Biosynthesis of acorane sesquiterpenes by Trichoderma†
Christian A.
Citron
,
Ramona
Riclea
,
Nelson L.
Brock
and
Jeroen S.
Dickschat
*
Institut für Organische Chemie, Technische Universität Braunschweig, Hagenring 30, 38106 Braunschweig, Germany. E-mail: j.dickschat@tu-bs.de; Fax: +49 (0)531 3915272; Tel: +49 (0)531 3915264
First published on 3rd August 2011
Abstract
The volatiles of three Trichoderma strains have been collected by use of a closed-loop stripping apparatus (CLSA) and analysed by GC-MS. Several biosynthetically related sesquiterpenes have been identified in the complex headspace extracts. Additionally, the absolute configurations of some sesquiterpenes were determined by chiral GC-MS. The biosynthesis of the main compound tricho-acorenol was investigated by feeding of deuterated mevalonolactone isotopomers and contradicts a previously published pathway for Fusidium.
Introduction
Terpenes form one of the largest and structurally most intriguing classes of natural products with more than 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 known representatives. The carbon backbones of these, in most cases, cyclic compounds are generated from only a handful of linear precursors such as geranyl diphosphate (GPP, monoterpenes), farnesyl diphosphate (FPP, sesquiterpenes), and geranylgeranyl diphosphate (GGPP, diterpenes) by the action of terpene cyclases.1 Terpene biosynthesis generally proceeds via cationic intermediates either formed by abstraction of the precursor's diphosphate or by protonation of an olefinic double bond. During the cyclisation highly complex carbon skeletons with several stereogenic centres can be formed. Termination mechanisms include deprotonations or capture of late cationic intermediates by nucleophiles such as water. To date, several plant terpene cyclases are known, but only a few bacterial (e.g. the pentalenene synthase from Streptomyces sp. UC53192) or fungal enzymes (e.g. the aristolochene synthase from Penicillium roquefortii3) have been investigated.
000 known representatives. The carbon backbones of these, in most cases, cyclic compounds are generated from only a handful of linear precursors such as geranyl diphosphate (GPP, monoterpenes), farnesyl diphosphate (FPP, sesquiterpenes), and geranylgeranyl diphosphate (GGPP, diterpenes) by the action of terpene cyclases.1 Terpene biosynthesis generally proceeds via cationic intermediates either formed by abstraction of the precursor's diphosphate or by protonation of an olefinic double bond. During the cyclisation highly complex carbon skeletons with several stereogenic centres can be formed. Termination mechanisms include deprotonations or capture of late cationic intermediates by nucleophiles such as water. To date, several plant terpene cyclases are known, but only a few bacterial (e.g. the pentalenene synthase from Streptomyces sp. UC53192) or fungal enzymes (e.g. the aristolochene synthase from Penicillium roquefortii3) have been investigated. 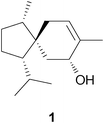
A promising approach to new microbial terpene sources are the frequently overlooked volatiles. Trichoderma, opportunistic, avirulent plant symbionts used in agriculture to increase crop productivity4 and well known for an extensive secondary metabolism,5 was chosen as part of our screening program towards fungal volatiles. Previous reports have demonstrated that several strains of this genus release volatiles. Recently, a complex mixture of volatile lipids, mono- and sesquiterpenes was identified in T. atroviride,6 while already in 1995 the spirocyclic sesquiterpene alcohol tricho-acorenol (1) was identified in culture extracts of Trichoderma koningii.7 Initially, this compound was reported to be a new natural product, but indeed it is identical to coccinol from Fusidium coccineum.5 A biosynthetic pathway to tricho-acorenol or coccinol, respectively, was developed from feeding experiments with 13C-labelled precursors, that involves the cyclisation of FPP via nerolidyl diphosphate (NPP) to the bisabolyl cation, a 1,2-hydride shift to the homobisabolyl cation, a second ring closure to form the spirocyclic acorenyl cation, and, most important for the discussion below, a 1,5-hydride shift that was, however, not experimentally proven, followed by final nucleophilic capture with water8 (reviewed in ref. 9). We reinvestigated the biosynthesis of tricho-acorenol in Trichoderma by feeding of several deuterated mevalonolactone isotopomers10 and demonstrate here that this previously proposed biosynthetic pathway to tricho-acorenol is incorrect at least for Trichoderma.
Results and discussion
The volatiles released by three Trichoderma strains were collected by use of a closed-loop stripping apparatus (CLSA) and the obtained headspace extracts were analysed by GC-MS.11 The results of these analyses are summarised in Table 1 of the ESI† and the gas chromatograms are shown in Fig. 1. All three strains emitted a complex bouquet of sesquiterpenes with tricho-acorenol (1) as the principal component accompanied by acorenone (2, Fig. 2). A large number of structurally related cyclic sesquiterpene hydrocarbons with acora-2,4-diene (3), acora-3,5-diene (4), α-neocallitropsene (5), α-alaskene (6), (–)-β-cedrene (7), (–)-β-funebrene (8), and β-duprezianene (9) as main compounds were identified from their mass spectra and retention indices by comparison to published data. The linear compound (E)-nerolidol (10) was also found in major amounts. Beside these main components the sesquiterpenes 11–38 were identified and occurred in minor percentage. These side-products were (−)-α-funebrene (11), sesquisabinene B (12), epi-isozizaene (13), myltayl-8(12)-ene (14), cadina-4,11-diene (15), (E)-β-farnesene (16), muurola-4,11-diene (17), amorpha-4,11-diene (18), muurola-4(15),5-diene (19), (+)-β-microbiotene (20), γ-muurolene (21), α-curcumene (22), bicyclosesquiphellandrene (23), zingiberene (24), β-alaskene (25), 10-epi-zonarene (26), α-cuprenene (27), (Z)-α-bisabolene (28), α-chamigrene (29), (S)-cuparene (30), (S)-β-bisabolene (31), cis-calamenene (32), β-sesquiphellandrene (33), zonarene (34), (S)-γ-cuprenene (35), trans-calamenene (36), and δ-cuprenene (37) (Fig. 2, Table 1 of ESI†). The peak representing compound 17 may also be 10-epi-muurola-4,11-diene (38) that has the same mass spectrum and retention index as muurola-4,11-diene.12 The absolute configurations of (−)-β-cedrene (7), (−)-β-funebrene (8), (−)-α-funebrene (11), (+)-β-microbiotene (20), (S)-cuparene (30), (S)-β-bisabolene (31), and (S)-γ-cuprenene (35) were determined by chiral GC-MS using the essential oils of different plants that contained reference compounds with known absolute configurations or commercial standards (Fig. 1–5 of ESI†). These sesquiterpenoids were produced by all three Trichoderma strains in nearly the same relative amounts, with the exception that some of the trace components were occasionally missing in one or a few extracts, pointing to a highly conserved sesquiterpene cyclase encoded in their genomes. Additionally, a few monoterpenes (linalool, linalool oxide, α-terpineol, and camphor) and a few unidentified sesqui- and diterpenes were released. Further compounds emitted in strain-specific proportions were 2,3-butanediol, the alcohols 3-methylbutan-1-ol and 2-methylbutan-1-ol, their acetate esters together with isobutyl acetate, the lavender lactone 4-methylhex-5-en-4-olide, the aromatic compounds 2-phenylethanol and 3,4-dimethoxystyrene, and the typical fungal volatiles oct-1-en-3-ol, octan-3-one, octan-3-ol, and 2-acetyl-4-hydroxy-6-methyl-2H-pyran-2-one.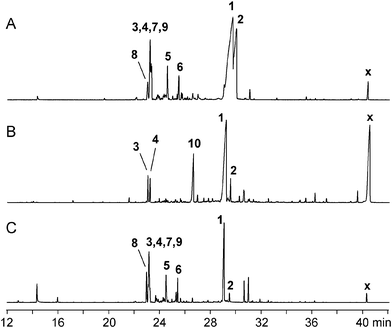 | ||
| Fig. 1 Total ion chromatograms of the headspace extracts of (A) Trichoderma harzianum 714, (B) T. longibrachiatum 594, and (C) T. viride 54. Compound x is an unidentified diterpene. | ||
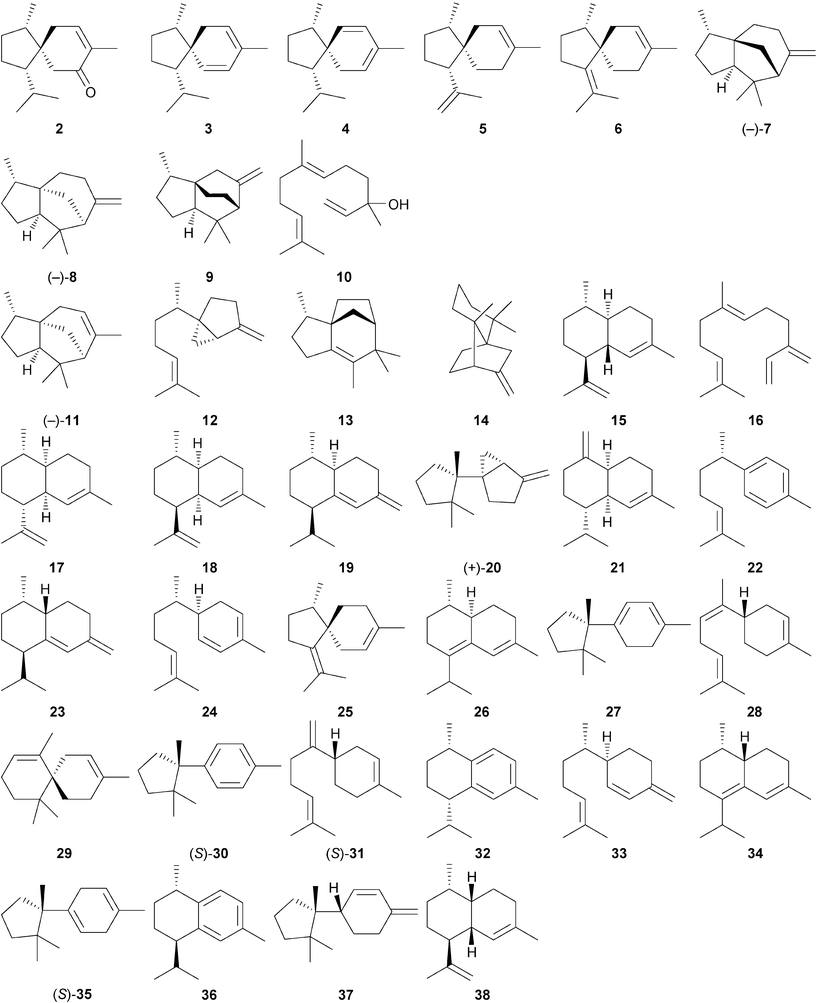 | ||
| Fig. 2 Volatile sesquiterpenes from Trichoderma. | ||
All sesquiterpenes except the linear molecules 10 and 16 have a six-membered ring in common, suggesting a joint biosynthetic pathway via the (S)-bisabolyl cation as can be deduced from the absolute configuration of 31. In addition, the cyclic main compounds 1–9 coincide in a spirocentre. A plausible biosynthetic route to these sesquiterpenes is presented in Scheme 1 (Scheme 1 of ESI† shows a full scheme to all sesquiterpenes). The pathway includes the initial isomerisation of (E,E)-farnesyl diphosphate (39) to (E)-nerolidyl diphosphate (41) by diphosphate abstraction and reattack to the allylic cation 40, whereas capture by water results in 10. Reionisation of NPP to 42, also a possible precursor for 10, and subsequent cyclisation yields the (S)-bisabolyl cation (43) followed by the (S)-homobisabolyl cation (44) via a 1,2-hydride shift. Cyclisations with different stereochemical courses can in theory result in four diastereomeric acorenyl cations (45) all with (10S) configuration as constituted in 44, but only three of these cyclisations to 45a, 45b, and 45c happen to occur since no terpenes generated via45d could be identified in the Trichoderma extracts.
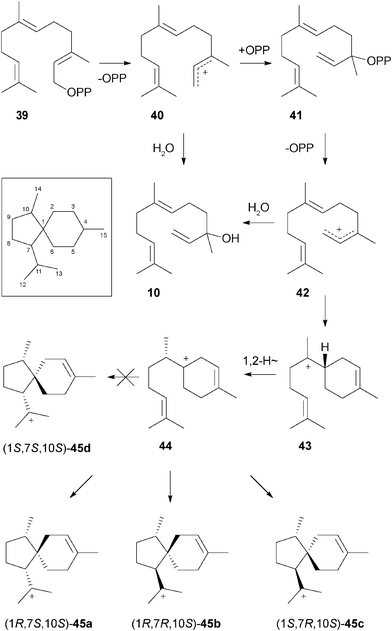 | ||
| Scheme 1 Formation of diastereomeric acorenyl cations from FPP. Carbon numbering of the acoranes is shown in the box. | ||
Terpenes arising through one of the other three cations 45a–c were instead found in large amounts. The cation 45a is the precursor for 1 and 2 and the sesquiterpene hydrocarbons 3–6. The compounds 5 and 6 require deprotonation of 45a (Scheme 2A). Two possible pathways exist for the formation of the allylic cation 47 as direct precursor of 1 by the attack of water, or 3 and 4 by loss of a proton, either via a 1,4- followed by a 1,2-hydride shift, or directly by 1,5-hydride migration, respectively (Scheme 2B). The 1,4-hydride shift would formally produce a secondary cation 46, but the two consecutive hydride shifts may be highly concerted. Such combinations of two or even more mechanistical steps including hydride shifts, proton transfers, rearrangements of the carbon skeleton, and deprotonations to avoid secondary cation intermediates have been suggested for several terpene biosynthetic pathways based on quantum chemical calculations.13–19 The alternative pathway via 1,5-hydride transfer as suggested previously for the biosynthesis of 1 in F. coccineum8 would omit the secondary cation without the requirement of concerted steps, but it seems questionable if this process is sterically possible. The oxidation of 1 affords 2.
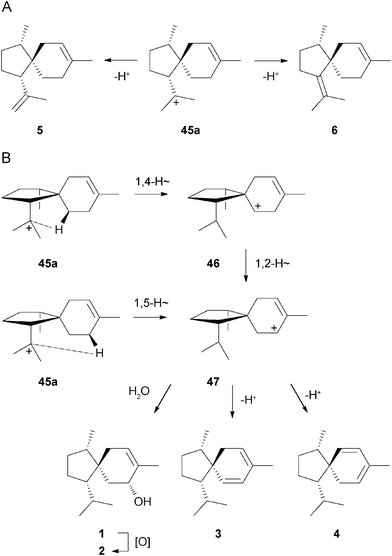 | ||
| Scheme 2 Biosynthesis of 1 and related sesquiterpenes. | ||
The alternatively formed cation 45b can undergo a third cyclisation to the cedryl cation (48) that generates (−)-7 by deprotonation (Scheme 3A). A similar cyclisation of 45c produces cation 49 as precursor for (−)-8 (Scheme 3B). Rearrangement of 49 by two sequential Wagner–Meerwein shifts via cations 50 and 51 followed by deprotonation results in 9. In summary, the biosynthesis of all these sesquiterpenes proceeds in the early steps via common intermediates, and then branches via structurally similar cations, suggesting that they are all produced by a single terpene cyclase.
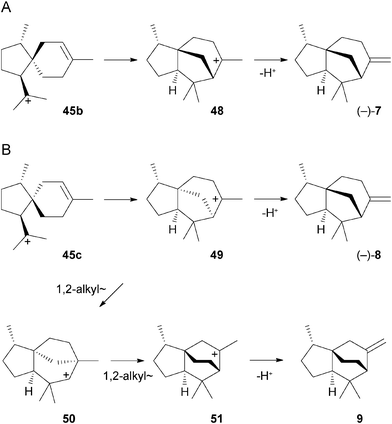 | ||
| Scheme 3 Biosynthesis of (–)-7, (–)-8, and 9. | ||
It is well known that some terpenoids can undergo reactions in the injection port of a GC due to its thermal instability and form analytical artifacts; e. g. germacrene A can undergo a thermal Cope rearrangement towards β-elemene.20 To rule out that some of the identified volatiles are not generated by the biosynthetic pathways described here, but are instead artifacts arising by such reactions in the GC injection port that was heated to 300 °C in our analyses, an authentic sample of the main compound 1 was analysed by GC-MS. Traces of 3 and 4 were found in the GC-MS analysis of an analytically pure (1H NMR spectroscopy) sample of 1 (Fig. 6 of ESI†). However, the total amount of 3 and 4 was below 0.2%, whereas the amount of these compounds in the natural extracts was much higher (compare Fig. 1) accounting for their formation mainly by biosynthesis rather than during the analytical procedure.
Feeding experiments with synthesised deuterated mevalonolactones10 were carried out to test the suggested biosynthetic pathways to 1. The incorporation of deuterium into 1 was followed by GC-EI-MS and GC-EI-HRMS measurements. The EI mass spectrum of natural 1 (Fig. 3A) is characterised by a molecular ion at m/z = 222 and diagnostic fragment ions at m/z = 151, 138, and 84. Plausible fragmentation pathways to these ions were developed (Scheme 4). The ion 56 (m/z = 88, C5H8O+) arises by retro-Diels–Alder fragmentation21via ionisation of the olefinic double bond to 52 and two successive cleavage reactions to 53 and 56, or, alternatively, by ionisation of the hydroxyl group to 54, α-cleavage to 55 and fragmentation to 56. The formation of the ions m/z = 138 and 151 is initiated by secondary cleavage reactions from the radical cation 55 that is converted into 57 by a ring-opening α-fragmentation. A 1,2-hydrogen shift to 58 followed by a third α-fragmentation results in the neutral loss of C6H12 to form 60 (m/z = 138, C9H14O+). The radical cation 57 can also undergo a 1,4-hydrogen shift to 59 that generates the fragment 61 (m/z = 151, C10H15O+) by α-cleavage under neutral loss of C5H11. These proposed fragmentation pathways were corroborated by the elemental compositions of the diagnostic fragment ions that were determined by GC-EI-HRMS (Table 1, entry 1). Using this method, each fragment was shown to contain the correct number of carbons, hydrogens, and oxygens, therefore establishing the proposed fragmentation mechanisms.
| Ions | EI-HRMS | Formula | calc.mass | Δm (mmu) |
|---|---|---|---|---|
| Molecular ion and diagnostic fragment ions ofa unlabelled 1,b labelled [2H12]-1 obtained after feeding of [4,4,6,6,6-2H5]MVA (62a),c labelled [2H15]-1 obtained after feeding of [2,2,6,6,6-2H5]MVA (62b),d labelled [2H15]-1 obtained after feeding of [5,5,6,6,6-2H5]MVA (62c),e labelled [2H9]-1 obtained after feeding of [6,6,6-2H3]MVA (62d). | ||||
| Entry 1 | ||||
| [1]+ | 222.19799 | C15H26O+ | 222.19781 | +0.18 |
| 61 | 151.11280 | C10H15O+ | 151.11174 | +1.06 |
| 60 | 138.10708 | C9H14O+ | 138.10391 | +3.16 |
| 56 | 84.05824 | C5H8O+ | 84.05696 | +1.28 |
| Entry 2 | ||||
| [[2H12]-1]+ | 234.27361 | C15H14D12O+ | 234.27312 | +0.49 |
| [2H7]-61 | 158.15678 | C10H8D7O+ | 158.15567 | +1.11 |
| [2H8]-60 | 146.15558 | C9H6D8O+ | 146.15413 | +1.45 |
| [2H4]-56 | 88.08409 | C5H4D4O+ | 88.08207 | +2.02 |
| Entry 3 | ||||
| [[2H15]-1]+ | 237.29519 | C15H11D15O+ | 237.29197 | +3.22 |
| [2H10]-61 | 161.17684 | C10H5D10O+ | 161.17451 | +2.33 |
| [2H8]-60 | 146.15789 | C9H6D8O+ | 146.15413 | +3.76 |
| [2H4]-56 | 88.08537 | C5H4D4O+ | 88.08207 | +3.30 |
| Entry 4 | ||||
| [[2H15]-1]+ | 237.28898 | C15H11D15O+ | 237.29197 | –2.99 |
| [2H9]-61 | 160.16819 | C10H6D9O+ | 160.16822 | –0.03 |
| [2H9]-60 | 147.15996 | C9H5D9O+ | 147.16040 | –0.44 |
| [2H5]-56 | 89.09086 | C5H3D5O+ | 89.08835 | +2.51 |
| Entry 5 | ||||
| [[2H9]-1]+ | 231.25511 | C15H17D9O+ | 231.25430 | +0.81 |
| [2H6]-61 | 157.15007 | C10H9D6O+ | 157.14940 | +0.67 |
| [2H6]-60 | 144.14069 | C9H8D6O+ | 144.14157 | –0.88 |
| [2H3]-56 | 87.07857 | C5H5D3O+ | 87.07580 | +2.77 |
![EI mass spectra and diagnostic ions determined by GC-EI-MS and GC-EI-HRMS of 1 (A), of [2H12]-1 after feeding of 62a (B), of [2H15]-1 after feeding of 62b (C), of [2H15]-1 after feeding of 62c (D), and of [2H9]-1 after feeding of 62d. Isotopomers of 1 obtained in feeding experiments and their fragment ions. Asterisks indicate completely deuterated carbons.](/image/article/2011/RA/c1ra00212k/c1ra00212k-f3.gif) | ||
| Fig. 3 EI mass spectra and diagnostic ions determined by GC-EI-MS and GC-EI-HRMS of 1 (A), of [2H12]-1 after feeding of 62a (B), of [2H15]-1 after feeding of 62b (C), of [2H15]-1 after feeding of 62c (D), and of [2H9]-1 after feeding of 62d. Isotopomers of 1 obtained in feeding experiments and their fragment ions. Asterisks indicate completely deuterated carbons. | ||
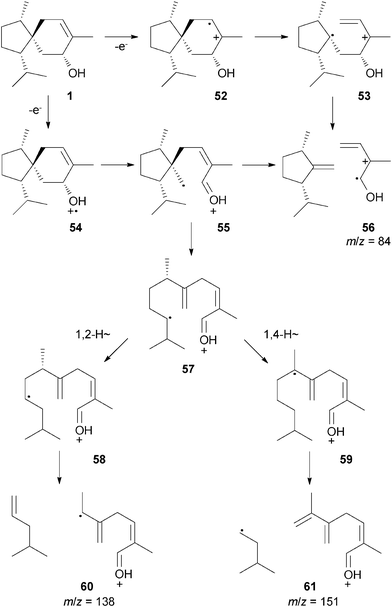 | ||
| Scheme 4 Proposed MS fragmentation pathways to diagnostic fragment ions of 1. | ||
A first feeding experiment using 62a was carried out to investigate the suggested biosynthesis of 1. The deuterium incorporation observed by GC-EI-MS showed a shift of the molecular ion to m/z = 234 indicating the uptake of twelve deuteriums from three mevalonolactone precursor molecules (Fig. 3B). The diagnostic fragment ions shifted to m/z = 158 (representing [2H7]-61), m/z = 146 ([2H8]-60), and m/z = 88 ([2H4]-56), and their exact masses determined by GC-EI-HRMS established the expected elemental compositions (Table 1, entry 2). This information was used to localise the deuterium incorporated into 1 and then the biosynthetic pathway was tested for its consistency with these EI-MS and EI-HRMS data. Following the biosynthesis of FPP from mevalonolactone via the well known mevalonate pathway (not shown) and the biosynthesis of 1 from FPP as outlined above, 62a is converted into [2H12]-1 in full agreement with the observed molecular ion. The biosynthetic pathway should further result in the occurrence of the labelling in [2H12]-1 in specific positions as indicated in Fig. 3B, and by taking the fragmentation mechanism into account the diagnostic fragment ions [2H4]-56, [2H8]-60, and [2H7]-61 were expected. This matches exactly the observations made by mass spectrometry and, conclusively, the suggested biosynthetic pathway is fully consistent with the experimental data. This is true, regardless of which pathway from 45a to 47, either a long-distance 1,5-hydride transfer or two sequential hydride migrations, is used. In particular, these data also demonstrate the 1,2-hydride shift from the bisabolyl to the homobisabolyl cation resulting in a labelled 10-position of 1. During the fragmentation process this deuterium is transferred and lost by a 1,4-hydrogen shift and subsequent α-cleavage, and is consequently missing in 61.
A second experiment was designed to investigate which alternative from 45a, either a 1,4- followed by a 1,2-hydride shift or a 1,5-hydride shift, is used in the downstream steps to 1. Feeding of 62b resulted in the incorporation of 15 deuteriums as indicated by a shift of the molecular ion to m/z = 237. The diagnostic fragment ions were observed at m/z = 161 ([2H10]-61), m/z = 146 ([2H8]-60), and m/z = 88 ([2H4]-56). Their elemental compositions were confirmed by EI-HRMS (Table 1, entry 3). Following the biosynthetic pathway including the sequence of 1,4- plus 1,2-hydride migration or the alternative pathway via a long-distance 1,5-hydride shift, in both cases the incorporation of 15 deuteriums to give [2H15]-1 from three units of mevalonolactone 62b would be expected, in agreement with the obtained mass spectrum. However, the two alternative pathways would result in a different distribution of the deuterium atoms in [2H15]-1, but only the isotopomer obtained via the sequence of a 1,4- plus a 1,2-hydride transfer can produce fragment ions that coincide with the observations, whereas the isotopomer obtained via a 1,5-hydride migration should result in the fragment ions [2H4]-56 (m/z = 88), [2H7]-60 (m/z = 145), and [2H9]-61 (m/z = 160).
Although these experiments are sufficient to distinguish between the possible pathways to 1 further feedings were conducted to test the robustness of the suggested MS fragmentations and the concluded biosynthetic pathway. Feeding of 62c and 62d resulted in [2H15]-1 and [2H9]-1, respectively, and their observed mass spectra (Fig. 3D and 3E) as well as the determined elemental compositions of the fragment ions (Table 1, entries 4 and 5) were in full agreement with the expectations following the established biosynthetic pathway to 1 including the two sequential hydride shifts.
The deduced biosynthetic pathway via the bisabolyl, homobisabolyl and acorenyl cations followed by two successive 1,4- and 1,2-hydride migrations and capture with water contradicts a published alternative that included a 1,5-hydride shift instead,8 but in this work no experimental proof was presented for this proposal. Although it cannot be ruled out that in the previously investigated species F. coccineum a 1,5-hydride transfer is operative, it was clearly demonstrated by our experimental approach that this is not true for Trichoderma.
Alternative pathways in terms of sequential hydride shifts versus long-distance hydride transfers have previously been discussed for the biosynthesis of other terpenes. For example, feeding experiments with deuterated isotopomers of FPP demonstrated that the biosynthesis of amorphadiene, the hydrocarbon precursor of artemisinin, involves a 1,3-hydride shift instead of two sequential 1,2-hydride migrations,22 whereas the results of quantum chemical calculations could not clearly distinguish between these alternatives.23 Most interestingly, in this theoretical work a 1,5-hydride migration was proposed for the biosynthesis of α-amorphene that branches from the amorphadiene pathway at a late intermediate, but this has not yet been experimentally confirmed. One of the rare examples of long-distance 1,5-hydride shifts was, however, experimentally established in the biosynthesis of a neo-epi-verrucosane diterpene from Fossombronia alaskana.24
Conclusions
In summary, the volatiles from three Trichoderma strains have been analysed. A large group of sesquiterpenes was found with tricho-acorenol (1) as the main compound. The absolute configurations of seven sesquiterpenes were elucidated by chiral GC and, together with their general structural resemblance, point to a common biosynthesis via the (S)-bisabolyl cation. The published8 biosynthesis of 1 was reinvestigated in feeding experiments with deuterated mevalonolactones and proceeds via the acorenyl cation 45a, followed by a 1,4- and a subsequent 1,2-hydride shift and not via a direct 1,5-hydride transfer, followed by nucleophilic capture with water. Our results obtained for Trichoderma disagree with the suggested pathway for F. coccineum that included a 1,5-hydride migration. The structural relationship of all sesquiterpenes produced by Trichoderma suggests the action of only a single terpene cyclase and allows for a prediction of the absolute configurations of the sesquiterpenes that have not been investigated under this aspect. Future experiments in our laboratory will include the identification of the terpene cyclase for the biosynthesis of 1 in Trichoderma.Experimental section
Strains, growth conditions, and feeding experiments
All three strains of Trichoderma viride 54, Trichoderma harzianum 714, and Trichoderma longibrachiatum 594 were obtained from Gabriele König (University of Bonn, Germany). T. viride was cultivated at 28 °C in BM liquid medium (20 g malt extract per L of deionised water), and T. harzianum and T. longibrachiatum were cultivated at 28 °C in BM-ASW liquid medium (20 g malt extract per L of artificial sea water: 23.5 g NaCl, 10.6 g MgCl2·6H2O, 3.92 g Na2SO4, 1.47 g CaCl2·2H2O, 0.66 g KCl, 0.19 g NaHCO3, 0.10 g KBr, 0.04 g SrCl2·6H2O, 0.03 g H3BO3, 1 L of deionised water) for 10–14 d, then inoculated on agar plates of BM or BM-ASW medium, respectively, amended with 10 mM of the deuterated mevalonolactones ([4,4,6,6,6-2H5]MVA (62a), [2,2,6,6,6-2H5]MVA (62b), [5,5,6,6,6-2H5]MVA (62c), or [6,6,6-2H3]MVA (62d), respectively). The agar plates were incubated for 1 d at 28 °C and then analysed by closed loop stripping analysis (CLSA).Collection of volatiles
The volatile products emitted by the agar plate cultures were collected by use of a closed-loop stripping apparatus (CLSA). In a closed apparatus containing the agar plate a circulating air stream was passed through a charcoal filter (Chromtech GmbH, Idstein, Precision Char Coal Filter 5 mg) for 18–24 h. The absorbed volatiles were eluted with analytically pure dichloromethane (40-50μl) and the extract was immediately analysed by GC-MS and stored at −80 °C.Extraction of plant materials
A sample of Microbiota decussata was obtained in May 2010 from Thomas Stützel (University of Bochum, Germany, botanical gardens). The plant material was cut into small pieces and crushed with a blender. An amount of 200 g was subjected to hydrodistillation, and the distillate was extracted with analytically pure ethyl acetate. The organic portion was dried and concentrated to yield the pale yellow essential oil (1.2 g). Samples of Mannia fragrans were obtained from Herrmann Muhle (University of Ulm, Germany). The plant material was collected in June 2010 near Kelheim (Germany). The leaves were carefully separated with a pair of tweezers, washed, and dried. The dried leaves (0.8 g) were crushed with a mortar and extracted by hydrodistillation. Extraction of the distillate with analytically pure ethyl acetate, drying of the organic layer with MgSO4, and evaporation of the solvent yielded the colourless essential oil (20 mg).GC-MS
GC-MS analyses were carried out on an Agilent 7890A connected with an Agilent 5975C inert mass detector fitted with a HP-5 fused silica capillary column (30 m, 0.25 mm i. d., 0.25 μm film, Agilent). GC conditions were as follows: inlet pressure 77.1 kPa, He 23.3 mL min−1, injection volume 1.5 μL, transfer line 300 °C, electron energy 70 eV. The operation mode was splitless (60 s valve time) and the carrier gas was He at 1.2 mL min−1. The GC was programmed as follows: 5 min at 50 °C increasing with 5 °C min−1 to 320 °C. Chiral GC analyses were performed using a Hydrodex-6-TBDMS fused silica capillary column (50 m, 0.25 mm i. d., 0.25 μm film, Macherey-Nagel). The GC was programmed as follows: 10 min at 50 °C increasing with 1 °C min−1 to 100 °C and then with 10 °C min−1 to 220 °C.GC-HRMS
GC-HRMS measurements were carried out using an Agilent 6890 gas chromatograph equipped with a ZB5-MS fused silica capillary column (30 m, 0.25 mm i. d., 0.25 μm film, Phenomenex). A split injection port at 250 °C was used for sample introduction and the split ratio was set to 10:1. The GC was programmed as follows: 3 min at 70 °C increasing with 10 °C min−1 to 310 °C. The helium carrier gas was set to a flow rate of 1.0 ml min−1 (constant flow mode). The transfer line was kept at 270 °C. A JMS-T100GC (GCAccuTOF, JEOL, Japan) time of flight mass spectrometer in electron ionization (EI) mode at 70 eV and JEOL MassCenter workstation software was used. The source and transfer line temperature were set at 200 °C and 270 °C respectively. The detector voltage was set at 2000 V. The acquisition range was from m/z 41 to 700 with a recoding interval of 0.4 s. The system was tuned with PFK to achieve a resolution of 5.000 (FWHM) at m/z 292.9824.
Acknowledgements
Funding by the Deutsche Forschungsgemeinschaft (DFG) by an Emmy Noether scholarship (to JSD) and the Fonds der Chemischen Industrie with a stipend (to NLB) is gratefully acknowledged. We thank T. Stützel (University of Bochum) and Herrmann Muhle (University of Ulm) for the plant materials and G. König (University of Bonn) for the Trichoderma strains. We thank Stefan Schulz (TU Braunschweig) for generous support.References
- D. W. Christianson, Chem. Rev., 2006, 106, 3412 CrossRef CAS.
- (a) C. A. Lesburg, G. Zhai, D. E. Cane and D. W. Christianson, Science, 1997, 277, 1820 CrossRef CAS; (b) M. Seemann, G. Zhai, J.-W. de Kraker, C. M. Paschall, D. W. Christianson and D. E. Cane, J. Am. Chem. Soc., 2002, 124, 7681 CrossRef CAS.
- (a) J. M. Caruthers, I. Kang, M. J. Rynkiewicz, D. E. Cane and D. W. Christianson, J. Biol. Chem., 2000, 275, 25533 CrossRef CAS; (b) B. Felicetti and D. E. Cane, J. Am. Chem. Soc., 2004, 126, 7212 CrossRef CAS.
- G. E. Harman, C. R. Howell, A. Viterbo, I. Chet and M. Lorito, Nat. Rev. Microbiol., 2004, 2, 43 CrossRef CAS.
- J. L. Reino, R. F. Guerrero, R. Hernandéz-Galán and I. G. Collado, Phytochem. Rev., 2008, 7, 89 CrossRef CAS.
- N. Stoppacher, B. Kluger, S. Zeilinger, R. Krska and R. Schuhmacher, J. Microbiol. Methods, 2010, 81, 187 CrossRef CAS.
- Q. Huang, Y. Tezuka, Y. Hatanaka, T. Kikuchi, A. Nishi and K. Tubaki, Chem. Pharm. Bull., 1995, 43, 1035 CAS.
- S. E. Gotfredsen, Dissertation ETH Zürich, 1978 Search PubMed.
- D. E. Cane, Tetrahedron, 1980, 36, 1109 CrossRef CAS.
- J. S. Dickschat, C. A. Citron, N. L. Brock, R. Riclea and H. Kuhz, Eur. J. Org. Chem., 2011, 3339 CrossRef CAS.
- J. S. Dickschat, S. C. Wenzel, H. B. Bode, R. Müller and S. Schulz, ChemBioChem, 2004, 5, 778 CrossRef CAS.
- Y. Saritas, M. M. Sonwa, H. Iznaguen, W. A. König, H. Muhle and R. Mues, Phytochemistry, 2001, 57, 443 CrossRef CAS.
- Y. J. Hong and D. J. Tantillo, J. Am. Chem. Soc., 2010, 132, 5375 CrossRef CAS.
- B. A. Hess, J. Am. Chem. Soc., 2002, 124, 10286 CrossRef CAS.
- Y. J. Hong and D. J. Tantillo, J. Org. Chem., 2008, 73, 6570 CrossRef.
- Y. J. Hong and D. J. Tantillo, J. Am. Chem. Soc., 2009, 131, 7999 CrossRef CAS.
- Y. J. Hong and D. J. Tantillo, Org. Lett., 2006, 8, 4601 CrossRef CAS.
- S. C. Wang and D. J. Tantillo, Org. Lett., 2008, 10, 4827 CrossRef CAS.
- S. P. T. Matsuda, W. K. Wilson and Q. Xiong, Org. Biomol. Chem., 2006, 4, 530 CAS.
- P. J. Teisseire, Chemistry of fragrant substances, VCH Publishers Inc, New York, 1994. Search PubMed.
- F. Turecek and V. Hanus, Mass Spectrom. Rev., 1984, 3, 85 CrossRef CAS.
- S.-H. Kim, K. Heo, Y.-J. Chang, S.-H. Park, S.-K. Ree and S.-U. Kim, J. Nat. Prod., 2006, 69, 758 CrossRef CAS.
- Y. J. Hong and D. J. Tantillo, Chem. Sci., 2010, 1, 609 RSC.
- W. Eisenreich, C. Rieder, C. Grammes, G. Heßler, K.-P. Adam, H. Becker, D. Arigoni and A. Bacher, J. Biol. Chem., 1999, 274, 36312 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Full results of the analyses of the Trichoderma headspace extracts, total ion chromatograms of chiral GC analyses for the determination of the absolute configurations of several terpenoid volatiles, biosynthetic scheme towards all identified sesquiterpenoids from Trichoderma, HR-MS data of unlabelled and deuterated 1 obtained in feeding experiments, and experimental details. See DOI: 10.1039/c1ra00212k. |
| This journal is © The Royal Society of Chemistry 2011 |