DOI:
10.1039/C1RA00234A
(Paper)
RSC Adv., 2011,
1, 191-198
Synthesis of Pd on porous hollow carbon spheres as an electrocatalyst for alcohol electrooxidation
Received
27th May 2011
, Accepted 2nd June 2011
First published on 28th July 2011
Abstract
Porous hollow carbon spheres (PHCSs) are prepared with glucose as the carbon source and a solid core microporous shell silica (SCMSS) as the template. The PHCSs are composed of broken and complete hollow carbon spheres cemented to each other. There are big gaps between the PHCSs which make it possible to utilize the inner wall of the hollow carbon spheres as an electrocatalyst support. The PHCSs have a high Brauner–Emett–Teller (BET) surface area of 998.1 m2 g−1, and a pore volume of 1.88 cm3 g−1. The Pd nanoparticles supported on PHCS electrocatalysts are highly active for methanol, ethanol and isopropanol electrooxidation. Pd/PHCS has a 3.1 times higher in peak current density and a 80 mV negatively shifted onset potential compared with that of a Pd/C electrocatalyst at the same Pd loadings for ethanol electrooxidation. The porous structure of Pd/PHCS is favorable for mass transfer and remains high activity in higher concentration of ethanol. The Pd/PHCS electrocatalyst is a potential candidate for application in direct liquid alcohol fuel cells.
1. Introduction
Carbon materials with different structures and functions such as carbon spheres,1–3carbon nanotubes,4–7carbon nanowires,8,9carbon nanofibers10 honeycomb-like carbon,11,12diamond-like carbon,13 bamboo-like carbon,14 onion-like carbon15 and graphene16 have been of interest of the research community for various potential applications as adsorbents,17 for gas/energy storage18,19 and as catalyst supports20–22 because of their large specific surface area, low weight, chemical inertia and excellent electronic conductivity. Due to their high specific surface area and porous structure, the use of hollow carbon materials23–28 as electrocatalyst supports can improve the dispersion of noble metal and the mass transfer in the electrochemical processes. However, most inner wall surface of the hollow carbon materials could hardly be used. We have previously prepared hollow carbon hemispheres using solid core mesoporous shell silica templates.29 The hemispherical structure made reactants transport freely into and out of the inner and outer wall of the hollow carbon hemispheres.
Here, we report the preparation of the porous hollow carbon spheres (PHCSs) using glucose as the carbon source and a solid core microporous shell silica (SCMSS) as the template. The PHCSs show high Brauner–Emett–Teller (BET) surface area, owing to the accessibility of the inner wall surface and porous structure. The PHCSs have a different shape to the honeycomb-like carbon,11,12 and their formation mechanism is different from that of hollow carbon hemispheres.29 Moreover, The PHCSs have good contact with each other to reduce resistance, which could further increase the performance of the electrocatalyst. Ethylene glycol was used to reduce Pd salt into Pd nanoparticles as commonly used by the Xia group30–33 to prepare noble metals. Pd nanoparticles supported on PHCSs show enhanced catalytic activity for alcohol oxidation.
2. Experimental
2.1 Synthesis of the core/shell silica template
In this study, the solid core microporous shell silica (SCMSS) was prepared as the template. In a typical synthesis of the silica template, 25.0 ml tetraethoxysilane (TEOS, A.R., Guangzhou Chemical Reagent Co., China) was added to a solution containing 27.0 ml ethanol (A.R., Tianjin Fuyu Fine Chemical Co., Ltd, China), 3.7 ml deinoized water and 7.0 ml aqueous ammonia (A.R., 30 wt.%, Guangdong Guanghua Chemical Co., Ltd, China) at 303 K with vigorous stirring. The mixture was stirred continuously for 1 h to get uniform silica spheres. A mixed solution containing 2.5 ml TEOS and 1.5 ml Phenyltriethoxysilane (PhTES, A.R., Shanghai Citailong Co., Ltd, China) was added into the above colloidal solution and further stirred for 1 h. The resulting nanocomposite was dried in atmosphere at 363 K for 12 h and further calcined at 873 K for 5 h under atmosphere to obtain the SCMSS.
2.2 Synthesis of the PHCSs
For the synthesis of the PHCSs, 1.0 g SCMSS, 0.5 g glucose (A.R., Tianjin Damao Chemical Reagent Co., China) and 100 ml deionized water were mixed in a flask and stirred vigorously in a 333 K water bath until the mixture turned into a dry solid sample. Subsequently, the sample was heated at 5 K min−1 to 1123 K and held for 3 h to carbonize the glucose. The silica template was removed by etching in 10% HF solution (A.R., Guangzhou Chemical Reagent Co., China) to obtain the PHCSs.
2.3 Preparation of Pd/PHCS electrocatalysts
Pd supported on PHCSs was prepared and used as an electrocatalyst for alcohol oxidation. The preparation of Pd/PHCS and Pd/C electrocatalysts was as follows. 50 mg PHCSs or Vulcan XC-72 carbon (Carbot Co., USA) were mixed with a mixture of 16.67 mg PdCl2 and 50 ml ethylene glycol (A.R., Tianjin Fuyu Fine Chemicals Co., Ltd, China) to form a uniform ink in an ultrasonic bath for 30 min. The pH of the ink was then adjusted to 10 using a 5 wt% NaOH (A.R., Guangzhou Chemical Reagent Factory, China)/ethylene glycol solution, the mixture was heated in a microwave oven (1000 W, 2.45 GHz), 10 s on and 10 s off for a total of 10 cycles. After cooling, the mixture was washed with deionized water 4–5 times, then dried in vacuum at 353 K for 2 h to get Pd/PHCS and Pd/C. The weight percentage of Pd in all the electrocatalysts was kept at 20 wt%, which was proved by the inductively coupled plasma-atomic emission spectrometry (ICP, IRIS(HR), USA).
2.4 Preparation of the electrode
For electrode preparation, Pd/PHCS or Pd/C (5 mg) were dispersed in a mixture of ethanol (1 ml) and Nafion suspension (1 ml, 0.5 wt%, DuPont, USA) under ultrasonic stirring to form an ink. 10 μl ink was then deposited onto the surface of the glassy carbon rod (4 mm in diameter) and dried at room temperature overnight. The Pd loading was 0.02 mg cm−2.
2.5 Characterization
All electrochemical measurements were performed in a three-electrode cell on a potentiostat (IM6e, Zahner-Electrik, Germany) at 303 K. Platinum foil (1.0 cm2) and Hg/HgO were used as counter and reference electrodes, respectively.
The morphologies and sizes of the templates, Pd/PHCS and Pd/C electrocatalysts were characterized by scanning electron microscopy (SEM, LEO 1530VP, Germany) and transmission electron microscopy (TEM, JEM-2010HR, JEOL Ltd., Japan) operating at 200 kV. The structure of the PHCSs and electrocatalysts were determined by an X-ray diffractometer (D/Max-IIIA, RigakuCo., Japan, CuK1, λ = 1.54056 Å radiation). The BET surface area, pore volume and pore diameter were determined on a physical adsorption instrument (ASAP 2400, Micrometeritics Co., USA).
3. Results and discussion
3.1 Morphologies of the SCMSS template and the PHCSs
Fig. 1a–b show the TEM and HRTEM images of the SCMSS templates. The diameter of the SCMSS is about 330 nm as shown in Fig. 1a. Even in the HRTEM image shown in Fig. 1b it is difficult to tell the difference between the microporous shell of SCMSS and its solid core. This is because the diameter of the micropores in the shell is extremely small. The small pore size makes it difficult for the glucose to infiltrate into the micropores, which is the determining factor in the formation of PHCSs. Fig. 1c is the SEM image of the PHCSs. It shows that the PHCSs are composed of broken and complete hollow carbon spheres cemented to each other. The internal diameters were around 290 nm and the wall thickness ranged from 5 to 15 nm. Each broken hollow carbon sphere has an independent carbon shell with several mesopores, which is quite different from the morphology of honey-comb carbon.11,12 The TEM image in Fig. 1d shows the same information on the structure of the PHCSs as the SEM micrograph (Fig. 1c) but also shows that the PHCSs are composed of broken and complete hollow carbon spheres and have lots of mesopores widely distributed, making the inner walls of the spheres easy to use for the loading of metal nanoparticles.
3.2 Synthesis mechanism of PHCSs
The possible formation of the spherical morphology of the PHCSs is schematically illustrated in Fig. 2. The first step is to cover SCMSS with glucose which is realized by mixing glucose, water and SCMSS. With the evaporation of water, the glucose was absorbed on the surface of the SCMSS and partly infiltrated into the microsporous shell of the SCMSS. The small size of the micropores in the shell of SCMSS makes it difficult for the glucose to enter the micropores, parts of the shell can not be filled with glucose, thus resulting in lots of glucose breaches on the shell surface after the evaporation of water. The second step is to carbonize the glucose. At the carbonization temperature, the glucose on the surface of SCMSS melts and flows down, this process enlarged the glucose breaches on the surface of the SCMSS, then these large glucose breaches become large carbon breaches after carbonization. Finally, the SCMSS was removed by HF etching and the PHCSs with lots of large breaches resulted. These breaches exposed the inner wall surface of hollow carbon materials which could greatly increase the specific surface area.
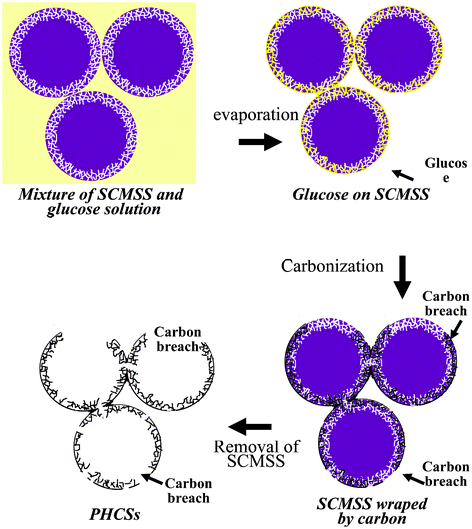 |
| | Fig. 2 Schematic illustration of the synthesis procedure of PHCSs. | |
The formation of glucose breaches on the surface of the SCMSS is a determining factor in the formation of the PHCSs. So it is necessary to study the parameters during the water evaporation process to control the formation of breaches. Fig. 3 shows PHCSs synthesized at different evaporation temperatures after the SCMSS templates and the glucose solution are mixed. At higher temperatures, water evaporates faster, leaving less time for the glucose to infiltrate into the micropores of the surface of the SCMSS. The shell could be partially filled with glucose which results in one-third or even larger breaches formed on a sphere after carbonization and removal of SCMSS. Fig. 3a shows a honeycomb-like structure composed by hemispheres. In this structure, both the inner and outer walls are exposed. The honeycomb-like carbon structure is formed because the glucose did not have enough time to enter the micropores of the SCMSS template, resulting in an independent carbon layer. In contrast, the low evaporation temperature provided adequate time for the glucose to enter the micropores so the whole surface and inside of the wall of the SCMSS could be covered or filled with glucose. In that case, the inner wall surface could hardly contribute to the total surface area since almost all carbon particles have a spherical but hollow sturcture after carbonization as shown in Fig. 3b. This is not a favorable structure for electrocatalyst loading. By comparing these two cases, the evaporation temperature was chosen to be 333 K, as this is a suitable temperature for the formation of the optimized carbon material.
 |
| | Fig. 3
SEM micrographs of PHCSs synthesized at evaporation temperature of (a) 363 K and (b) 313 K and PHCSs with 1 g SCMSS and 1.5 g glucose (c). | |
As discussed above, the coverage of glucose on the surface of the SCMSS could influence the morphology of the final product. The temperature of water evaporation is one of the critical factors. Another factor affecting the final product is the ratio of glucose to SCMSS. An excess of glucose would result in a similar structure to that prepared at the low evaporation temperature. Fig. 3c shows the SEM micrograph of PHCSs prepared with 1 g SCMSS and 1.5 g glucose. Obviously, more glucose leads to an interconnected macroporous carbon block, i.e. honeycomb-like carbon. It is important to optimize the ratio of glucose to SCMSS to get the best structure that exposes both the inner and outer surfaces of the hollow carbon spheres. To sum up, to get PHCSs with not only the outer wall but also the inner wall which could be utilized for metal loading, the SCMSS/glucose ratio and the evaporation rate are the key factors. In this work, the PHCSs were synthesized under optimized conditions and the morphology of the product is as shown in Fig. 1.
3.3 Surface structure of PHCSs
The surface area and pore volume of the PHCSs were measured and summarized in Table 1. The PHCSs have a high BET surface area of 998.1 m2 g−1, and a large pore volume of 1.88 cm3 g−1. The micropore area was 450.3 m2 g−1, which was contributed to from the inverse replica of the microporous shell of the silica template and the decomposition of glucose during carbonization. Those micropores contributed 45.1% of the total surface area. The micropore volume was 0.27 cm3 g−1 which contributes merely 14.4% to the total volume. It is clear that the surface area and pore volume mainly come from outer surface and big pores which are accessible for molecules. The pore size distribution was measured by the Barrett–Joyner–Halenda (BJH) method as shown in Fig. 4, indicating that the mesopores and macropores produce were the mainly contributors to the pore volume of the PHCSs.
 |
| | Fig. 4
Pore size distribution of PHCSs. | |
Table 1 Surface area and pore volume of the PHCSs
| Total surface area/m2 g−1 |
Micropore area/m2 g−1 |
Total pore volume/cm3 g−1 |
Micropore volum/cm3 g−1 |
| 998.1 |
450.3 |
1.88 |
0.27 |
3.4 Physical characterization of Pd/PHCS
The high BET surface area and porous structure of the PHCSs are favorable for the uniform dispersing noble metal nanoparticles and mass transport needed for supports of fuel cell electrocatalysts. Pd was loaded onto the PHCSs to form a Pd/PHCS electrocatalyst for alcohol oxidation. The distirbution of Pd on PHCSs was observed by TEM as shown in Fig. 5a. It reveals that the distribution of the Pd nanoparticles was uniform and very narrow in size. Based on 100 Pd nanoparticles randomly selected, the average particle size was 5.9 nm. Fig. 5b is the histogram of Pd particle size with a Gaussian distribution. Fig. 5c is the TEM image of Pd/C with an uneven distribution and conglomeration, which would affect its activity for use as an electrocatalyst. Since both the Pd/PHCS and Pd/C were prepared via the same procedure, the difference in Pd distribution on different supporting materials could only be explained by the structure of the loading material. The porous and high surface area structure of the PHCSs resulted in a uniform distribution compared with the low surface area and solid carbon particles. Fig. 5d shows the XRD patterns of the Pd/PHCS and Pd/C electrocatalysts. The diffraction peaks observed at 2θ of 39.8°, 46.1° and 67.5° correspond to the (111), (200) and (220) facets of the face-centered cubic structure of a palladium crystal. The Pd(220) peak was used to calculate the particle size according to the Scherrer's equation:
where d denotes the average diameter in nm, K denotes the Scherrer constant (0.89), λ denotes the wavelength of the X-rays (λ = 0.154056 nm), B denotes the corresponding full width at half maximum (FWHM) of the (220) diffraction peak, and θ denotes the Bragg diffraction angle. The Pd particle size in Pd/PHCS was calculated to be 6.3 nm which is consistent with the TEM results. The Pd particle size in Pd/C was 7.0 nm.
 |
| | Fig. 5 (a) TEM image of Pd/PHCS, (b) corresponding Pd particle size distribution on Pd/PHCS, (c) TEM image of Pd/C, (d) XRD patterns of Pd/PHCS and Pd/C. | |
3.5 Electrochemistry properties of Pd/PHCS
Fig. 6a displays the cyclic voltammograms of methanol, ethanol and isopropanol oxidation on a Pd/PHCS electrode. Pd/PHCS shows very high activities for the oxidation of the three alcohols. In particular, the ethanol oxidation gave the most negative onset potential and the highest peak current density. Fig. 6b compares the cyclic voltammograms of ethanol oxidation on Pd/PHCS and Pd/C electrodes. The onset potential on Pd/PHCS has a negative shift of 80 mV compared with that of the Pd/C electrode. Consider the good contact between the carbon spheres in the PHCSs, the electronic resistance may be much smaller than that of carbon particles. This would be one of the origins of the negative shift in onset potential. The peak current density on the Pd/PHCS electrode was 3.1 times of that on the Pd/C electrode, which is due to the high BET surface area and porous structure for increasing Pd utilization and mass transport of the reactants. Fig. 6c compares the cyclic voltammograms of Pd/PHCS and Pd/C in a background solution of 1 mol L−1KOH. The results indicate that the Pd/PHCS has a much higher electrochemical active surface area than Pd/C.
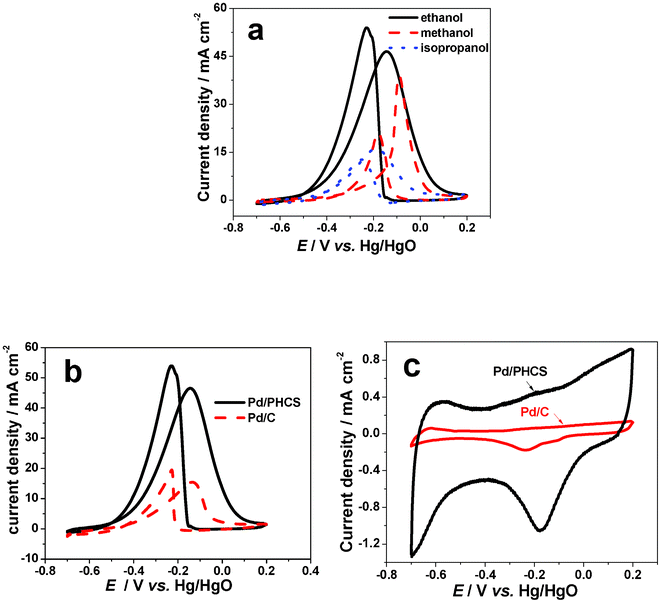 |
| | Fig. 6
Cyclic voltammograms of (a) different alcohol oxidation on Pd/PHCS electrodes in 1.0 mol l−1KOH/1.0 mol l−1alcohol solution, (b) cyclic voltammograms of ethanol oxidation on Pd/C and Pd/PHCS electrodes, in 1.0 mol l−1KOH/1.0 mol l−1ethanol solution and (c) cyclic voltammograms of Pd/C and Pd/PHCS in 1.0 mol l−1KOH solution at 303 K, scan rate: 50 mV s−1. | |
For the application in direct alcohol fuel cells, higher concentrations of alcohol are desired to enhance the power to volume densities. However, higher concentrations of alcohol also brings severe mass transport problem. The cyclic voltammogram curves of the oxidation of ethanol at different concentrations on Pd/PHCS and Pd/C electrodes are shown in Fig. 7. When the concentration of ethanol was increased from 1 mol l−1 to 2 mol l−1, the peak current density of Pd/C was reduced by 40%, however, the peak current density of Pd/PHCS is hardly reduced. The results further prove that the PHCS structure is favorable for mass transfer and, consequently, can increase the energy densities of direct ethanol fuel cells by using higher concentration alcohol.
 |
| | Fig. 7 (a) Cylic voltammograms of ethanol oxidation on Pd/C and Pd/PHCS in 1.0 mol l−1KOH/different ethanol concentrations at 303 K, scan rate: 50 mV s−1 and (b) chronopotentiometric curves of ethanol oxidation on Pd/PHCS and Pd/C at different current densities in 1.0 mol l−1ethanol/1.0 mol l1KOH solution, at 303 K. | |
The steady state performance of the Pd/PHCS and Pd/C electrocatalysts for ethanol oxidation was compared. The chronopotentiometric characterization proved that the Pd/PHCS electrocatalyst could sustain larger current densities for ethanol oxidation than Pd/C as shown in Fig. 7b. The oxidation remained continuously at lower potential under 1.2 mA cm−2 polarization on the Pd/PHCS electrode. The degradation of the electrode performance was fast at the higher polarization current density of 8 mA cm−2. The loss of the electrode activity could be the poisoning of the electrode by the accumulation of the poisoning species produced during the oxidation of ethanol. A fast increase in the potential means a fast loss of activity. The Pd/C electrode could only be sustained for a very short time at a constant current density of 1.2 mA cm−2 due to the lower active surface area. The Pd/PHCS is more stable than Pd/C, showing a much improved stability.
4. Conclusions
Porous hollow carbon spheres (PHCSs) were prepared using glucose as the carbon source and a solid core microporous shell silica as the template. SEM and TEM measurements revealed that the PHCSs were composed of partially broken hollow carbon spheres (or hollow carbon hemispheres). This exact shape depends on the SCMSS/glucose ratio and the evaporation temperature of water in the glucose solution. High surface area spherical carbon of 998.1 m2 g−1 could be formed under suitable conditions. Pd nanoparticles supported on PHCSs (Pd/PHCS) were used as the electrocatalyst for ethanol oxidation. The Pd/PHCS electrocatalyst gave 3.1 times higher current density and 80 mV negative shift of the onset potential compared with Pd/C at the same Pd loadings. Moreover, the Pd/PHCS electrocatalyst showed advantages in the oxidation of higher concentrations of ethanol and improved stability at larger current densities. The origin of these advantages are contributed to from the high surface area of the PHCSs, the hollow porous structure and good contact between the carbon spheres and the resulting uniform distribution of Pd to improve the mass transfer and electronic conductivity.
Acknowledgements
The work was supported by the National Natural Science Foundation of China (21073241, U1034003) and the China National 863 Program (2009AA034400). Dr H. Meng thanks the Doctoral Fund of Ministry of Education of China (20100171120022). Mr. Z. X. Yan thanks the support by Yat-sen Innovative Talents Cultivation Program for Excellent Tutors.
References
- Z. Wang, L. Yu, W. Zhang, Z. Zhu, G. He, Y. Chen and G. Hu, Phys. Lett. A, 2003, 307, 249–252 CrossRef CAS.
- G. Y. Chen, V. Stolojan, S. R. P. Silva, H. Herman and S. Haq, Carbon, 2005, 43, 704–708 CrossRef CAS.
- L. Wang, M. Fujita, M. Toyoda and M. Inagaki, New Carbon Mater., 2007, 22, 102–108 CrossRef CAS.
- Z. Xu, J. Lin, V. A. L. Roy, Y. Ou and D. Liao, Mater. Sci. Eng., B, 2005, 123, 102–106 CrossRef.
- S. Huang, Carbon, 2003, 41, 2347–2352 CrossRef CAS.
- Y. C. Choi and W. Choi, Carbon, 2005, 43, 2737–2741 CrossRef CAS.
- S. N. Bondi, W. J. Lackey, R. W. Johnson, X. Wang and Z. L. Wang, Carbon, 2006, 44, 1393–1403 CrossRef CAS.
- Z. Ni, Q. Li, L. Yan, J. Gong and D. Zhu, Diamond Relat. Mater., 2008, 17, 365–371 CrossRef CAS.
- H. Li, F. W. Sun, Y. F. Li, X. Liu and K. M. Liew, Scr. Mater., 2008, 5, 479–482 CrossRef.
- A. Genaidy, R. Sequeira, M. Rinder and A. A-Rehim, Sci. Total Environ., 2009, 407, 5825–5838 CrossRef CAS.
- S. X. Chen, X. Zhang and P. K. Shen, Electrochem. Commun., 2006, 8, 713–719 CrossRef CAS.
- Z. Yan, G. He, G. Zhang, H. Meng and P. K. Shen, Int. J. Hydrogen Energy, 2010, 35, 3263–3269 CrossRef CAS.
- A. Yonezu and X. Chen, Diamond Relat. Mater., 2010, 19, 40–49 CrossRef CAS.
- F. Wang, L. Lang, B. Li, W. Liu, X. Li and Z. Xu, Mater. Lett., 2010, 64, 86–88 CrossRef CAS.
- M. Bystrzejewski, M. H. Rummeli, T. Gemming, H. Lange and A. Huczko, Carbon, 2010, 48, 21–23 Search PubMed.
- T. V. Cuong, V. H. Pham, Q. T. Tran, J. S. Chung, E. W. Shin, J. S. Kim and E. J. Kim, Mater. Lett., 2010, 64, 765–767 CrossRef CAS.
- V. Y. Yakovlev, A. A. Fomkin and A. V. Tvardovski, J. Colloid Interface Sci., 2003, 268, 33–36 CrossRef CAS.
- H. Cheng, Q. Yang and C. Liu, Carbon, 2001, 39, 1447–1454 CrossRef CAS.
- M. W. Verbrugge, P. Liu and S. Soukiazian, J. Power Sources, 2005, 141, 369–385 CrossRef CAS.
- T. V. Reshetenko, L. B. Avdeeva, Z. R. Ismagilov and A. L. Chuvilin, Carbon, 2004, 42, 143–148 CrossRef CAS.
- A. Guha, T. A. Zawodzinski Jr and D. A. Schiraldi, J. Power Sources, 2007, 172, 530–541 CrossRef CAS.
- M. Gurrath, T. Kuretzky, H. P. Boehm, L. B. Okhlopkova, A. S. Lisitsyn and V. A. Likholobov, Carbon, 2000, 38, 1241–1255 CrossRef CAS.
- F. P. Hu, Z. Wang, Y. Li, C. Li, X. Zhang and P. K. Shen, J. Power
Sources, 2008, 177, 61–66 CrossRef CAS.
- Y. M. Chang, Y. C. Hsieh and P. W. Wu, Diamond Relat. Mater., 2009, 18, 501–504 CrossRef CAS.
- K. H. Lim, H. S. Oh and H. Kim, Electrochem. Commun., 2009, 11, 1131–1134 CrossRef CAS.
- K. Nam, D. Jung, S. K. Kim, D. Peck and S. Ryu, J. Power Sources, 2007, 173, 149–155 CrossRef CAS.
- B. Fang, M. Kim and J. S. Yu, Appl. Catal., B, 2008, 84, 100–105 CrossRef CAS.
- Z. Yan, Z. Hu, C. Chen, H. Meng, P. K. Shen, H. Ji and Y. Meng, J. Power Sources, 2010, 195, 7146–7151 CrossRef CAS.
- Z. Yan, H. Meng, L. Shi, Z. Li and P. K. Shen, Electrochem. Commun., 2010, 12, 689–692 CrossRef CAS.
- Y. Xia, Y. J. Xiong and B. Lim, Angew. Chem., Int. Ed., 2009, 48, 60–103 CrossRef CAS.
- B. Lim, T. Yu and Y. Xia, Angew. Chem., Int. Ed., 2010, 49, 9819–9820 CrossRef CAS.
- Q. Zhang, W. Li, L. P. Wen, J. Chen and Y. Xia, Chem.–Eur. J., 2010, 16, 10234–10239 CrossRef CAS.
- T. Yu, J. Zeng, B. Lim and Y. Xia, Adv. Mater., 2010, 22, 5188–5192 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here. 