Temperature-dependent adsorption of cellulose ethers on silica and hydrophobized silica immersed in aqueous polymer solution
Rasmus
Bodvik
a,
Esben
Thormann
a,
Leif
Karlson
b and
Per M.
Claesson
*a
aDepartment of Chemistry, Surface and Corrosion Science, Royal Institute of Technology, Drottning Kristinas väg 51, SE-100 44 Stockholm, Sweden and Institute for Surface Chemistry, Box 5607, SE-114 86 Stockholm, Sweden. E-mail: percl@kth.se; Tel: +46 8 790 99 04;
bAkzo Nobel Functional Chemicals AB, SE-444 85 Stenungsund, Sweden
First published on 4th August 2011
Abstract
The influence of temperature on adsorption and the adsorbed layer properties of methylcellulose (MC) and hydroxypropylmethylcellulose (HPMC) were investigated on silica and hydrophobized silica surfaces immersed in aqueous polymer solution. To achieve a concise understanding a quartz crystal microbalance with dissipation, ellipsometry, and atomic force microscopy imaging were employed. These techniques provide complimentary information on the structure, mass and viscoelastic properties of the polymer layers. Adsorption was first allowed at 25 °C. Next, the temperature was increased step-wise up to 50 °C and then decreased again. This procedure highlights the temperature dependence of the adsorbed material, as well as the hysteresis in the adsorption due to temperature cycling. A change in temperature not only affects the adsorbed amount, but also the properties of the layer as illustrated by measurements of its water content, thickness and viscoelasticity.
1. Introduction
Development of temperature responsive surfaces receives increasing interest in fields as diverse as food, medical and paint technology. To this end polymer structures containing e.g.poly(N-isopropylacrylamide),1–2 poly(ethylene oxide)3 and poly(propylene oxide)4 chains, exhibiting negative temperature response, can be utilized. Such polymers are water soluble at lower temperatures, but phase separate above their respective lower critical solution temperature. Several cellulose ethers share the same behaviour and find use in many applications due to their adsorption properties and due to the fact that their temperature response can be modified by small changes in the degree of substitution. Examples include ethyl(hydroxyethyl)cellulose (EHEC),5–14methylcellulose (MC)15–16 and hydroxypropylmethylcellulose (HPMC).17MC has been used to enhance drug release from microcrystals,15 and functionalized by laminin it has been utilized in neural tissue engineering.18–19HPMC may form chelate complexes with Ca2+, which improves its properties for use in injectable biomaterials for bone and dental surgery.18HPMC in combination with ethylcellulose has also been used as a coating on drug-loaded silica gels to control the drug release profile.19 In this application it was found that the HPMC undercoating suppressed the drug release by making the core surface softer and facilitating the formation of a continuous ethylcellulose film.Bulk properties of aqueous solutions of MC and HPMC have been investigated intensely20–25 due to the marked temperature-response of their viscoelastic properties. In a recent contribution, we showed by cryo-TEM26 that MC forms a very dense network of fibrils at high temperatures, and also at a relatively low polymer concentration in the bulk. In contrast, HPMC adopts an ordered, but less dense, network structure and requires a higher polymer concentration and higher temperatures for network formation compared to MC. The transition takes place at a higher temperature in HPMC due to the large hydroxypropyl groups, which also are less hydrophobic than the methoxy groups. Earlier studies have shown the immobility of substituents in MC at high temperatures, which contrasts with the mobility of the substituents in HPMC or the non-gelling hydroxypropylcellulose (HPC).22–23 The inhibition of intermolecular association, due to hydroxypropyl groups, has also been indicated by NMR data.21
The effect of temperature on adsorption and layer structure has been investigated for some thermo-responsive polymers, including poly(N-isopropylacrylamide) and EHEC, using techniques such as quartz crystal microbalance measurements,2,27–30ellipsometry9,3 and surface force techniques.8,11–12,14 In earlier studies, the conformation of EHEC on hydrophobized mica was shown to be relatively flat, and the main driving force for the adsorption was suggested to be hydrophobic interactions.11 The adsorbed amount increased upon temperature increase, and then decreased as the temperature was subsequently decreased.13 The forces between two such EHEC-coated surfaces stayed repulsive but became less long-ranged upon heating to several degrees above the cloud point, suggesting that the most hydrophilic segments were oriented towards the solution.12 Further above the cloud point, an attractive force was observed.
In another study HPMC was found to form very elastic films at the air–water interface, even at low surface pressures.31–32 High coverage of the interface was reached at very low concentrations in the bulk phase. With increasing bulk concentration the monolayer structure finally collapsed. Differences in surface activity between different HPMC samples were attributed to different hydroxypropyl molar substitution and molecular weight. The similar viscoelasticity and elastic dilatational moduli of the investigated samples was explained by the presence of a similar degree of methyl substitution.
In a previous manuscript we have described the temperature-response of pre-adsorbed layers of MC and HPMC on hydrophobized silica.33 The situation explored in this manuscript—the temperature response in the presence of these polymers in solution—is considerably more complex since the adsorbed amount is allowed to vary. Thus, the layer properties are affected by both possible compaction/expansion of the adsorbed polymers and the adsorption/desorption of polymers. By comparing with results from our previous work33 we distinguish the role of adsorption/desorption in the change of layer properties observed during temperature changes.
We utilized two substrate surfaces, hydrophilic silica and hydrophobized silica, and investigated the adsorption of MC and HPMC as the temperature was cycled from 25 °C to 50 °C, and down to 25 °C again. An ellipsometer and a quartz crystal microbalance with dissipation (QCM-D) were used to obtain the amount adsorbed, layer thickness and water content of the layer and to elucidate the shear viscoelastic properties in the MHz frequency range, while an atomic force microscope (AFM) provided images of the adsorbed layers.
2. Materials and Methods
The two modified cellulose ethers used in this study were obtained from Akzo Nobel, Stenungsund, Sweden. The methylcellulose sample identified as M115 and the hydroxypropylmethylcellulose sample with code HPM116 at Akzo Nobel will be referred to as M1.6C and HP0.12M1.57C throughout this report. In this nomenclature the substitution degrees of the cellulose ethers are highlighted. Their composition, molecular weight, and the viscosity of 2 wt% solutions in water are provided in Table 1. The molecular weight was obtained from size exclusion chromatography at 30 °C using 0.04 M sodium acetate and 0.02% sodium azide at pH 6 as a mobile phase. Refractive index, light scattering (at 7° and 90°) and viscosity measurements were employed in the analysis.| Our Name | Viscosity at 20 °C, 2% in water (Pa s) | Degree of Methoxy Substitution | Hydroxypropyl Molar Substitution | Molecular Weight Mw |
|---|---|---|---|---|
| M1.6C | 20 | 1.6 | 0 | 530![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
| HP0.12M1.57C | 20 | 1.57 | 0.12 | 620000 |
The methoxy content of the two polymers is very similar. The viscosity at 20 °C is also similar, indicating similar molecular weights. The major difference is that HP0.12M1.57C contains hydroxypropyl with a molar substitution of 0.12, whereas no such groups exist in M1.6C.
2.1. Rheology
An AR-G2 rheometer (TA Instruments) equipped with a bob and cup system was utilized for oscillation strain control measurements. A temperature sweep was carried out starting at 10 °C, heating to 85–90 °C, and then cooling to 10 °C again. Heating/cooling rates between 0.1–1.5 °C min−1 were used. The temperature control was assured by Peltier concentric cylinders, which allow a maximum heating rate of 13 °C min−1. Four heating elements are placed in contact with the lower cup geometry, providing efficient heat transfer up the walls of the cup. The elements are held in place by an insulated jacket. A platinum resistance thermocouple is placed close to the top of the cup ensuring accurate temperature measurement and control.2.2. Quartz Crystal Microbalance with Dissipation Monitoring (QCM-D)
A q-sense E4 microbalance (q-sense, Gothenburg), utilizing AT-cut quartz crystals with 5 MHz fundamental frequency, was employed in this investigation. The sensors were coated with a 50 nm silica layer. They were cleaned with a 2% Hellmanex (Hellma GmbH) solution for 30 min, placed in water over night, and then stored in ethanol until use. Hydrophobic substrates were obtained by silanization of the silica-coated crystals by exposing the surfaces to (3,3-dimethylbutyl)dimethylchlorosilane (DDS) vapour in a desiccator over night. The hydrophobized surfaces were then rinsed with water and stored in ethanol. The contact angle with water was checked to be larger than 90° before use (see also Kjellin et al.34).Temperature effects were investigated by increasing the temperature in steps from 25 °C to 50 °C and then back to 25 °C. The upper limit of 50 °C corresponds to the maximum temperature allowed for the QCM-D instrument. Two temperature ramps were carried out: the first was executed prior to adsorption with only water in the cell and the second after allowing 2–3 h of adsorption from a 40 ppm polymer solution. The frequency and dissipation were allowed to stabilize for 100 min at each temperature. The changes in frequency and dissipation were recorded at six overtones, out of which the third, fifth and seventh (15, 25 and 35 MHz) were used to obtain viscoelastic data from the Voigt model,35 which is briefly described below. For each temperature the difference in frequency and dissipation between the bare substrate and the crystal with an adsorbed layer was used in the evaluation of the data. This time consuming procedure, including two similar temperature ramps, enables us to correct for changes in frequency and dissipation that are due to changes in the viscosity and density of the bulk solvent. All measurements were performed with a temperature control of ±0.02 °C.
The sensed mass can for rigid layers be calculated from the Sauerbrey relation, which states that the change in the oscillation frequency is linearly related to the mass added to the crystal, including water associated with the adsorbed layer.36 However, the Sauerbrey relation does not hold for viscoelastic layers.37 This situation should be indicated by a difference in the Sauerbrey mass calculated from the different overtones, and in such a case a viscoelastic model is normally needed.38 The Voigt model is one such viscoelastic model. It consists of two elements, connected in parallel: a viscous damper, where the stress is directly proportional to the rate of strain, and an elastic spring, where the stress is directly proportional to the strain.39 While the shear strain is the same for both elements of the model, the total shear stress is the sum of both contributions:
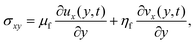 | (1) |
The viscoelastic properties of the film are related to the changes in frequency and dissipation through the β parameter,38–39 which is obtained by solving the wave equation for bulk shear waves propagating in the medium:
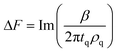 | (2) |
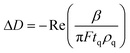 | (3) |
If needed, the Voigt model can be extended, taking into account the frequency dependence of the viscosity and/or elasticity.35 In this study the extended model was used in the analysis.
2.3. Ellipsometry
The ellipsometry measurements were performed with a thin film ellipsometer, type 43603-200E (Rudolph Research with modification by Lund University), using the null ellipsometry method, where the instrument components are set to minimize the effect of the refracted beam (i.e. make the beam linearly polarized). A xenon arc lamp with a filtered wavelength of 401.5 nm was utilized as the light source. The angle of incidence was set to 67.5°. A thorough description of the instrumental set up has been published elsewhere.40
Silicon wafers coated with 34 nm silica were used as substrates. They were cleaned for 5 min in 80 °C 5:1:1 (w/w/w) H2O:NH3:H2O2, rinsed in water, then cleaned for 5 min in 80 °C 5:1:1 (w/w/w) H2O:HCl:H2O2, and rinsed again. The wafers were stored in ethanol until use. Hydrophobic substrates, using the same type of wafers, were obtained by the same procedure as described above for the surfaces used in QCM-D.
The adsorption was measured in situ in a cuvette with a solution volume of 5 mL, using continuous stirring. Two temperature cycles were executed from 25 °C up to 50 °C and then down to 25 °C. The first cycle was conducted with only water in the cuvette, and the second cycle was executed after 40–60 min of adsorption from a 40 ppm polymer solution. The polymer solution was kept in the cell during the entire second temperature cycle. The time needed for the change of temperature and stabilization was around 30 min for each heating step and around 45 min for each cooling step.
The ellipsometric angles Ψ and Δ were measured continuously and used to calculate the mean thickness (df) and refractive index (nf) of the adsorbed layer.41 The baseline values of Ψ and Δ used in the evaluation of the adsorption were those at the corresponding temperatures during cooling of the bare substrate. While both the thickness and refractive index are very sensitive to small changes in the ellipsometer angles, the variations in these parameters are coupled.42 This explains why the adsorbed mass, which is proportional to the thickness and the difference in refractive index between the layer and the bulk, is obtained with a better accuracy than either of these two parameters.
The adsorbed mass, Γ, was calculated using the de Feijter formula43:
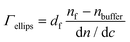 | (4) |
A dn/dc value of 0.135 mL g−1 was used for M1.6C and 0.132 mL g−1 for HP0.12M1.57C. These values were determined using an Optilab DSP refractometer (Wyatt).
2.4. Analysis
The effective hydrodynamic thickness, deff, of the adsorbed layer was calculated, using the data obtained by ellipsometry and QCM-D, from the expression:35,44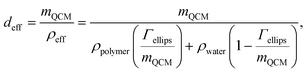 | (5) |
Since the adsorbed mass obtained by ellipsometry is due to the adsorbing species only, the water content of the layer can be calculated from:
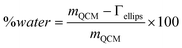 | (6) |
2.5. Atomic Force Microscopy
Topographical images of hydrophobized surfaces with an adsorbed polymer layer and polymers present in solution were recorded using an atomic force microscope (AFM) Nanoscope Multimode V (Veeco, USA) operating in PeakForce mode using ScanAsyst, silicon nitride cantilevers (ScanAsyst Air, Veeco probes) and a fluid cell with in- and outlets allowing for solvent exchange. PeakForce is a relatively new imaging mode, which allows high imaging resolution with a low feedback force and limited shear forces between the tip and the sample. This makes PeakForce very suitable for imaging a soft layer in liquid, which can be challenging with other existing imaging modes.46 The temperature was set by use of a thermal application controller attached to a Bioheater element (Veeco) mounted under the sample. The temperature on the sample surface was calibrated by an external thermostat and controlled with an accuracy of ±1 °C. Hydrophobic substrates were prepared as described for the ellipsometry measurements and the experiments were conducted as follows. The cell was loaded with water and the hydrophobic surface was imaged at 25 °C. Thereafter 40 ppm polymer solution was injected and the system was left for 30 min to allow adsorption before the surface was imaged. Next, the temperature was increased to first 35 °C and then 45 °C. At both temperatures images were acquired 30 min after the temperature had stabilized to allow the system to equilibrate.3. Results
3.1. Rheology
The complex viscosity, μ*, is defined as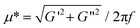 , where G′ and G′′ are the storage and the loss moduli respectively, and f is the frequency.10 The temperature dependence of the complex viscosity is strikingly different for aqueous solutions of HPMC and MC.20–21,47 Both systems studied in this work (see Fig. 1a) undergo a large increase in viscosity at a temperature that is denoted T2.26 From Fig. 1a it is clear that a low amount of hydroxypropyl substitution is sufficient to increase the T2 temperature significantly. Further, a small quantity of such groups introduces typical HPMC behaviour with a drop in viscosity, before reaching T2, at a temperature that is denoted T1. The continuous viscosity decrease upon heating from room temperature to the transition point (T2 for M1.6C and T1 HP0.12M1.57C) can be attributed to the decrease in viscosity of water with increasing temperature. The transition temperatures are observed to almost linearly depend on the temperature sweep rate (see Fig. 1b) and the most accurate values for T1 and T2 are thus obtained by extrapolation of the results to zero sweep rate. For a 0.75 wt% M1.6C solution the extrapolated value of T2 is around 51 °C, while the T1 and T2 values obtained for a 0.75 wt% HP0.12M1.57C solution are around 61 °C and 67 °C, respectively.
, where G′ and G′′ are the storage and the loss moduli respectively, and f is the frequency.10 The temperature dependence of the complex viscosity is strikingly different for aqueous solutions of HPMC and MC.20–21,47 Both systems studied in this work (see Fig. 1a) undergo a large increase in viscosity at a temperature that is denoted T2.26 From Fig. 1a it is clear that a low amount of hydroxypropyl substitution is sufficient to increase the T2 temperature significantly. Further, a small quantity of such groups introduces typical HPMC behaviour with a drop in viscosity, before reaching T2, at a temperature that is denoted T1. The continuous viscosity decrease upon heating from room temperature to the transition point (T2 for M1.6C and T1 HP0.12M1.57C) can be attributed to the decrease in viscosity of water with increasing temperature. The transition temperatures are observed to almost linearly depend on the temperature sweep rate (see Fig. 1b) and the most accurate values for T1 and T2 are thus obtained by extrapolation of the results to zero sweep rate. For a 0.75 wt% M1.6C solution the extrapolated value of T2 is around 51 °C, while the T1 and T2 values obtained for a 0.75 wt% HP0.12M1.57C solution are around 61 °C and 67 °C, respectively.
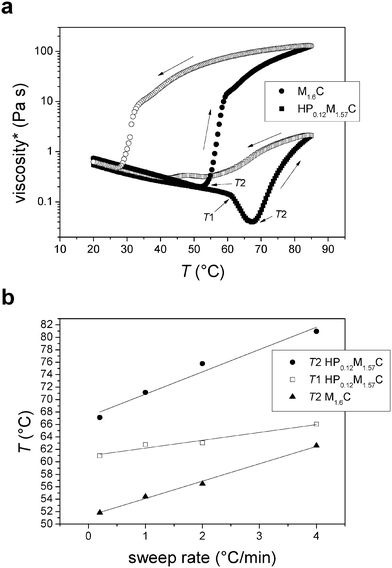 | ||
| Fig. 1 (a) Complex viscosity as a function of temperature (sweep rate 0.2 °C min−1) for aqueous 0.75 wt% M1.6C and HP0.12M1.57C solutions. Filled and unfilled symbols correspond to data obtained on heating and cooling, respectively. Arrows along the curve indicate the direction of the temperature sweep. The T1 and T2 temperatures are also shown. (b) T1 and T2 versus sweep rate for 0.75 wt% M1.6C and HP0.12M1.57C solutions. The lines are linear fits to the data. | ||
3.2. Adsorption on silica
QCM-D was used to investigate the adsorption of both M1.6C and HP0.12M1.57C on hydrophilic silica surfaces. After the temperature cycle had been executed once, at about 17 h, the frequency and dissipation did not change on addition of M1.6C at 25 °C for 2–3 h (see Fig. 2a). The changes in frequency and dissipation reported in Fig. 2 have been normalized by the overtone number. When a second temperature cycle was carried out with the polymer remaining in solution there was no distinguishable change in frequency and dissipation in comparison with the first cycle (in absence of polymer). We conclude that no measurable adsorption occurs, and that the changes in viscosity and density of the solution containing 40 ppm polymer are indistinguishable from that of pure water. The same result was obtained for HP0.12M1.57C. Thus, for both polymers the adsorption to silica is negligible up to a temperature of 50 °C.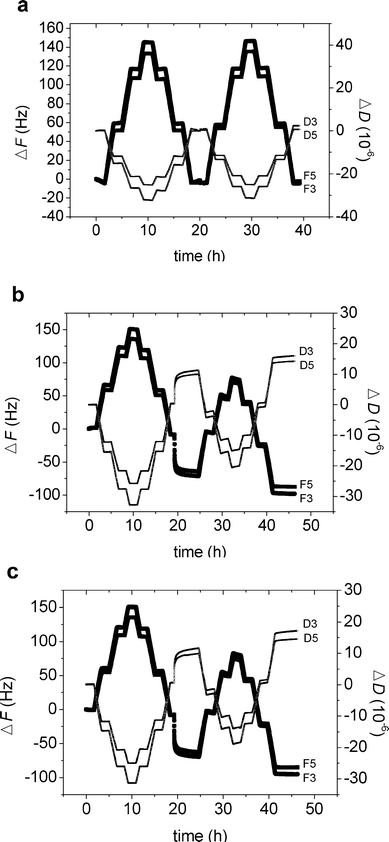 | ||
| Fig. 2 (a–c) Change in frequency (ΔF) and dissipation (ΔD) versus time for M1.6C on silica (a), M1.6C (b) and HP0.12M1.57C (c) on silanized silica. First the bare crystal was subjected to a temperature cycle, where it was allowed to stabilize for 100 min at 25 °C, 35 °C, 45 °C, 50 °C, 45 °C, 35 °C and 25 °C. Next, the polymer was injected into the measuring cell (after about 17 h), and allowed to adsorb for 2–3 h (40 ppm in solution). After adsorption at 25 °C, the polymer was kept in the solution during a similar temperature ramp as for the bare crystal. The figures illustrate the data normalized by the overtone number obtained for the third (F3, D3) and fifth (F5, D5) overtones. | ||
3.3. Adsorption on hydrophobized silica
The adsorption of M1.6C and HP0.12M1.57C on hydrophobized silica surfaces was investigated with QCM-D and ellipsometry. First a temperature cycle was conducted on the bare substrate, and then the polymer was adsorbed for 2–3 h. The polymer was then kept in the solution during the second temperature cycle.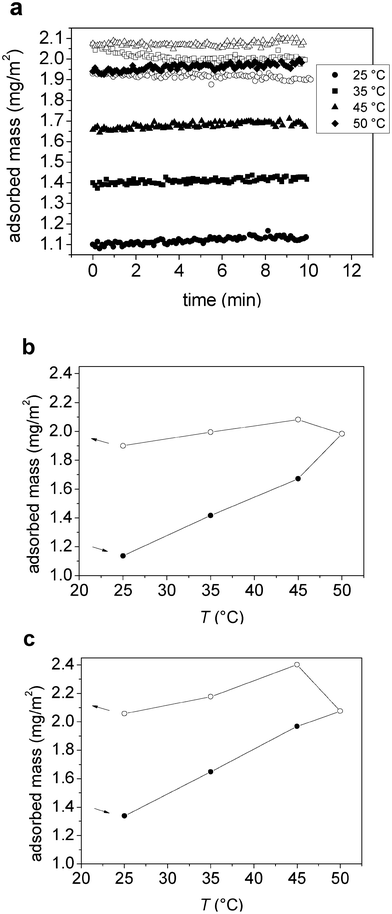 | ||
| Fig. 3 (a) Adsorbed mass (from ellipsometry) versus time after temperature stabilization in the measuring cell at each temperature for M1.6C on silanized silica. Filled and unfilled symbols correspond to data obtained on heating and cooling, respectively. (b–c) Adsorbed mass (from ellipsometry) as a function of temperature for (b) M1.6C and (c) HP0.12M1.57C on silanized silica. After adsorption at 25 °C (40 ppm polymer solution), the polymer was kept in the solution during the entire temperature ramp. Filled and unfilled symbols correspond to data obtained on heating and cooling, respectively. At each temperature the data was evaluated after 30–45 min of equilibration. The arrows indicate the direction of the temperature ramp. | ||
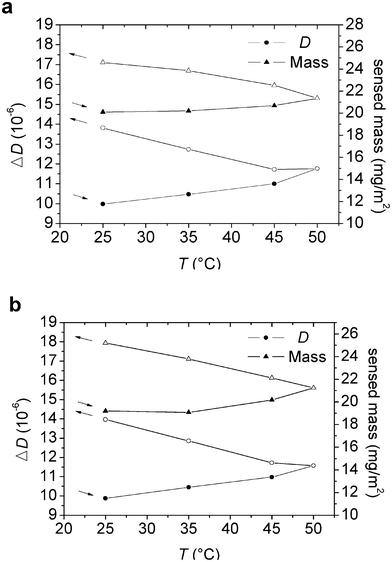 | ||
| Fig. 4 (a–b) Change in dissipation (at the fifth overtone) and sensed mass (from Voigt simulation) versus temperature for an adsorbed layer of (a) M1.6C and (b) HP0.12M1.57C on silanized silica. After adsorption at 25 °C (40 ppm polymer solution), the polymer was kept in the solution during the entire temperature ramp. Filled and unfilled symbols correspond to data obtained on heating and cooling, respectively. At each temperature the data was evaluated after 100 min of equilibration. The arrows indicate the direction of the temperature ramp. | ||
The shear viscosity and shear elasticity of the layer in the 15–35 MHz frequency range obtained from the Voigt modelling are shown in Fig. 5a and 5b. Both the shear modulus and the shear viscosity decrease with increasing temperature, and the hysteresis is small. Thus, the initial values are approached after the temperature cycle. The water content of the adsorbed layer in contact with the polymer solution was found to be in the range of 89–95% (see Fig. 6). The trend is that the water content decreases on heating and then increases again on cooling. There is a hysteresis in the water content data, with higher water content at a given temperature during the initial heating stage than during the subsequent cooling.
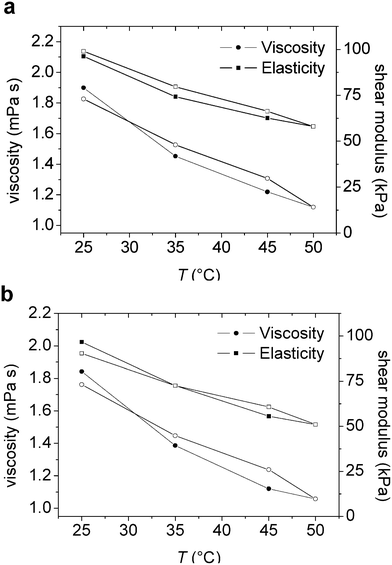 | ||
| Fig. 5 (a–b) Viscosity and elasticity shear modulus versus temperature from the Voigt simulation of an adsorbed layer of (a) M1.6C and (b) HP0.12M1.57C on silanized silica. After adsorption at 25 °C (40 ppm polymer solution), the polymer was kept in the solution during the entire temperature ramp. Filled and unfilled symbols correspond to data obtained on heating and cooling, respectively. At each temperature the data was evaluated after 100 min of equilibration. | ||
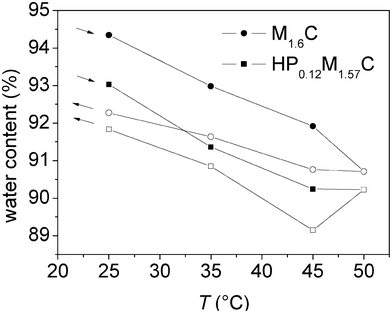 | ||
| Fig. 6 Water content, calculated from the adsorbed mass (ellipsometry) and the measured mass (QCM-D), versus temperature for an adsorbed layer of M1.6C and HP0.12M1.57C on silanized silica. After adsorption at 25 °C (40 ppm polymer solution), the polymer was kept in the solution, followed by a temperature ramp. Filled and unfilled symbols correspond to data obtained on heating and cooling, respectively. At each temperature the data was evaluated after 100 min of equilibration. The arrows indicate the direction of the temperature ramp. | ||
The thickness obtained from the Voigt modelling was very similar to the effective thickness calculated using eqn (5), as shown in Fig. 7a and 7b. The thickness values were increasing marginally during the heating stage, but increased more markedly during the subsequent cooling stage, from about 19 to 24 nm for M1.6C and from about 18 to 24 nm for HP0.12M1.57C.
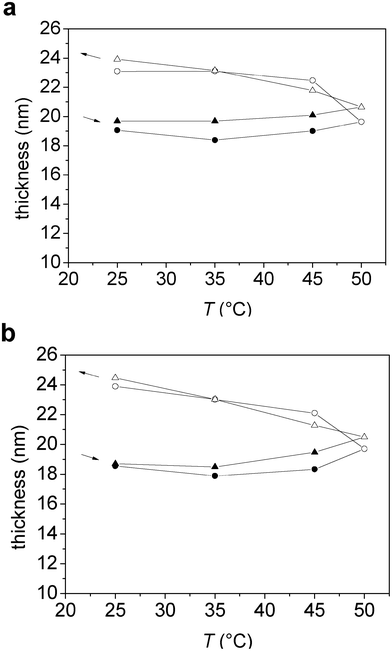 | ||
| Fig. 7 (a–b) Thickness versus temperature obtained from Voigt simulations (circles), and calculated from the measured and adsorbed mass according to eqn (5) (triangles), for an adsorbed layer of (a) M1.6C and (b) HP0.12M1.57C on silanized silica. After adsorption at 25 °C (40 ppm polymer solution), the polymer was kept in the solution during the entire temperature ramp. Filled and unfilled symbols correspond to data obtained on heating and cooling, respectively. At each temperature the data was evaluated after 100 min of equilibration. The arrows indicate the direction of the temperature ramp. | ||
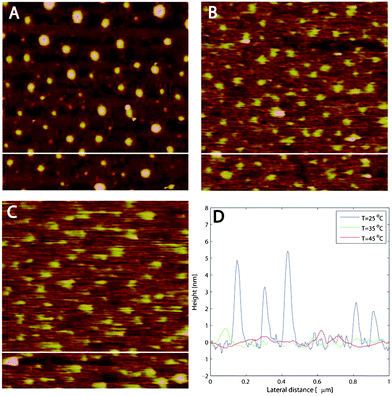 | ||
| Fig. 8 (a–c) AFM images (1 μm × 1 μm) of M1.6C adsorbed onto silanized silica at (a) 25, (b) 35 and (c) 45 °C. (d) Blob sizes for the different temperatures. At each temperature the image was acquired after 30 min of equilibration. The white horizontal lines in (a–c) correspond to the height distributions in (d). | ||
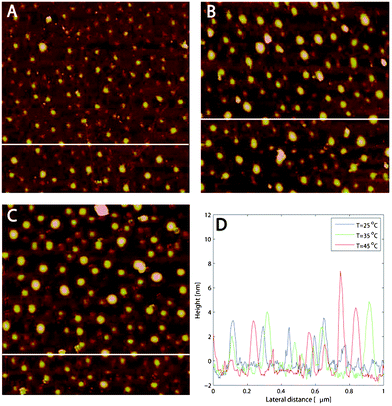 | ||
| Fig. 9 (a–c) AFM images (1 μm × 1 μm) of HP0.12M1.57C adsorbed onto silanized silica at (a) 25, (b) 35 and (c) 45 °C. (d) Blob sizes for the different temperatures. At each temperature the image was acquired after 30 min of equilibration. The white horizontal lines in (a–c) correspond to the height distributions in (d). | ||
As the temperature is increased to first 35 °C and then 45 °C (Fig. 8b and 8c) the polymer blobs gradually vanish. A line analysis, shown in Fig. 8d, demonstrates that the height variations decrease to around 0.5 nm. The increased blurriness in the images at 35 and 45 °C further suggests that more loosely bound M1.6C polymers are present on the surface and that these polymers are dragged more easily in the scan direction. A similar smearing of the images at 45 °C did not occur for pre-adsorbed layers of M1.6C,33 suggesting that this feature is due to the additional polymers that are adsorbed when the temperature is increased with polymers present in solution.
The AFM images of the hydrophobic surface in HP0.12M1.57C solution at 25, 35 and 45 °C are shown in Fig. 9a–c. As in the case of M1.6C, polymer blobs are appearing as spherical caps. However, opposite to the case of M1.6C, the line analysis presented in Fig. 9d does not show any significant changes in the height variations, while the apparent diameter of the blobs increases from around 40 nm at 25 °C to around 60 nm at 45 °C. It should be noted that the same behaviour was seen in several consecutive scans at different length scales and the change with temperature is thus a real effect and not an imaging artefact.
4. Discussion
4.1. The effects of polymers in solution during the temperature scan
In a previous study we have reported the effect of temperature on pre-adsorbed layer of M1.6C and HP0.12M1.57C on hydrophobized silica.33 For the pre-adsorbed layers the adsorbed mass does not change with temperature, in contrast to the more complex situation dealt with in this report where the adsorbed mass is allowed to vary. We find that the adsorbed mass for the two modified cellulose esters increase significantly (>60%), Fig. 3, with increasing temperature due to the worsening of the solvent quality, which is consistent with previous studies of adsorption of polymers with a lower critical solution temperature 11, 48 as well as with theoretical predictions.49 Only limited desorption occurs on cooling, and for any given temperature the adsorbed mass is lower during the initial heating stage than during the subsequent cooling stage. Thus, equilibration with the bulk solution is slow compared to the time scale of our experiment (about 17 h). By considering the data shown in Fig. 3a, we note that equilibrium is approached within less than one hour at low temperatures (≤40 °C), whereas this is not the case at higher temperatures. This occurs for both M1.6C and HP0.12M1.57C despite only M1.6C approaching the T2 value at the highest temperature being explored in this investigation. We propose that this is a consequence of the large heterogeneity of the MC and HPMC samples, and that in both polymer samples the most hydrophobic polymers (or regions of polymer chains) experience sufficiently poor solvency conditions to accumulate at the interface with time. It further implies that at a given temperature the mass of the adsorbed layer can be enhanced by allowing adsorption to proceed from an elevated temperature, and then cooling the system to the desired temperature. This may be of importance from an application point of view.By comparing the data presented in this report to corresponding data for pre-adsorbed layers,33 it is possible to draw conclusions on how the additional mass acquired during the heating stage affects the layer properties. For pre-adsorbed layers of M1.6C and HP0.12M1.57C on hydrophobized silica the layer thickness was observed to decrease with increasing temperature, and for a given temperature the thickness was smaller on cooling than on heating.33 In contrast, when polymers are present in solution during the temperature cycle the additional adsorption (Fig. 3) leads to a nearly constant thickness during heating, followed by an increase in thickness during the subsequent cooling (Fig. 7). Both for the pre-adsorbed layers33 and for the present case with polymer in solution a worsening of the solvent quality due to heating results in removal of water from the layer, and a partial reincorporation of water on cooling. This process, in both cases, is not fully reversible, but heat treatment results in a structural change towards a more compact state that persists for more than 17 h.
The energy dissipation value (Fig. 4) increases marginally as the temperature is increased, and then more significantly as the temperature subsequently is reduced. This suggests that even though the inner part of the layer becomes more compact on heating, as indicated by the reduced value of the energy dissipation for pre-adsorbed layers,33 the additional adsorption that occurs in the presence of polymers in solution compensates for this effect. Upon cooling the adsorption decreases only marginally and the swelling of the layer (Fig. 6 and 7) explains the increased energy dissipation.
Both the layer elasticity and layer viscosity decrease with increasing temperature (Fig. 5), as was also observed for pre-adsorbed layers.33 This result could be explained provided the polymers in the adsorbed layers aggregated at higher temperatures, whereby forming water rich domains with low viscosity and elasticity. However, the AFM images provided in Fig. 8 and 9 do not confirm this interpretation and further studies, preferably at even higher temperatures, are needed in order to rationalize these findings.
4.2. Temperature-dependent adsorption of polymers with a lower critical solution temperature
An increase in adsorbed amount with increasing temperature has been observed for poly(ethylene oxide),48poly(N-isopropylacrylamide),50 and ethyl(hydroxyethyl)cellulose, EHEC,11 due to the decreasing solvent quality with increasing temperature. However, compared to the results reported for EHEC the increase in layer thickness with increasing temperature for M1.6C and HP0.12M1.57C is considerably smaller, which can be attributed to the temperature not exceeding T2 (or T1 in the case of HP0.12M1.57C) in our experiments. For EHEC it has also been shown that the adsorption is considerably higher on a hydrophobic 11–12 rather than hydrophilic14silica surface. This is qualitatively similar to the data presented here, but quantitatively the adsorption (if any) of M1.6C and HP0.12M1.57C on silica is significantly smaller and below the detection limit of our instruments. This suggests that the adsorption of EHEC to silica is driven by the favourable interactions between the oligo(ethylene oxide) chains attached to this polymer and the silica surface.4.3. Comparison between MC and HPMC
The results obtained for M1.6C and HP0.12M1.57C in this study are in many aspects similar, which is in contrast to the case of pre-adsorbed layers without polymers in solution.33 The ΔD values are similar for the two polymers, indicating the same degree of compaction upon temperature increase and subsequent de-compaction with decreasing temperature. The bulkiness of the substituents in HP0.12M1.57C compared to M1.6C could be expected to infer a less compact configuration of HP0.12M1.57C at elevated temperatures, as was observed for pre-adsorbed layers. Thus, since this does not occur when polymer is present in solution, the additional adsorption from the bulk is suggested to explain why the M1.6C layer does not become more compact than the HP0.12M1.57C layer at temperatures up to 50 °C. The thickness, water content, dissipation and viscoelastic changes that occur during the temperature cycle are also similar for the two polymer layers. It remains to be seen if this also holds if the temperature is increased above T2 for M1.6C.Additional information on the temperature-dependent properties of the M1.6C and HP0.12M1.57C layers can be extracted from the AFM images in Fig. 8 and 9, and here a difference is seen between the two polymers. The decrease in the height variations with increasing temperature, for M1.6C, but not for HP0.12M1.57C, is attributed to the proximity to the T2 temperature in the case of M1.6C. For HP0.12M1.57C, despite the additional adsorption that occurs at elevated temperature, no changes in the height features of the outermost layer can be detected. We speculate that the more loosely bound polymer layers indicated by the stripes in the scan direction for M1.6C, but not for HP0.12M1.57C, is a consequence of a tip-induced structural change in the layer when the temperature approaches T2, presumably towards formation of fibrils as observed in bulk solution above T2.26 However, it still remains to be shown that a similar change in layer structure takes place in the HP0.12M1.57C layer at temperatures close to T2 for this system.
5. Conclusions
Methylcellulose and hydroxypropylmethylcellulose find use as viscosity modifiers in dispersions, e.g. paints, where it comes into contact with colloidal particles as well as surfaces. Due to the temperature-dependent solvency one can expect that in general adsorption of these polymers to particles and surfaces will be affected by a temperature change. Even though the details of the adsorption process will be strongly dependent on the surface chemistry of the particles/surfaces present we can draw some general conclusions from our investigation: I) The adsorption of these polymers is larger on hydrophobic surfaces than on hydrophilic ones. II) The adsorption increases with increasing temperature due to the worsening of the solvent quality. III) Adsorption equilibrium is established very slowly, particularly upon cooling. IV) The adsorption hysteresis observed during temperature cycling is significant and may well strongly affect properties of dispersions containing these polymers. More specifically we summarize our results as follows: M1.6C and HP0.12M1.57C adsorb significantly on hydrophobized silica surfaces, even at very low bulk concentrations. In contrast, no measurable adsorption was found on bare silica surfaces up to a temperature of 50 °C. The maximum temperature investigated, 50 °C, was close to the T2 temperature for M1.6C, but well below T2 for HP0.12M1.57C. For both polymers the amount adsorbed increases with increasing temperature, and despite the large difference in T2 temperature for M1.6C and HP0.12M1.57C the temperature dependence of the adsorption was found to be similar. The temperature-induced changes in energy dissipation, layer thickness, water content and viscoelastic properties for M1.6C and HP0.12M1.57C were also found to be similar. This contrasts to what was found for pre-adsorbed layers, where M1.6C layers became more compact with increasing temperature compared to HP0.12M1.57C layers. Thus, the additional adsorption that occurs at elevated temperature influences the layer properties significantly, and masks the temperature-response of the initially adsorbed polymer layer. AFM imaging showed that the layers are predominately flat with some parts of the polymers protruding into solution. For M1.6C, the height variations decreased considerably when approaching the T2 temperature, whereas for HP0.12M1.57C, no change in height variations was observed when the temperature was increased. This is rationalized by the large difference between the highest temperature used for AFM imaging (45 °C) and the T2 temperature for HP0.12M1.57C (67 °C). We note that the additional polymers that adsorb at elevated temperature are only to a small degree desorbed on cooling. This suggests a convenient way for enhancing the amount adsorbed at low temperature by allowing adsorption to proceed at high temperatures, and then cooling the system.Acknowledgements
This work was supported by the institute excellent centre CODIRECT. PC acknowledges financial support from the Swedish Research Council, VR.References
- P. W. Zhu and D. H. Napper, J. Colloid Interface Sci., 1994, 164, 489 CrossRef CAS.
- A. Dedinaite, E. Thormann, G. Olanya, P. M. Claesson, B. Nystrom, A. L. Kjoniksen and K. Z. Zhu, Soft Matter, 2010, 6, 2489 RSC.
- S. Verbrugghe, K. Bernaerts and F. E. D. Prez, Macromol. Chem. Phys., 2003, 204, 1217 CrossRef CAS.
- E. Thormann, P. M. Claesson and O. G. Mouritsen, Phys. Chem. Chem. Phys., 2010, 12, 10730 RSC.
- J. H. Elam, C. Karlsson and H. Nygren, Biomaterials, 1993, 14, 233 CrossRef CAS.
- C. Karlsson, M. Braide and H. Nygren, Colloids Surf., B, 2000, 17, 95 CrossRef CAS.
- C. Karlsson, A. Carlsson, M. Stenberg and H. Nygren, Colloid Polym. Sci., 1992, 270, 377 CAS.
- P. M. Claesson, M. Malmsten and B. Lindman, Langmuir, 1991, 7, 1441 CrossRef CAS.
- F. Joabsson, K. Thuresson and B. Lindman, Langmuir, 2001, 17, 1499 CrossRef CAS.
- L. Karlson, F. Joabsson and K. Thuresson, Carbohydr. Polym., 2000, 41, 25 CrossRef CAS.
- M. Malmsten and P. M. Claesson, Langmuir, 1991, 7, 988 CrossRef CAS.
- M. Malmsten, P. M. Claesson, E. Pezron and I. Pezron, Langmuir, 1990, 6, 1572 CrossRef CAS.
- M. Malmsten and B. Lindman, Langmuir, 1990, 6, 357 CrossRef CAS.
- I. Pezron, E. Pezron, P. M. Claesson and M. Malmsten, Langmuir, 1991, 7, 2248 CrossRef CAS.
- N. Rasenack, H. Hartenhauer and B. W. Muller, Int. J. Pharm., 2003, 254, 137 CrossRef CAS.
- S. E. Stabenfeldt, A. J. Garcia and M. C. LaPlaca, J. Biomed. Mater. Res., Part A, 2006, 77A, 718 CrossRef CAS.
- S. Baumgartner, O. Planinsekc, S. Srcic and J. Kristl, Eur. J. Pharm. Sci., 2006, 27, 375 CrossRef.
- S. Bohic, P. Weiss, P. Roger and G. Daculsi, J. Mater. Sci.: Mater. Med., 2001, 12, 201 CrossRef CAS.
- K. M. Ohta, M. Fuji, T. Takei and M. Chikazawa, Eur. J. Pharm. Sci., 2005, 26, 87 CrossRef CAS.
- A. Haque and E. R. Morris, Carbohydr. Polym., 1993, 22, 161 CrossRef CAS.
- A. Haque, R. K. Richardson, E. R. Morris, M. J. Gidley and D. C. Caswell, Carbohydr. Polym., 1993, 22, 175 CrossRef CAS.
- R. N. Ibbett, K. Philp and D. M. Price, Polymer, 1992, 33, 4087 CrossRef CAS.
- N. Onoda-Yamamuro, O. Yamamuro, Y. Inamura and H. Nomura, Phys. B, 2007, 393, 158 CrossRef CAS.
- N. Sarkar, J. Appl. Polym. Sci., 1979, 24, 1073 CrossRef CAS.
- S. M. C. Silva, F. V. Pinto, F. E. Antunes, M. G. Miguel, J. J. S. Sousa and A. Pais, J. Colloid Interface Sci., 2008, 327, 333 CrossRef CAS.
- R. Bodvik, A. Dedinaite, L. Karlson, M. Bergstrom, P. Baverback, J. S. Pedersen, K. Edward, G. Karlsson, I. Varga and P. M. Claesson, Colloids Surf., A, 2010, 354, 162 CrossRef CAS.
- G. Z. Zhang, Macromolecules, 2004, 37, 6553 CrossRef CAS.
- M. A. Plunkett, Z. Wang, M. W. Rutland and D. Johannsmann, Langmuir, 2003, 19, 6837 CrossRef CAS.
- G. M. Liu and G. Z. Zhang, J. Phys. Chem. B, 2005, 109, 743 CrossRef CAS.
- Y. K. Jhon, R. R. Bhat, C. Jeong, O. J. Rojas, I. Szleifer and J. Genzer, Macromol. Rapid Commun., 2006, 27, 697 CrossRef CAS.
- O. E. Perez, C. C. Sanchez, J. M. R. Patino and A. M. R. Pilosof, Biomacromolecules, 2006, 7, 388 CrossRef CAS.
- O. E. Perez, C. C. Sanchez, A. M. R. Pilosof and J. M. R. Patino, Food Hydrocolloids, 2008, 22, 387 CrossRef CAS.
- R. Bodvik, E. Thormann, L. Karlson and P. M. Claesson, Phys. Chem. Chem. Phys., 2011, 13, 4260 RSC.
- U. R. M. Kjellin, P. M. Claesson and P. Linse, Langmuir, 2002, 18, 6745 CrossRef CAS.
- J. Iruthayaraj, G. Olanya and P. M. Claesson, J. Phys. Chem. C, 2008, 112, 15028 CAS.
- G. Sauerbrey, Z. Phys., 1959, 155, 206 CrossRef CAS.
- F. Hook, M. Rodahl, B. Kasemo and P. Brzezinski, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 12271 CrossRef CAS.
- M. Rodahl and B. Kasemo, Sens. Actuators, A, 1996, 54, 448 CrossRef.
- M. V. Voinova, M. Rodahl, M. Jonson and B. Kasemo, Phys. Scr., 1999, 59, 391 CrossRef CAS.
- M. Landgren and B. Joensson, J. Phys. Chem., 1993, 97, 1656 CrossRef CAS.
- F. L. McCrackin, H. L. Steinberg, R. R. Stromberg and E. Passaglia, J. Res. Natl. Bur. Stand. (U.S), 1963, 67A, 363 Search PubMed.
- P. A. Cuypers, J. W. Corsel, M. P. Janssen, J. M. M. Kop, W. T. Hermens and H. C. Hemker, J. Biol. Chem., 1983, 258, 2426 CAS.
- J. A. D. Feijter, J. Benjamins and F. A. Veer, Biopolymers, 1978, 17, 1759 CrossRef.
- C. Larsson, M. Rodahl and F. Hook, Anal. Chem., 2003, 75, 5080 CrossRef CAS.
- Methocel Cellulose Ethers Technical Handbook, Dow Chemical Company, USA, 2002 Search PubMed.
- M. Lundin, F. Solaqa, E. Thormann, L. Macakova and E. Blomberg, Langmuir, 2011, 27, 7537 CrossRef CAS.
- G. Csoka, S. Marton, A. Gelencser and I. Klebovich, Eur. J. Pharm. Sci., 2005, 25, S74 Search PubMed.
- P. M. Claesson and C.-G. Gölander, J. Colloid Interface Sci., 1987, 117, 366 CrossRef CAS.
- G. J. Fleer, M. A. Cohen Stuart, J. M. H. M. Scheutjens, T. Cosgrove and B. Vincent, Polymers at Interfaces, Chapman & Hall, London, p. 1993 Search PubMed.
- S. Balamurugan, S. Mendez, S. S. Balamurugan, M. J. O'Brien and G. P. Lopez, Langmuir, 2003, 19, 2545 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |