 Hz, 1H), 6.88 (t, J = 7.6 Hz, 1H), 6.64 (d, J = 8.0 Hz, 1H), 4.60–4.55 (m, 1H), 3.46 (s, 3H), 3.42–3.37 (m, 1H), 3.25–3.21 (m, 1H), 2.25–2.15 (m, 1H), 2.09–1.99 (m, 1H). 13C NMR (100 MHz, CD3OD): δ (ppm) 158.8, 133.4, 133.0, 132.7, 132.5, 132.4, 132.0, 131.9, 130.4, 130.2, 130.1, 129.6, 129.0, 128.9, 124.4, 121.7, 111.5, 60.5, 55.7, 34.5, 32.8. 31P NMR (CD3OD): δ (ppm) 35.3 (s). HRMS (EI): exact mass calculated for M+ (C22H23O3P) requires m/z 366.1385, found m/z 366.1383. The enantiomeric excess was determined by HPLC. [IA-H column, 220 nm, Hexane:EtOH = 8
 :1, 0.8 mL/min]: 15.763 min (major), 1.978 min (minor), ee = 86%.
:1, 0.8 mL/min]: 15.763 min (major), 1.978 min (minor), ee = 86%.
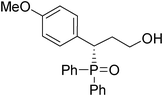
(S)-3-(4-methoxylphenyl)-3-(diphenylphosphoryl)propan-1-ol (Table 3, Entry 12)
The product was obtained in 41% yield, yellow solid. [α]D15.8 = −41.2 (c 0.43, CH3OH); mp = 201–203 °C. 1H NMR (400 MHz, CD3OD): δ (ppm) 8.02–7.98 (m, 2H), 7.65–7.61 (m, 3H), 7.55–7.50 (m, 2H), 7.43–7.32 (m, 4H), 7.22–7.20 (m, 2H), 6.76 (d, J = 8.4 Hz, 1H), 4.02–3.96 (m, 1H), 3.73 (s, 3H), 3.52–3.46 (m, 1H), 3.31–3.25 (m, 1H), 2.23-2.14 (m, 1H), 2.09–2.00 (m, 1H), 2.04 (brs, 1H). 13C NMR (100 MHz, CD3OD): δ (ppm) 159.0, 131.9, 131.4, 131.0, 130.9, 130.8, 130.7, 130.6, 130.5, 128.8, 128.6, 128.3, 128.0, 127.9, 126.4, 125.5, 113.4, 58.6, 54.2, 41.2, 31.6. 31P NMR (CD3OD): δ (ppm) 37.0 (s). HRMS (EI): exact mass calculated for M+ (C22H23O3P) requires m/z 366.1385, found m/z 366.1390. The enantiomeric excess was determined by HPLC. [IA-H column, 220 nm, Hexane:EtOH = 8
 :1, 0.8 mL/min]: 20.474 min (major), 32.508 min (minor), ee = 90%.
:1, 0.8 mL/min]: 20.474 min (major), 32.508 min (minor), ee = 90%.
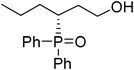
(R)-3-(diphenylphosphoryl)hexan-1-ol (Table 3, Entry 13)
The product was obtained in 77% yield, yellow solid. [α]D20.1 = −68.6 (c 0.56, CH3OH); mp = 116 °C. 1H NMR (400 MHz, CD3OD): δ (ppm) 7.90–7.84 (m, 4H), 7.62–7.53 (m, 6H), 3.62–3.49 (m, 2H), 2.76–2.68 (m, 1H), 1.99–1.87 (m, 1H), 1.80–1.60 (m, 2H), 1.57–1.46 (m, 2H), 1.28–1.24 (m, 1H), 0.82 (t, J = 7.2 Hz, 3H). 13C NMR (100 MHz, CD3OD): δ (ppm) 132.2, 131.8, 131.7, 131.2, 130.8, 130.7, 130.6, 130.5, 128.7, 128.6, 128.5, 128.4, 59.1, 33.1, 30.1, 29.4, 20.8, 13.0. 31P NMR (CD3OD): δ (ppm) 40.8 (s). HRMS (EI): exact mass calculated for M+ (C18H23O2P) requires m/z 302.1436, found m/z 302.1433. The enantiomeric excess was determined by HPLC. [IC-H column, 220 nm, Hexane:EtOH = 8
 :1, 0.6 mL/min]: 42.028 min (major), 38.146 min (minor), ee = 82%.
:1, 0.6 mL/min]: 42.028 min (major), 38.146 min (minor), ee = 82%.
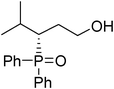
(S)-3-(diphenylphosphoryl)-4-methylpentan-1-ol (Table 3, Entry 14)
The product was obtained in 36% yield, yellow solid. [α]D20.3 = −2.453 (c 0.53, CH3OH); mp = 158–160 °C. 1H NMR (400 MHz, CD3OD): δ (ppm) 7.92–7.88 (m, 4H), 7.60–7.52 (m, 6H), 3.34–3.30 (m, 2H), 2.66–2.60 (m, 1H), 2.17–2.09 (m, 1H), 1.99–1.82 (m, 2H), 1.05 (d, J = 6.8 Hz, 3H), 1.00 (d, J = 7.2 Hz, 3H),. 13C NMR (100 MHz, CD3OD): δ (ppm) 133.3, 132.4, 131.6, 131.5, 130.5, 130.4, 128.7, 128.6, 128.5, 128.4, 60.7, 47.2, 38.3, 27.2, 25.6, 21.8, 17.5. 31P NMR (CD3OD): δ (ppm) 39.5 (s). HRMS (MALDI): exact mass calculated for M+ (C18H23O2P) requires m/z 302.1436, found m/z 302.1437. The enantiomeric excess was determined by HPLC. [IC-H column, 220 nm, Hexane:EtOH = 8
 :1, 0.6 mL/min]: 26.154 min (major), ee = 99%.
:1, 0.6 mL/min]: 26.154 min (major), ee = 99%.
Acknowledgements
We are grateful for the financial support from National Natural Science Foundation of China (20902018), Shanghai Pujiang Program (08PJ1403300), the Fundamental Research Funds for the Central Universities and 111 Project (B07023).References
- (a) B. K. Alnasleh, W. M. Sherrill and M. Rubin, Org. Lett., 2008, 10, 3231 CrossRef CAS; (b) V. Devreux, J. Wiesner, H. Jomaa, J. Rozenski, J. Van der Eycken and S. Van Calenbergh, J. Org. Chem., 2007, 72, 3783 CrossRef CAS; (c) V. D. Romanenko and V. P. Kukhar, Chem. Rev., 2006, 106, 3868 CrossRef CAS; (d) T. Haemers, J. Wiesner, S. Van Poecke, J. Goeman, D. Henschker, E. Beck, H. Jomaa and S. Van Calenbergh, Bioorg. Med. Chem. Lett., 2006, 16, 1888 CrossRef CAS; (e) T. Kurz, K. Schlüter, U. Kaula, B. Bergmann, R. D. Walter and D. Geffken, Bioorg. Med. Chem., 2006, 14, 5121 CrossRef CAS; (f) F. Palacios, C. Alonso and J. M. de los Santos, Chem. Rev., 2005, 105, 899 CrossRef CAS; (g) K. Moonen, E. Van Meenen, A. Verwee and C. V. Stevens, Angew. Chem., Int. Ed., 2005, 44, 7407 CrossRef CAS; (h) K. Moonen, I. Laureyn and C. V. Stevens, Chem. Rev., 2004, 104, 6177 CrossRef CAS; (i) J. L. Methot and W. R. Roush, Adv. Synth. Catal., 2004, 346, 1035 CrossRef CAS; (j) W. Tang and X. Zhang, Chem. Rev., 2003, 103, 3029 CrossRef CAS; (k) R. Noyori, Angew. Chem., Int. Ed., 2002, 41, 2008 CrossRef CAS; (l) W. S. Knowles, Angew. Chem., Int. Ed., 2002, 41, 1998 CrossRef CAS.
- (a) R. Schuecker, K. Mereiter, F. Spindler and W. Weissensteiner, Adv. Synth. Catal., 2010, 352, 1063 CrossRef CAS; (b) K. Barta, G. Franciò, W. Leitner, G.C. Lloyd-Jones and I. R. Sheppersonb, Adv. Synth. Catal., 2008, 350, 2013 CrossRef CAS; (c) X. Liu, T. N. Le, Y. Lu, Y. Xiao, J. Ma and X. Li, Org. Biomol. Chem., 2008, 6, 3997 RSC; (d) B. Wiese, G. Knühl, D. Flubacher, J. W. Prieß, B. Ulriksen, K. Brödner and G. Helmchen, Eur. J. Org. Chem., 2005, 3246 CrossRef CAS; (e) Y. Li, S. D. Aubert, E. G. Maes and F. M. Raushel, J. Am. Chem. Soc., 2004, 126, 8888 CrossRef CAS; (f) K. M. Pietrusiewicz and M. Zablocka, Chem. Rev., 1994, 94, 1375 CrossRef; (g) G. Castelot-Deliencourt, E. Roger, X. Pannecoucke and J.-C. Quirion, Eur. J. Org. Chem., 2001, 3031 CrossRef CAS.
- (a) Ł. Albrecht, A. Albrecht, H. Krawczyk and K. A. Jøgensen, Chem.–Eur. J., 2010, 16, 28 CrossRef CAS; (b) D. Enders, A. S. Dizier, M. I. Lannou and A. Lenzen, Eur. J. Org. Chem., 2006, 29 CrossRef CAS.
- (a) J.-J. Feng, X. –F. Chen, M. Shi and W. –L. Duan, J. Am. Chem. Soc., 2010, 132, 5562 CrossRef CAS; (b) A. D. Sadow and A. Togni, J. Am. Chem. Soc., 2005, 127, 17012 CrossRef CAS; (c) C. Scriban, I. Kovacik and D. S. Glueck, Organometallics, 2005, 24, 4871 CrossRef CAS; (d) A. D. Sadow, I. Haller, L. Fadini and A. Togni, J. Am. Chem. Soc., 2004, 126, 14704 CrossRef CAS.
- (a) I. Ibrahem, P. Hammar, J. Vesely, R. Rios, L. Eriksson and A. Córdova, Adv. Synth. Catal., 2008, 350, 1875 CrossRef CAS; (b) E. Maerten, S. Cabrera, A. Kjærsgaard and K. A. Jørgensen, J. Org. Chem., 2007, 72, 8893 CrossRef CAS; (c) A. Carlone, G. Bartoli, M. Bosco, L. Sambri and P. Melchiorre, Angew. Chem., Int. Ed., 2007, 46, 4504 CrossRef CAS; (d) I. Ibrahem, R. Rios, J. Vesely, P. Hammar, L. Eriksson, F. Himo and A. Córdova, Angew. Chem., Int. Ed., 2007, 46, 4507 CrossRef CAS.
- G. Bartoli, M. Bosco, A. Carlone, M. Locatelli, A. Mazzanti, L. Sambri and P. Melchiorre, Chem. Commun., 2007, 722 RSC.
- (a) L. Hong, W. Sun, C. Liu, D. Zhao and R. Wang, Chem. Commun., 2010, 46, 2856 RSC; (b) X. Fu, W. -T. Loh, Y. Zhang, T. Chen, T. Ma, H. Liu, J. Wang and C. –H. Tan, Angew. Chem., Int. Ed., 2009, 48, 7387 CrossRef CAS; (c) F. Wang, X. Liu, X. Cui, Y. Xiong, X. Zhou and X. Feng, Chem.–Eur. J., 2009, 15, 589 CrossRef CAS; (d) D. Zhao, Y. Yuan, A. S. C. Chan and R. Wang, Chem.–Eur. J., 2009, 15, 2738 CrossRef CAS; (e) D. Zhao, Y. Wang, L. Mao and R. Wang, Chem.–Eur. J., 2009, 15, 10983 CrossRef CAS; (f) P. Merino, E. Marqués-López and R. P. Herrera, Adv. Synth. Catal., 2008, 350, 1195 CrossRef CAS; (g) J. P. Abell and H. Yamamoto, J. Am. Chem. Soc., 2008, 130, 10521 CrossRef CAS; (h) N. Ajellal, C. M. Thomas and J. -F. Carpentier, Adv. Synth. Catal., 2006, 348, 1093 CrossRef CAS; (i) L. Tedeschi and D. Enders, Org. Lett., 2001, 3, 3515 CrossRef CAS.
- (a) Y. Zhu, J. P. Malerich and V. H. Rawal, Angew. Chem., Int. Ed., 2010, 49, 153 CAS; (b) M. Terada, T. Ikehara and H. Ube, J. Am. Chem. Soc., 2007, 129, 14112 CrossRef CAS; (c) X. Fu, Z. Jiang and C.-H. Tan, Chem. Commun., 2007, 5058 RSC; (d) J. Wang, L. D. Heikkinen, H. Li, L. Zu, W. Jiang, H. Xie and W. Wang, Adv. Synth. Catal., 2007, 349, 1052 CrossRef CAS; (e) D. Enders, L. Tedeschi and J.W. Bats, Angew. Chem., Int. Ed., 2000, 39, 4605 CrossRef CAS.
- (a) S. Nakamura, M. Hayashi, Y. Hiramatsu, N. Shibata, Y. Funahashi and T. Toru, J. Am. Chem. Soc., 2009, 131, 18240 CrossRef CAS; (b) D. Pettersen, M. Marcolini, L. Bernardi, F. Fini, R. P. Herrera, V. Sgarzani and A. Ricci, J. Org. Chem., 2006, 71, 6269 CrossRef CAS; (c) T. Akiyama, H. Morita, J. Itoh and K. Fuchibe, Org. Lett., 2005, 7, 2583 CrossRef CAS; (d) G. D. Joly and E. N. Jacobsen, J. Am. Chem. Soc., 2004, 126, 4102 CrossRef CAS; (e) I. Schlemminger, Y. Saida, H. Gróger, W. Maison, N. Durot, H. Sasai, M. Shibasaki and J. Martens, J. Org. Chem., 2000, 65, 4818 CrossRef CAS; (f) H. Gróger, Y. Saida, H. Sasai, K. Yamaguchi, J. Martens and M. Shibasaki, J. Am. Chem. Soc., 1998, 120, 3089 CrossRef; (g) H. Sasai, S. Arai, Y. Tahara and M. Shibasaki, J. Org. Chem., 1995, 60, 6656 CrossRef CAS.
- S. Wen, P. Li, H. Wu, F. Yu, X. Liang and J. Ye, Chem. Commun., 2010, 46, 4806 RSC.
- (a) M. Marigo, T. C. Wabnitz, D. Fielenbach and K. A. Jørgensen, Angew. Chem., Int. Ed., 2005, 44, 794 CrossRef CAS; (b) Y. Hayashi, H. Gotoh, T. Hayashi and M. Shoji, Angew. Chem., Int. Ed., 2005, 44, 4212 CrossRef CAS.
- The crystallographic data of 7ag for this paper has been deposited in the Cambridge Structural Database, Cambridge Crystallographic Data Centre, Cambridge CB2 1EZ, United Kingdom (CSD reference no. 818724).
Footnote |
| † Electronic supplementary information (ESI) available: NMR and HPLC spectra of the products. CCDC reference numbers 818724. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1ra00269d |
| This journal is © The Royal Society of Chemistry 2011 |