Adsorption of proteins to thin-films of PDMS and its effect on the adhesion of human endothelial cells†
Karin Y.
Chumbimuni-Torres
a,
Ramon E.
Coronado
b,
Adelphe M.
Mfuh
a,
Carlos
Castro-Guerrero
c,
Maria Fernanda
Silva
d,
George R.
Negrete
a,
Rena
Bizios
b and
Carlos D.
Garcia
*a
aDepartment of Chemistry, The University of Texas at San Antonio, TX 78249, USA. E-mail: carlos.garcia@utsa.edu; Tel: (210) 458-5774
bDepartment of Biomedical Engineering, The University of Texas at San Antonio, San Antonio, USA
cDepartment of Physics and Astronomy, The University of Texas at San Antonio, San Antonio, USA
dSchool of Agronomic Sciences - IBAM-CONICET, National University of Cuyo, Mendoza, Argentina
First published on 31st August 2011
Abstract
This paper describes a simple and inexpensive procedure to produce thin-films of poly(dimethylsiloxane). Such films were characterized by a variety of techniques (ellipsometry, nuclear magnetic resonance, atomic force microscopy, and goniometry) and used to investigate the adsorption kinetics of three model proteins (fibrinogen, collagen type-I, and bovine serum albumin) under different conditions. The information collected from the protein adsorption studies was then used to investigate the adhesion of human dermal microvascular endothelial cells. The results of these studies suggest that these films can be used to model the surface properties of microdevices fabricated with commercial PDMS. Moreover, the paper provides guidelines to efficiently attach cells in BioMEMS devices.
1. Introduction
Recent developments in fabrication procedures and instrumentation1 have enabled the development and application of microfluidic devices to chemical, biomedical,2,3pharmaceutical,4 environmental, and forensic sciences.5 Among other advantages, these devices have the potential to combine sample-handling capabilities, custom design, low-power requirements, and portability while providing similar performance to their standard bench-top counterparts. Additionally, various well-established laboratory techniques can be easily integrated in microfluidic devices, increasing the versatility and throughput of these systems.6Although microfluidic devices were initially constructed using glass, a wide variety of polymeric materials have been recently used.7–10 Among them, poly(dimethylsiloxane) (PDMS) has been one of the most widely used materials because it allows rapid fabrication of devices using relatively simple and inexpensive instrumentation.11–14 Although the general attributes of PDMS and their molecular bases were recognized many decades ago,15 it is worth highlighting its chemical inertness, low electrical conductivity, elasticity,6 and optical transparency.7,16 PDMS does not swell or dissolve in a number of solvents17 and is permeable to most gases, including oxygen.18 Despite several advantages of PDMS for microfluidic devices, several drawbacks still limit the applicability of this material.19 Probably one of the most noteworthy characteristics of PDMS is its hydrophobic nature (contact angle ∼110°) and porosity, allowing the absorption20,21 and adsorption22 of a wide variety of molecules. Because such processes can have negative effects in devices used for separations,23,24 several procedures have been developed to control the surface properties of PDMS.25–28 Taking advantage of the low surface energy of PDMS,15 similar procedures have been used to produce patterns and arrays by exposing the surface of this material to target proteins.29–34 In this regard, controlling not only the amount of adsorbed protein, but also the orientation and conformation of the protein layer is particularly important when proteins (such as fibronectin35) mediate interactions with other biological entities such as cells.36–40 Despite the advantages and the intriguing nature of the studies reported in literature, only few research groups41,42 have investigated the influence of adsorption kinetics on the biological activity of proteins adsorbed to PDMS. Because the adsorption rate can have a significant influence on the conformation and subsequent biological activity of the adsorbed protein layer, obtaining such information is critical to rationally design micro-electro mechanical systems for biological applications (BioMEMS).
For the aforementioned reasons, and aiming to address this gap in knowledge, thin-films of two n-dimethylsiloxanes were deposited on silicon substrates and characterized by a variety of complementary techniques. This approach developed to deposit thin-films of PDMS proved to be simpler and faster than others previously reported,22,43–47 some of which did not render uniform layers of PDMS and thus were incompatible with ellipsometric measurements. The deposited thin-films, that have identical chemical composition and similar macroscopic properties than commercial PDMS (e.g., Sylgard 184), were then used to investigate the adsorption kinetics of three model proteins: fibrinogen (Fib), collagen type I (Col), and bovine serum albumin (BSA) under different protein concentrations and pH values. Spectroscopic ellipsometry was used to characterize the optical properties of the films and to follow the adsorption process of each protein in real time. Finally, the selected substrates were used to evaluate the role of the characterized adsorbed protein layer on the adhesion and morphology of human dermal endothelial cells.
2. Experimental
2.1. Reagents and solutions
All chemicals were analytical reagent grade and used as received. Hydrogen peroxide, sodium hydroxide, and sodium dodecyl sulfate (SDS) were purchased from Fisher Scientific (Fair Lawn, NJ). All aqueous solutions were prepared using 18 MΩ cm water (NANOpure Diamond, Barnstead; Dubuque, IA). The pH of the solutions was adjusted using either 1 M NaOH or 1 M HCl and measured using a glass electrode and a digital pH meter (Orion 420A+, Thermo; Waltham, MA). Two chlorine-terminated n-dimethylsiloxanes were selected for these studies: 1,3-dicholoro-1,1,3,3-tetramethyldisiloxane (n = 2) and 1,7-dicholoro-octamethyltetrasiloxane (n = 4). These chemicals were purchased from Sigma-Aldrich (St. Louis, MO) and used as received. Dichloromethane (DCM) was also purchased from Sigma Aldrich and isopropanol (analytical grade) was obtained from Fisher Scientific. Unless otherwise stated, solutions of either bovine serum albumin (Fraction V, Fisher Scientific) or fibrinogen (Fraction I, type 1-S from bovine plasma, Sigma-Aldrich) were prepared in 10 mM phosphate buffer pH = 7.0. Collagen type-I (from rat tail) was purchased from Invitrogen (Grand Island, NY) and dissolved in acetate buffer (0.04 M, pH = 4.8) following manufacturer's instructions, ensuring complete dissolution. The most relevant properties of the chosen proteins are summarized in Table 1. Isoelectric points (IEP) were obtained from the literature. Data related to the temperature at which the denaturation transition is half completed (Tm) were also obtained from the literature and included to provide information regarding the structural stability of the chosen molecules in comparison to the control protein, BSA (which is typically considered a softprotein prone to denaturation upon adsorption).48,49 Unless otherwise stated, all experiments were conducted at room temperature (22 ± 1 °C).2.2. Synthesis and characterization of nanostructured films
Standard <111> silicon wafers (Si/SiO2, Sumco; Phoenix, AZ) were initially scored using a computer-controlled engraver (Gravograph IS400, Gravotech; Duluth, GA). The process defined substrates of 1 cm in width and 3 cm in length that were then manually cut and cleaned in piranha solution (30% hydrogen peroxide and 70% sulfuric acid) at 90 °C for 30 min. After thorough rinsing with water, the substrates were immersed and stored in ultrapure water until use. In order to deposit the thin-films on the substrates, the clean wafers were dried at 80 °C for 4 h and immersed in solutions containing the corresponding n-dimethylsiloxane (dissolved in dichloromethane) for 3 h, under gentle stirring (100 rpm; Innova 2000; New Brunswick Sci.). Subsequently, the coated wafers were sequentially rinsed with isopropanol and water, dried in a convection oven, and stored until use. Under the selected conditions, the attachment reaction proceeds rather quickly leading to the deposition of a layer of n-dimethylsiloxane covalently linked to the substrate by a head-to-surface arrangement.53,54Films produced by the deposition reaction of 1,7-dicholoro-octamethyltetrasiloxane were characterized by nuclear magnetic resonance (1H-NMR and 13C-NMR in CDCl3) using a Varian INOVA 500 MHz Spectrometer. For comparison purposes, the 1H-NMR of 1,7-dicholoro-octamethyltetrasiloxane was also obtained in CDCl3. In order to analyze the reaction products, silica beads (>15 nm) were modified with 1,7-dicholoro-octamethyltetrasiloxane, suspended in CDCl3, and analyzed under conditions similar to those of the precursors in solution.
Contact angle measurements, used to evaluate the surface hydrophobicity of the prepared substrates, were performed using a VCA-Optima surface analysis system (Ast Products, Inc.; Billerica, MA) and analyzed using the software provided by the manufacturer, 30 s after dispensing 2 μL of deionized water. Atomic force microscopy (AFM) images were obtained using a Veeco diMultiMode Nanoscope V scanning probe microscope operating in tapping and non-contact mode. The samples were analyzed without any coating.
2.3. Spectroscopic ellipsometry
Experiments were performed using a variable angle spectroscopic ellipsometer (WVASE, J.A. Woollam Co., Lincoln, NE) following a procedure described elsewhere.55–58 Under these conditions, spectroscopic ellipsometry has proven suitable to study the kinetics of protein adsorption processes59 and to calculate the optical constants, thickness, and microstructure of the adsorbed film. The sensitivity of the technique, critically evaluated elsewhere,60 was also considered appropriate for the purpose of the present study. Collected data (ellipsometric angles as function of time, angle, and/or wavelength) were modeled using the WVASE software package (J. A. Woollam Co., Lincoln, NE). Differences between the experimental and model-generated data were assessed by the mean square error (MSE),61 a built-in function in WVASE based on eqn (2),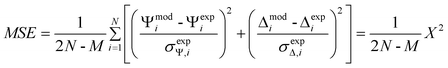 | (2) |
Before each protein adsorption experiment, the thickness of the deposited layer was measured by placing the substrate in the ellipsometry cell59 and by performing a spectroscopic scan in the 300 to 800 nm range (with 10 nm steps) using the corresponding aqueous buffer as the ambient medium. Then, the dynamic experiment was initiated by pumping background electrolyte through the cell at a rate of 1 mL min−1 to establish the baseline. Next, the protein solution was introduced, and the adsorption process initiated. An initial fast process, followed by a slower one, was always observed. After a plateau in the signal was observed, the dynamic scan was stopped, and a spectroscopic scan was collected to verify the thickness of the adsorbed protein layer. Experiments performed in this way provided data for calculating the initial protein adsorption rate and the saturation amount. Subsequently to protein adsorption, a desorption experiment was performed using the corresponding buffer (∼10 min) and then 4 mmol L−1 SDS (30 min). In between experiments, the flow cell and tubing were thoroughly rinsed (with 0.1 mM SDS and water) to avoid cross-contamination.
2.4. Optical models
One of the limitations of ellipsometry is the requirement for an optical model that describes the properties of the substrates in terms of optical constants (refractive index, n, and extinction coefficient, k) and thickness (d).62 In the present study, the model used to represent the optical properties of the substrates was composed of a layer of Si (bulk; d = 1 mm), a layer of SiO2 (d = 2.5 ± 0.5 nm), and a transparent layer (representing the n(dimethylsiloxane) film), represented by a Cauchy function (eqn (1)),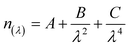 | (1) |
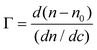 | (2) |
2.5. Cell culture, cell adhesion and cell morphology experiments
Human dermal microvascular endothelial cells (HDMEC) were purchased from Sciencell (Carlsbad, CA) and cultured under standard conditions (i.e., a humidified, 37 °C, 5% CO2/95% air environment) in endothelial-cell complete medium (Sciencell; the composition and concentration of the supplements contained in this complete medium are proprietary vendor information). When confluent, the cells were passaged after a short (6 min) exposure to a trypsin/EDTA solution (BioCell; Rancho Dominguez, CA), and re-suspended in fresh serum-free basal endothelial-cell media (without supplements). Cells at passage number 3 were used for the experiments.For these studies, substrates (1 cm × 1 cm) were modified with 1,7-dicholoro-octamethyltetrasiloxane according to the described procedure and then immersed (under constant agitation at 100 rpm) in solutions containing each one of the proteins tested under the chosen experimental conditions for two hours. Next, the protein-modified substrates were thoroughly rinsed with buffer (to remove loosely-bound proteins) and placed one each in individual wells of polystyrene tissue-culture plates (12-wells/plate, 22.1 mm internal diameter).
Human dermal microvascular endothelial cells were seeded (48![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells/well containing one substrate) in Dulbecco's Phosphate Buffered Saline with neither calcium nor magnesium (DPBS) and allowed to interact under standard conditions for 3 h. The adhered cells were then fixed in situ using 4% formaldehyde in DPBS for 15 min, rinsed twice with fresh DPBS, treated with 0.1% Triton-X, and finally stained with Alexa Fluor 568® Phalloidin (to visualize the F-actin filaments of the cytoskeleton) and/or 4′,6-diamidino-2-phenylindole dilactate (DAPI) (to visualize the cell nuclei). Both fluorescent stains were purchased from Invitrogen (Carlsbad, CA) and were used following procedures provided by the vendor. A fluorescent microscope (LEICA DM 5500B) was used to visualize the F-actin filaments (excitation/emission of 578/600 nm, respectively) and the cell nuclei (excitation at 358 nm/emission at 461 nm). All experiments were run in duplicate and repeated at three separate times. In all cases, 20 micrographs/sample were examined to determine adhering cell morphology and number of attached cells.
000 cells/well containing one substrate) in Dulbecco's Phosphate Buffered Saline with neither calcium nor magnesium (DPBS) and allowed to interact under standard conditions for 3 h. The adhered cells were then fixed in situ using 4% formaldehyde in DPBS for 15 min, rinsed twice with fresh DPBS, treated with 0.1% Triton-X, and finally stained with Alexa Fluor 568® Phalloidin (to visualize the F-actin filaments of the cytoskeleton) and/or 4′,6-diamidino-2-phenylindole dilactate (DAPI) (to visualize the cell nuclei). Both fluorescent stains were purchased from Invitrogen (Carlsbad, CA) and were used following procedures provided by the vendor. A fluorescent microscope (LEICA DM 5500B) was used to visualize the F-actin filaments (excitation/emission of 578/600 nm, respectively) and the cell nuclei (excitation at 358 nm/emission at 461 nm). All experiments were run in duplicate and repeated at three separate times. In all cases, 20 micrographs/sample were examined to determine adhering cell morphology and number of attached cells.
3. Results and discussion
3.1. Characterization of nanofilms
An optical model was developed to represent the optical properties of the substrates and to interpret the adsorption experiments. In all cases, a very good agreement (MSE < 10) between the experimental and the model-calculated data was obtained, indicating that the proposed model enables the description of the properties of the substrates and that it can be used to calculate thickness of the films. As a representative example, Fig. 1A shows the data collected during a spectroscopic scan (dependence of Ψ and Δ as a function of λ) obtained at three different angles of incidence for a thin-film of PDMS (fabricated from the reaction of 1,7-dicholoro-octamethyltetrasiloxane). Fig. 1A also shows the data generated using the corresponding optical model. As can be observed, a very good agreement (MSE < 5) between the experimental (data points) and the model-generated data (lines) was obtained. The optical constants calculated from these experiments (data not shown) are also in agreement with previously reported values for PDMS,72 though measured in a narrower spectral interval.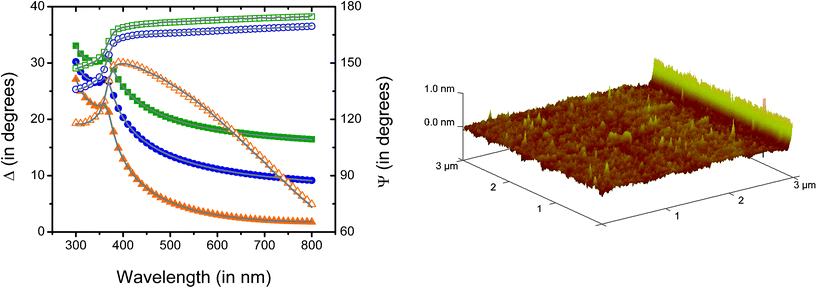 | ||
| Fig. 1 A (left): Spectroscopic scans corresponding to data experimentally collected (points) and calculated with the optical model (lines) corresponding to a Si/SiO2 substrate coated with a thin-film of PDMS of 2.01 ± 0.02 nm (MSE=4.3) fabricated from the reaction of 1,7-dicholoro-octamethyltetrasiloxane. Ψ and Δ values are represented with solid and open symbols, respectively. Angle of incidence: 65° (solid square and open square), 70° (solid circle and open circle), and 75° (solid triangle and open triangle). B (right): 3D AFM image corresponding to a Si/SiO2 substrate coated with a thin-film of PDMS of 2.01 ± 0.02 nm fabricated from the reaction of 1,7-dicholoro-octamethyltetrasiloxane. | ||
Additionally, reflective UV-Vis spectra (RP and RS; data not shown) confirmed the presence of a transparent film (measured in the 250–800 nm range) with isotropic properties, also in good agreement with the optical properties of PDMS.73,74 Furthermore, the aforementioned optical model also allowed calculation of the thickness of the n-dimethylsiloxane films deposited on the Si/SiO2 substrates. According to our results, treating the Si/SiO2 wafers with either 1,3-dicholoro-tetramethyldisiloxane or 1,7-dicholoro-octamethyltetrasiloxane produced uniform films with average thickness values of 1.3 ± 0.1 nm and 2.1 ± 0.2 nm (n = 3, independently prepared), respectively. Because the molecular dimensions of di- and tetra-(dimethylsiloxane) were calculated to be 0.65 and 1.33 nm, respectively (see ESI†) our results suggest that in both cases, the films are constituted by entangled oligomers (dimers and/or trimers) of the corresponding n-dimethylsiloxane covalently linked to the surface. Such arrangement closely resembles the porous structure of commercial PDMS. This conclusion is in good agreement with reports in the literature stating that many of the properties of PDMS are consequence of the static and dynamic structure of the siloxane backbone75 and the hydrophobicity of the methyl chain.76 In the case of the present study, these properties are indistinguishable from those of commercial PDMS. Also in agreement with previously reported values for commercial PDMS,77 the contact angle of the deposited films was 114 ± 2° (n = 3, independently prepared), indicating the presence of a rather hydrophobic surface. Furthermore, the topography of the substrates was investigated by atomic force microscopy (see representative image in Fig. 1B) and showed the presence of a smooth film on the silica wafer with abundant nanostructured features on the surface. The size of those features (as calculated form the roughness of the AFM images) was 0.2 ± 0.1 nm.
Films made with 1,3-dicholoro-1,1,3,3-tetramethyldisiloxane and 1,7-dicholoro-octamethyltetrasiloxane were then used to evaluate the dynamic adsorption of fibrinogen (0.1 mg mL−1 in 10 mM PBS, pH = 7.7). An unmodified wafer (Si/SiO2) was used as a control surface. According to our results (data not shown), fibrinogen attached onto both films and to the silica surface with almost identical initial adsorption rates (dΓ/dt), reaching ΓSAT values of 3.6 ± 0.1 mg m−2 and 3.4 ± 0.1 mg m−2, respectively. Rinsing the samples with buffer did not induce desorption of fibrinogen from the substrate surfaces tested. It was also observed that, while SDS induced desorption of 81% of the fibrinogen adsorbed onto Si/SiO2, a much smaller fraction (27% and 13%) was removed from the substrate surfaces coated with either 1,3-dicholoro-1,1,3,3-tetramethyldisiloxane or 1,7-dicholoro-octamethyltetrasiloxane, respectively. In line with previous reports,58,78 our results show that the three surfaces tested exhibited high fibrinogen adsorption, regardless of whether the surface was hydrophilic or hydrophobic.79 However, the binding strength of fibrinogen (as measured by elutability with SDS80–83) was significantly higher on the dimethylsiloxane-treated surface than on the plain silica surface. These results also support the hypothesis that 1,7-dicholoro-octamethyltetrasiloxane can coat the silica surface with a coverage higher than that of the 1,3-dicholoro-1,1,3,3-tetramethyldisiloxane, explaining the intermediate behavior observed during the protein desorption studies performed with SDS. Consequently, films made with 1,7-dicholoro-octamethyltetrasiloxane were considered more suitable for the scope of the present project, and consequently, these were further characterized, and used for the rest of the experiments described in the present manuscript. These films will be referred to as PDMS-like films for the remaining part of this paper.
NMR was used to gain insight on the structures of both the precursor and the deposited films (data included as ESI†). Two signals of identical intensity were observed for the precursor (1,7-dicholoro-octamethyltetrasiloxane): the signal observed at 0.14 ppm was assigned to the protons on the methyl groups attached to internal Si atoms, while the signal that appeared downfield (0.46 ppm) was assigned to the protons on the methyl groups in the vicinity of the chlorinated terminal Si atoms. In order to analyze the products of the reaction between the selected n-dimethylsiloxanes and silica by NMR, the glass inner surface of the NMR tube was modified according to the previously described procedure. However, the magnitude of the obtained signal was not considered appropriate. Consequently and aiming to increase the amount of material available, silica beads were modified with 1,7-dicholoro-octamethyltetrasiloxane under conditions identical to those used to modify the Si/SiO2 wafers, suspended in CDCl3 and analyzed using standard procedures. It is worth mentioning that a single peak (at 1.50 ppm) was observed in the 1H-NMR of the plain beads and was attributed to the protons in the SiOH groups of the surface. Conversely, the 1H-NMR of the modified beads showed a main peak (at 0.05 ppm), and a series of much smaller peaks at 1.57 and 4.84 ppm. The signal observed at 4.84 ppm was a rather small and broad peak, characteristic of protons in groups linked to surfaces. As expected, the 13C-NMR of the modified beads displayed a main peak at 1.00 ppm and a smaller peak at 0.74 ppm. Although a detailed description of the chemical connectivity of the deposited film was not possible from these experiments, the relative intensity of the peaks clearly demonstrates the presence of hydrogen and carbon atoms, therefore confirming the possibility to attach methyl groups to the silica surface.
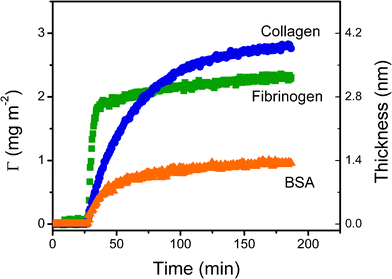 | ||
| Fig. 2 Dynamic adsorption experiments of BSA (in 10 mM PBS at pH = 7.0), fibrinogen (in 50 mM PBS at pH = 7.7) and collagen (in acetate buffer 40 mM at pH = 4.8) at a concentration of 0.01 mg mL−1 onto the nanostructured films. | ||
These experimental conditions were chosen to ensure complete dissolution of the proteins and to allow comparison of the results of the present studies with others reported for BSA,50fibrinogen,86 and collagen84. The first noticeable aspect is that, despite having the highest molecular weight, fibrinogen adsorbed to the substrate surface at the highest rate (0.33 ± 0.02 mg m−2 min−1). This result suggests that interactions with the substrate surface (and not only the flux of protein) played a fundamental role in the adsorption rate of Col and BSA. Conversely, it is important to note that the highest adsorbed amount of protein was obtained with collagen (2.6 ± 0.1 mg m−2). These results can be attributed to a combination of favorable electrostatic interactions (surface-to-protein) and slow rearrangements in the adsorbed layer. Probably the most important conclusion that can be extracted from these results is that similar conditions shall not be used if equivalent films of fibrinogen and collagen are to be adsorbed. While 82% of the saturation amount (ΓSAT) of fibrinogen can be achieved in 40 min, only 35% of the ΓSAT of collagen was adsorbed to the substrate surface during that period of time. This is a critical aspect to consider when adsorbing proteins because typically, there is a dynamic competition between the adsorption process and the structural rearrangements of the protein at the surface. While the former process increases the number of proteins adsorbed per unit area; the latter allows proteins to relax, maximize the interaction with the substrate surface, and leads to significant reductions in biological activity.
Considering the dimensions and structural rigidity of the selected proteins (Table 1) as well as the average thickness of the protein layers adsorbed onto the PDMS-like surface, it is reasonable to consider that, while BSA and fibrinogen formed a single (most likely incomplete) layer85,87,88 with side-on arrangement, collagen formed an entangled multilayer of linear fibers.
The effect of protein concentration on the adsorbed amount (Γ) onto the PDMS-coated surfaces was investigated in real-time for the three chosen proteins. The representative example of Fig. 3A shows the results obtained for fibrinogen. It was observed that both the amount of adsorbed fibrinogen and the initial adsorption rate increased as function of protein concentration. It is also interesting to note that, a secondary process was observed (at ∼60 min) when fibrinogen at 0.1 mg mL−1 was used, suggesting that post-adsorption rearrangements (from side-on to head-on) may be occurring. This observation is also in agreement with a molecular area of 2.4 mg m−2 of fibrinogen in a closely packed monolayer with side-on configuration, reported by Wertz and Santore.87 Post-adsorption processes such as tilting, rolling, and spreading have been reported for a number of proteins89–91 (including fibrinogen85,92,93) and are relevant because they may significantly affect the biological activity of the adsorbed molecules.
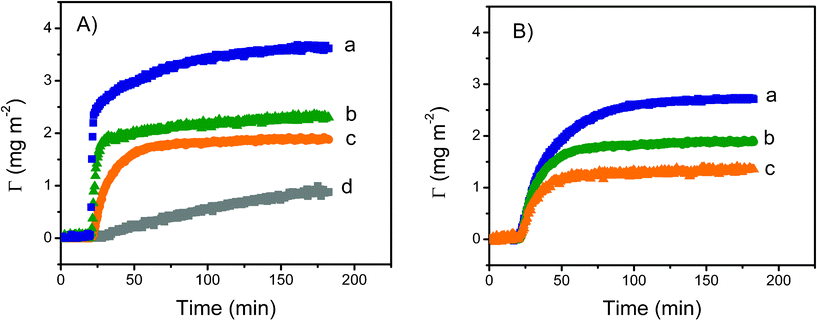 | ||
| Fig. 3 A: Effect of protein concentration on the dynamic adsorption of fibrinogen onto PDMS-like nanofilms. Conditions: (a) 0.1 mg mL−1, (b) 0.01 mg mL−1, (c) 0.001 mg mL−1 and (d) 0.0001 mg/mL. B: Effect of pH on the dynamic adsorption of fibrinogen (0.01 mg mL−1) onto PDMS-like nanofilms. Conditions: (a) pH = 6.6, (b) pH = 7.7 and (c) pH = 8.7. | ||
The amount of fibrinogen adsorbed on the PDMS film as a function of time and in response to changes in the pH of the buffer solution was also determined using spectroscopic ellipsometry. For these experiments, pH values were selected taking into consideration the isoelectric point of each protein (Table 1). These experiments enabled evaluation of the relative contribution of electrostatic and hydrophobic forces on the interaction of proteins with both the surface and the proteins already adsorbed to the substrate surface. Altering the charge of fibrinogen (0.01 mg mL−1) by changing the pH of the buffer solution affected protein adsorption onto the PDMS-like substrate (Fig. 3B). In all cases, a significant increase on the amount of protein adsorbed was observed as the solution pH approached the isoelectric point of each protein. Similarly, the initial adsorption rate was fastest at pH values around the isoelectric point of each protein tested but decreased as the pH of the solution moved further away from the isoelectric point of each protein. The results of the adsorption studies for the three selected proteins are summarized in Table 2.
| Protein | Adsorption conditions | |||
|---|---|---|---|---|
| Concentration | pH | dΓ/dt (mg m−2 min−1) | ΓSAT (mg m−2) | |
| BSA | 0.1 | 7.0 | 0.21 ± 0.03 | 1.3 ± 0.1 |
| 0.01 | 7.0 | 0.06 ± 0.01 | 1.0 ± 0.2 | |
| 0.001 | 7.0 | 0.006 ± 0.005 | 0.4 ± 0.1 | |
| Fib | 0.1 | 7.7 | 1.03 ± 0.11 | 3.6 ± 0.2 |
| 0.01 | 7.7 | 0.445 ± 0.06 | 2.3 ± 0.2 | |
| 0.001 | 7.7 | 0.07 ± 0.02 | 1.9 ± 0.3 | |
| 0.0001 | 7.7 | 0.01 ± 0.01 | 0.89 ± 0.09 | |
| 0.001 | 8.7 | 0.04 ± 0.004 | 1.35 ± 0.2 | |
| 0.001 | 6.7 | 0.08 ± 0.008 | 2.7 ± 0.2 | |
| Col | 0.01 | 4.8 | 0.10 ± 0.01 | 2.7 ± 0.3 |
| 0.001 | 4.8 | 0.04 ± 0.01 | 1.8 ± 0.1 | |
| 0.0001 | 4.8 | 0.01 ± 0.01 | 0.60 ± 0.08 | |
| 0.001 | 4.0 | 0.02 ± 0.01 | 0.92 ± 0.09 | |
The experiments of the present study provided unique insights into the amount and arrangement of proteins adsorbed onto the thin-films of PDMS. In agreement with literature reports, the highest amount of protein was adsorbed when the pH of the buffer solution was close to, or near, the isoelectric point of the respective protein. This observation is in agreement with literature reports stating that, due to minimal protein-to-protein electrostatic interactions, higher protein adsorption rates are usually observed at the IEP.49,94,95 When compared to results calculated from a purely diffusion-limited model,59 these results indicate that the attachment to the surface plays a fundamental role in the adsorption of the selected proteins. For that reason, maximizing the adsorption rate has proven to be an effective way to minimize structural rearrangements (such as spreading) of the adsorbing protein molecules. In addition, measurements of the initial adsorption rate only require a small amount of protein and can be completed in a relatively short timescale (∼20 min). On the other hand, measurements of the saturation amount can take significantly longer, allowing post-adsorption processes to influence the interpretation of the observed phenomena.
The importance of hydrophobic interactions in the adsorption of the chosen proteins is evidenced by the strong adsorption observed even under unfavorable electrostatic interactions. The results of the present study provide guidelines to assist other researchers to select the most favorable and time-efficient conditions to adsorb proteins onto PDMS.
| Protein | Favorable conditions | Unfavorable conditions |
|---|---|---|
| BSA | 0.01 mg mL−1PBS, pH = 7.0 | 0.0001 mg mL−1PBS, pH = 7.0 |
| Fib | 0.01 mg mL−1PBS, pH = 6.7 | 0.0001 mg mL−1PBS, pH = 7.7 |
| Col | 0.01 mg mL−1Acetate, pH = 4.8 | 0.0001 mg mL−1Acetate, pH = 6.0 |
In addition to unmodified silica substrates (Si/SiO2), substrates coated with the thin-films of PDMS but without pre-adsorbed proteins were used as controls. Details of the procedures followed in evaluating cell adhesion and morphology are given in the Experimental section of this manuscript.
The results provided evidence that HDMEC adhered to all substrates tested. Although the number of adherent cells was similar on all the substrate surfaces of interest to the present study, significant differences in cell morphology were observed. Cells did not spread out when adhering onto the unmodified silicon substrate (Fig. 4A). Only slight spreading was observed when cells adhered onto the plain PDMS-like substrates or the substrates modified with BSA at either condition tested (Fig. 4B–D). In contrast, spread-out cells were observed on all other substrates tested; however, the degree of cell spreading was dependent on the type and amount of adsorbed protein. In this respect, adhered cells exhibited moderate spread-out morphology onto PDMS-like substrates modified with either collagen type-I or fibrinogen under unfavorable conditions (Fig. 4E–F). Cells adhering onto PDMS-like substrates modified with either collagen or fibrinogen under the most favorable conditions, exhibited the most spread-out cell morphology (Fig. 4G–H). In addition, the adherent cells exhibited the typical F-actin arrangement for endothelial cells, specifically, a concentric arrangement along the cell periphery as well as filaments transversing the cell cytoplasm.
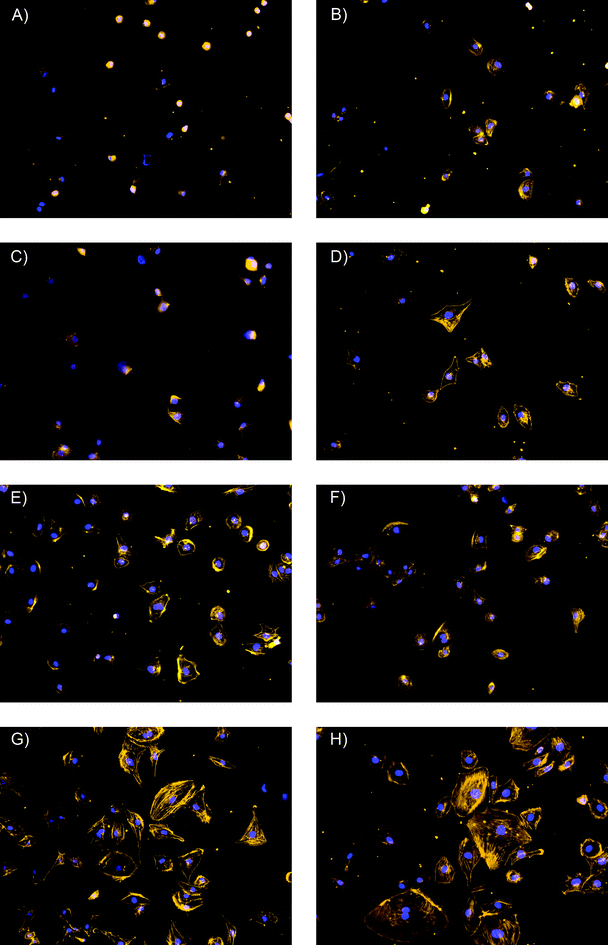 | ||
| Fig. 4 Fluorescent micrographs of human dermal microvessel endothelial cells after 3 h of adhesion on the selected substrates. Stains: Alexa Fluor 568® Phalloidin and 4′′,6-diamidino-2-phenylindole dilactate. Magnification: 20×. (A) Si/SiO2 substrate; (B) Unmodified substrate coated with PDMS-like film; (C) PDMS-like substrate modified with BSA under favorable conditions; (D) PDMS-like substrate modified with BSA under unfavorable conditions; (E) PDMS-like substrate modified with Fib under unfavorable conditions; (F) PDMS-like substrate modified with Col under unfavorable conditions; (G) PDMS-like substrate modified with Col under favorable conditions; (H) PDMS-like substrate modified with Fib under favorable conditions. | ||
4. Conclusions
This report described a simple procedure to fabricate films of n-dimethylsiloxane covalently attached to Si/SiO2 substrates. The films were characterized by ellipsometry, 1H-NMR, 13C-NMR, contact angle measurements, and atomic force microscopy. According to the presented results, exposing the surface of SiO2 to 1,7-dicholoro-octamethyltetrasiloxane leads to the deposition of homogeneous films of about 2 nm in thickness with characteristics similar to those of commercial PDMS. Dynamic adsorption experiments showed that the selected proteins (BSA, Fib, and Col) adsorbed onto the surface of the films with high affinity, that such adsorption process was determined by a combination of hydrophobic and electrostatic interactions, and that experimental conditions can be rationally selected to minimize protein spreading on the PDMS surface. Such knowledge of protein adsorption could lead to improved understanding of cell and tissue interactions on material surfaces pertinent to biomedical applications.Acknowledgements
Financial support for this project was provided by the University of Texas at San Antonio and the National Institutes of Health through the National Institute of General Medical Sciences (1SC3GM081085) and the Research Centers at Minority Institutions (2G12RR013646-11). The authors would also like to thank J. L. Felhofer and M. Penick for helpful discussions.References
- J. L. Felhofer, L. Blanes and C. D. Garcia, Electrophoresis, 2010, 31, 2469–2486 CrossRef CAS.
- J. R. Kraly, R. E. Holcomb, Q. Guan and C. S. Henry, Anal. Chim. Acta, 2009, 653, 23–35 CrossRef CAS.
- Y. Xu, X. Yang and E. Wang, Anal. Chim. Acta, 2010, 683, 12–20 CrossRef CAS.
- B. H. Weigl, R. L. Bardell and C. R. Cabrera, Adv. Drug Delivery Rev., 2003, 55, 349–377 CrossRef CAS.
- T. Vilkner, D. Janasek and A. Manz, Anal. Chem., 2004, 76, 3373–3386 CrossRef CAS.
- J. M. K. Ng, I. Gitlin, A. D. Stroock and G. M. Whitesides, Electrophoresis, 2002, 23, 3461–3473 CrossRef CAS.
- H. Becker and L. Locascio, Talanta, 2002, 56, 267–287 CrossRef CAS.
- M. Castano-Alvarez, M. T. Fernandez-Abedul and A. Costa-García, Electrophoresis, 2005, 26, 3160–3168 CrossRef CAS.
- J. A. Vickers, B. M. Dressen, M. C. Weston, K. Boonsong, O. Chailapakul, D. M. Cropek and C. S. Henry, Electrophoresis, 2007, 28, 1123–1129 CrossRef CAS.
- J. Liu, X. Sun and M. L. Lee, Anal. Chem., 2007, 79, 1926–1931 CrossRef CAS.
- C. D. García and C. S. Henry, in Microchip Capillary Electrophoresis: Methods and Protocols, ed. C. S. Henry, Humana Press, Totowa, NJ 2006, pp. pp. 27–36. Search PubMed.
- M. Zhang, J. Wu, L. Wang, K. Xiao and W. Wen, Lab Chip, 2010, 10, 1199–1203 RSC.
- W. Schrott, M. Svoboda, Z. Slouka, M. Pribyl and D. Snita, Microelectron. Eng., 2010, 87, 1600–1602 CrossRef CAS.
- E. W. K. Young, E. Berthier, D. J. Guckenberger, E. Sackmann, C. Lamers, I. Meyvantsson, A. Huttenlocher and D. J. Beebe, Anal. Chem., 2011, 83, 1408–1417 CrossRef CAS.
- P. Zheng and T. J. McCarthy, Langmuir, 2010, 26, 18585–18590 CrossRef CAS.
- D. K. Cai, A. Neyer, R. Kuckuk and H. M. Heise, Opt. Mater., 2008, 30, 1157–1161 CrossRef CAS.
- J. N. Lee, C. Park and G. M. Whitesides, Anal. Chem., 2003, 75, 6544–6554 CrossRef CAS.
- T. C. Merkel, V. I. Bondar, K. Nagai, B. D. Freeman and I. Pinnau, J. Polym. Sci., Part B: Polym. Phys., 2000, 38, 415–434 CrossRef CAS.
- R. Mukhopadhyay, Anal. Chem., 2007, 79, 3248–3253 CrossRef CAS.
- M. W. Toepke and D. J. Beebe, Lab Chip, 2006, 6, 1484–1486 RSC.
- K. Ren, Y. Zhao, J. Su, D. Ryan and H. Wu, Anal. Chem., 2010, 82, 5965–5971 CrossRef CAS.
- M. F. Mora, C. E. Giacomelli and C. D. Garcia, Anal. Chem., 2007, 79, 6675–6681 CrossRef CAS.
- H. Makamba, J. H. Kim, K. Lim, N. Park and J. H. Hahn, Electrophoresis, 2003, 24, 3607–3619 CrossRef CAS.
- B. Huang, S. Kim, H. Wu and R. N. Zare, Anal. Chem., 2007, 79, 9145–9149 CrossRef CAS.
- F. Abbasi, H. Mirzadeh and A.-A. Katbab, Polym. Int., 2001, 50, 1279–1287 CrossRef CAS.
- I. Wong and C.-M. Ho, Microfluid. Nanofluid., 2009, 7, 291–306 CrossRef CAS.
- J. Zhou, A. V. Ellis and N. H. Voelcker, Electrophoresis, 2010, 31, 2–16 CrossRef CAS.
- N. Maheshwari, A. Kottantharayil, M. Kumar and S. Mukherji, Appl. Surf. Sci., 2010, 257, 451–457 CrossRef CAS.
- G. Hu, Y. Gao, P. M. Sherman and D. Li, Microfluid. Nanofluid., 2005, 1, 346–355 CrossRef CAS.
- S. H. Chung and J. Min, Ultramicroscopy, 2009, 109, 861–867 CrossRef CAS.
- L. Wang, L. Lei, X. F. Ni, J. Shi and Y. Chen, Microelectron. Eng., 2009, 86, 1462–1464 CrossRef CAS.
- L. S. Roach, H. Song and R. F. Ismagilov, Anal. Chem., 2005, 77, 785–796 CrossRef CAS.
- W. Zhang, C.-Y. Xue and K.-L. Yang, J. Colloid Interface Sci., 2010, 353, 143–148 CrossRef.
- C. Volcke, R. P. Gandhiraman, L. Basabe-Desmonts, M. Iacono, V. Gubala, F. Cecchet, A. A. Cafolla and D. E. Williams, Biosens. Bioelectron., 2010, 25, 1295–1300 CrossRef CAS.
- G. K. Toworfe, R. J. Composto, C. S. Adams, I. M. Shapiro and P. Ducheyne, J. Biomed. Mater. Res., 2004, 71A, 449–461 CrossRef CAS.
- J. M. Anderson, N. P. Ziats, A. Azeez, M. R. Brunstedt, S. Stack and T. L. Bonfield, J. Biomater. Sci., Polym. Ed., 1996, 7, 159–169 CrossRef.
- M. N. de Silva, R. Desai and D. J. Odde, Biomed. Microdevices, 2004, 6, 219–222 CrossRef CAS.
- E. Leclerc, Y. Sakai and T. Fujii, Biotechnol. Prog., 2004, 20, 750–755 CrossRef CAS.
- M.-H. Wu, Surf. Interface Anal., 2009, 41, 11–16 CrossRef CAS.
- M. Ni, W. H. Tong, D. Choudhury, N. A. Rahim, C. Iliescu and H. Yu, Int. J. Mol. Sci., 2009, 10, 5411–5441 CrossRef CAS.
- K. E. Sapsford and F. S. Ligler, Biosens. Bioelectron., 2004, 19, 1045–1055 CrossRef CAS.
- L. Yu, Z. Lu, Y. Gan, Y. Liu and C. M. Li, Nanotechnology, 2009, 20, 285101 CrossRef.
- T. R. E. Simpson, B. Parbhoo and J. L. Keddie, Polymer, 2003, 44, 4829–4838 CrossRef CAS.
- S. L. Peterson, A. McDonald, P. L. Gourley and D. Y. Sasaki, J. Biomed. Mater. Res., 2005, 72A, 10–18 CrossRef CAS.
- K. Choonee, R. R. A. Syms, M. M. Ahmad and H. Zou, Sens. Actuators, A, 2009, 155, 253–262 CrossRef.
- D. P. Dowling, C. E. Nwankire, M. Riihimäki, R. Keiski and U. Nylén, Surf. Coat. Technol., 2010, 205, 1544–1551 CrossRef CAS.
- A. Zengin and T. Caykara, Appl. Surf. Sci., 2011, 257, 2111–2117 CrossRef CAS.
- C. E. Giacomelli and W. Norde, J. Colloid Interface Sci., 2001, 233, 234–240 CrossRef CAS.
- W. Norde, Colloids Surf., B, 2008, 61, 1–9 CrossRef CAS.
- H. Larsericsdotter, S. Oscarsson and J. Buijs, J. Colloid Interface Sci., 2005, 289, 26–35 CrossRef CAS.
- N. Hassan, J. M. Ruso and P. Somasundaran, Colloids Surf., B, 2011, 82, 581–587 CrossRef CAS.
- J. Mayne and J. J. Robinson, J. Cell. Biochem., 2002, 84, 567–574 CrossRef.
- S. Dash, S. Mishra, S. Patel and B. K. Mishra, Adv. Colloid Interface Sci., 2008, 140, 77–94 CrossRef CAS.
- M. Dion, M. Rapp, N. Rorrer, D. H. Shin, S. M. Martin and W. A. Ducker, Colloids Surf., A, 2010, 362, 65–70 CrossRef CAS.
- M. F. Mora, C. E. Giacomelli and C. D. Garcia, Anal. Chem., 2009, 81, 1016–1022 CrossRef CAS.
- J. L. Felhofer, J. Caranto and C. D. Garcia, Langmuir, 2010, 26, 17178–17183 CrossRef CAS.
- H. Soetedjo, M. F. Mora and C. D. Garcia, Thin Solid Films, 2010, 518, 3954–3959 CrossRef CAS.
- J. Wehmeyer, R. Bizios and C. D. Garcia, Mater. Sci. Eng., C, 2010, 30, 277–282 CrossRef CAS.
- M. F. Mora, M. Reza Nejadnik, J. L. Baylon-Cardiel, C. E. Giacomelli and C. D. Garcia, J. Colloid Interface Sci., 2010, 346, 208–215 CrossRef CAS.
- M. R. Nejadnik and C. D. Garcia, Colloids Surf., B, 2011, 82, 253–257 CrossRef CAS.
- S. A. Alterovitz and B. Johs, Thin Solid Films, 1998, 313–314, 124–127 CrossRef CAS.
- H. Fujiwara, Spectroscopic ellipsometry. Principles and applications, J. Wiley&Sons, West Sussex, England, 2007 Search PubMed.
- R. A. Synowicki, Thin Solid Films, 1998, 313–314, 394–397 CrossRef CAS.
- M. R. Nejadnik, L. Francis and C. D. Garcia, Electroanalysis, 2011, 23, 1462–1469 CrossRef CAS.
- S. Logothetidis, M. Gioti, S. Lousinian and S. Fotiadou, Thin Solid Films, 2005, 482, 126–132 CrossRef CAS.
- D. Filippini, F. Winquist and I. Lundstrom, Anal. Chim. Acta, 2008, 625, 207–214 CrossRef CAS.
- J. A. D. Feijter, J. Benjamins and F. A. Veer, Biopolymers, 1978, 17, 1759–1772 CrossRef.
- M. F. Mora, C. E. Giacomelli and C. D. Garcia, Anal. Chem., 2009, 81, 1016–1022 CrossRef CAS.
- R. Kurrat, J. E. Prenosil and J. J. Ramsden, J. Colloid Interface Sci., 1997, 185, 1–8 CrossRef CAS.
- C. E. Giacomelli, M. J. Esplandiu, P. I. Ortiz, M. J. Avena and C. P. D. Pauli, J. Colloid Interface Sci., 1999, 218, 404–411 CrossRef CAS.
- M. Vinnichenko, R. Gago, N. Huang, Y. X. Leng, H. Sun, U. Kreissig, M. P. Kulish and M. F. Maitz, Thin Solid Films, 2004, 455–456, 530–534 CrossRef CAS.
- S. M. Sirard, H. Castellanos, P. F. Green and K. P. Johnston, J. Supercrit. Fluids, 2004, 32, 265–273 CrossRef CAS.
- J. C. McDonald and G. M. Whitesides, Acc. Chem. Res., 2002, 35, 491–499 CrossRef CAS.
- D. Cai, A. Neyer, R. Kuckuk and H. M. Heise, J. Mol. Struct., 2010, 976, 274–281 CrossRef CAS.
- M. A. Hoque, Y. Kakihana, S. Shinke and Y. Kawakami, Macromolecules, 2009, 42, 3309–3315 CrossRef CAS.
- M. A. Brook, Silicon in organic, organometallic, and polymer chemistry, John Wiley and Sons, Inc., New York, 2000 Search PubMed.
- E. P. T. d. Givenchy, S. Amigoni, C. Martin, G. Andrada, L. Caillier, S. Geribaldi and F. Guittard, Langmuir, 2009, 25, 6448–6453 CrossRef.
- L. E. Valenti, P. A. Fiorito, C. D. Garcia and C. E. Giacomelli, J. Colloid Interface Sci., 2007, 307, 349–356 CrossRef CAS.
- R. J. Rapoza and T. A. Horbett, J. Biomed. Mater. Res., 1990, 24, 1263–1287 CrossRef CAS.
- B. Verzola, C. Gelfi and P. G. Righetti, J. Chromatogr., A, 2000, 874, 293 CrossRef CAS.
- M. A. Bos and T. van Vliet, Adv. Colloid Interface Sci., 2001, 91, 437–471 CrossRef CAS.
- A. Mackie and P. Wilde, Adv. Colloid Interface Sci., 2005, 117, 3–13 CrossRef CAS.
- J. J. Gray, Curr. Opin. Struct. Biol., 2004, 14, 110–115 CrossRef CAS.
- V. I. Shcheslavskiy, G. I. Petrov and V. V. Yakovlev, Chem. Phys. Lett., 2005, 402, 170–174 CrossRef CAS.
- Z. Adamczyk, J. Barbasz and M. Cieśla, Langmuir, 2010, 26, 11934–11945 CrossRef CAS.
- A. M. Brzozowska, B. Hofs, A. de Keizer, R. Fokkink, M. A. Cohen Stuart and W. Norde, Colloids Surf., A, 2009, 347, 146–155 CrossRef CAS.
- C. F. Wertz and M. M. Santore, Langmuir, 1999, 15, 8884–8894 CrossRef CAS.
- C. F. Wertz and M. M. Santore, Langmuir, 2001, 17, 3006–3016 CrossRef CAS.
- C. E. Giacomelli and W. Norde, in Encyclopedia of Surface and Colloid Science, ed. A. T. Hubbard, Marcel Dekker, New York, 2003 Search PubMed.
- M. M. Santore and C. F. Wertz, Langmuir, 2005, 21, 10172–10178 CrossRef CAS.
- M. van der Veen, M. C. Stuart and W. Norde, Colloids Surf. B, 2007, 54, 136–142 CrossRef CAS.
- S. Lousinian, S. Kassavetis and S. Logothetidis, Diamond Relat. Mater., 2007, 16, 1868–1874 CrossRef CAS.
- Y. Yu and G. Jin, J. Colloid Interface Sci., 2005, 283, 477–481 CrossRef CAS.
- W. Norde Proteins at Solid Surfaces. Physical Chenistry of Biological Interfaces, Marcel Dekker, New York, 2000 Search PubMed.
- W. Norde, in Biopolymers at Interfaces, ed. M. Malmsten, Marcel Dekker, New York, 2003 Search PubMed.
Footnote |
| † Electronic Supplementary Information (ESI) available. See DOI: 10.1039/c1ra00198a/ |
| This journal is © The Royal Society of Chemistry 2011 |