Synergistic interactions during thermosensitive chitosan-β-glycerophosphate hydrogel formation
Xueying
Qiu
a,
Yuhong
Yang
*b,
Liping
Wang
c,
Shanling
Lu
a,
Zhengzhong
Shao
a and
Xin
Chen
*a
aKey Laboratory of Molecular Engineering of Polymers of Ministry of Education, Department of Macromolecular Science, Laboratory of Advanced Materials, Fudan University, 220 Handan Road, Shanghai, China. E-mail: chenx@fudan.edu.cn
bResearch Center for Analysis and Measurement, Fudan University, 220 Handan Road, Shanghai, China. E-mail: yuhongyang@fudan.edu.cn
cDepartment of Chemistry, Fudan University, 220 Handan Road, Shanghai, China
First published on 2nd August 2011
Abstract
Time-evolved gelation behavior of a chitosan-β-glycerophosphate (CS/β-GP) system was elucidated from rheological investigation, NMR analysis, fluorescence measurement, and morphology observation. Urea and isobutanol were selected to assess the interactions between the two components during the gelation process of the CS/β-GP system. Urea was found to be detrimental to the gelation process by both disrupting hydrogen bonding and retarding the formation of hydrophobic domains. On the contrary, the addition of isobutanol accelerated the sol–gel transition by strengthening the hydrophobic interactions. These results reveal that both hydrogen bonding and hydrophobic interactions within chitosan or between chitosan and β-glycerophosphate molecules are the main reasons for gel formation. The results also indicate that firstly the formation of hydrogen bonds makes the hydrophobic sites more accessible, and then the synergistic hydrogen bonding and hydrophobic interactions lead to the final formation of CS/β-GP gel network. This work demonstrates the possibility of tuning the gelling ability as well as the mechanical properties of CS/β-GP hydrogels. Thus, it gives the opportunities to optimize the performance of this promising natural thermosensitive material for practical applications in the future.
Introduction
Injectable hydrogels have attracted much attention during the past decade, due to their rapid gelation from flowable aqueous solution when injected into the desired tissue or organ, thus giving rise to minimal surgical wounds as they are implanted in human body.1,2 As a specific injectable biomaterial, thermosensitive hydrogel that undergoes a sol–gel transition at body temperature has provoked great interest in recent years because it has many potential applications, such as drug delivery, tissue engineering and cell encapsulation, etc.3 Many polymeric systems are free-flowing sols at high temperature and become gels when the temperature decreases by normal thermosensitive sol–gel transitions, for example, like gelatin and some polysaccharides.4,5 However, reverse thermosensitive materials, which show spontaneous physical gelation during heating,6,7 may be more suitable for biomedical application by protecting the encapsulated proteins and cells before injection.As a biocompatible and biodegradable cationic amino-polysaccharide, chitosan (CS), prepared by alkaline deacetylation of chitin, has been extensively studied in medical, pharmaceutical, nutraceutical and cosmetic fields, etc.8 CS is typically soluble in acidic aqueous media with the charged amino groups interacting with water. However, it experiences phase separation when pH > 6 to form a gel-like precipitate, which limits its applications. In 2000, Cheniteet al.3 found that the addition of β-glycerophosphate (β-GP) could increase the pH value of CS solution to neutral without phase separation. Such a CS/β-GP system remained in solution state at room temperature but changed into a gel when heated to physiological temperature (37 °C). This finding is a breakthrough for injectable hydrogels, which implies such a natural polymer-based reverse thermosensitive gelling system has the great potential for applications in the pharmaceutical field9–12 and tissue engineering.13–17
Whilst the application of the CS/β-GP system has been extensively studied, the understanding of its gelation mechanism is still controversial.3,18–23 Several interactions were proposed for the contribution to the gelation process of CS/β-GP by investigating its morphology, physicochemical and rheological properties, etc. The hydrophobic or water-structuring character of the glycerol moiety of β-GP was thought to be the main source of the heat-induced increase in hydrophobic attraction between CS molecules from the investigations on the effect of temperature3,18,19 and urea21 during the gelation process. The effect of urea on the gelation was surmised through the measurements of physicochemical (pH and conductivity) and rheological properties of CS/β-GP solution, but only the influences of gelation time and gelation temperature were discussed. The structural changes with time evolution during the gelation upon the addition of urea have not been involved. The heat-induced transfer of protons from CS to β-GP was also suggested to result in the formation of gel by examining the polyelectrolyte properties of CS.22 In addition, the morphology observation of CS/β-GP hydrogel on micro-scale by laser scanning confocal microscopy (LSCM) showed a heterogeneous microstructure, so it was suggested that the gelation kinetics may be nucleation and growth.20,23 However, all the gelation mechanisms shown above were proposed from indirect evidence, the direct detection of the interactions corresponding to the gel formation is still not available yet.
NMR spectroscopy is widely used to explore intra- and inter-molecular interactions at atomic level.24–26 Variable-temperature 31P NMR techniques have been used to monitor the sol–gel transition of CS/β-GP system upon heating and tried to provide evidence for the speculation that heat-induced proton transfer from CS to β-GP occurs during the gelation process,27 in which the long-range electrostatic repulsion was considered as the main interaction of this system. However, the hydrogen bonding interactions that should exist in such a system were not mentioned at all in that reference. In our previous study,28 both 1H and 31P NMR analysis were used to monitor the gelation process of CS/β-GP system. The results showed that the chemical shifts of protons in CS and phosphorus in GP changed in the gelation process, indicating that the electrostatic interaction between CS and GP was broken down with the increase of temperature, followed by the formation of hydrogen bonds between CS macromolecular chains. In this article, in addition to the more extensive study with 31P NMR spectroscopy, rheological measurement was used to evaluate the change of micro-structure and molecular interactions during the gelation process of CS/β-GP system. We chose urea and isobutanol to investigate the role of hydrogen bonding and hydrophobic effect in CS/β-GP gelation process as urea is generally known as a hydrogen bonding disrupting agent29–31 and the hydrophobic domain of CS is considered to be increased by the addition of isobutanol.32,33
Experimental
Materials
CS (molecular weight = 40 kDa, deacetylation degree = 95%) in a powder form, was purchased from Jinan Haidebei Marine Biological Product Co. Ltd. (China). β-GP (C3H7Na2O6P·5H2O) was purchased from Aladdin Reagent Co. Ltd. (China). Acetic acid (Shanghai Chemical Reagent Co. Ltd, China), urea (Yixing Chemical Reagent Co. Ltd, China), and isobutanol (Shanghai Runjie Chemical Reagent Co. Ltd, China) were used without further purification.Sample preparation
CS powder was dissolved in 1.0 wt% aqueous acetic acid solution under magnetic stirring for 5 h at room temperature to make a 3.0 wt% CS solution. Thereafter, a given amount of β-GP and urea (or isobutanol) were dissolved in de-ionized water and then added to the CS solution in succession, drop by drop and under magnetic stirring in an ice bath, to provide a clear and homogeneous liquid solution with a pH value of 7.0 ± 0.1. The final samples had CS with the concentration of 1.0 wt%, 7.0 wt% β-GP and urea (or isobutanol) with different concentrations. The samples were incubated at 4 °C and subjected to characterization.Rheological measurements
Rheological measurements were performed in strain-controlled mode using a Physica MCR 301 rheometer (Anton Paar GmbH, Austria) with a torque transducer sensitivity of 0.1 μNm. Sample temperature and atmosphere were controlled by a Peltier temperature control device. A cone-and-plate geometry, with a cone angle of 1° and a diameter of 60 mm, was employed in all measurements. To minimize dehydration, a solvent trap (AT-CYL-C/Q1) was employed. In the oscillatory frequency sweep experiments, the values of the strain amplitude (5% in this work) were checked to ensure that all measurements were carried out within the linear viscoelastic regime, where the dynamic storage modulus (G′) and loss modulus (G′′) were independent of the strain amplitude. The frequency sweeps were carried out over a broad angular frequency (ω) interval.During the gelation process, the dynamic properties were monitored by time sweeps in isothermal conditions at 34 °C. A low oscillation frequency (1 Hz) and a small deformation (5%) were applied. The gelation time (tgel) was determined as the crossover point of the storage (G′) and loss (G′′) moduli (tanδ = 1). To determine gelation temperature, the non-isothermal tests were performed at 1 Hz with a small deformation (5%) as for isothermal tests, while the temperature was increased at the rate of 1 °C min−1 between 25 and 55 °C. The gelation temperature (Tgel) was also determined as the crossover point of G′ and G′′.
31P NMR analysis
Time-dependent 31P NMR spectra were obtained from a Bruker DMX 500 spectrometer using a 5 mm QMP probe. H3PO4 (85%) was used as an external reference. The one-pulse experiments were performed with the 90° pulse length of 5 μs. The delay before the application of pulse was 6.15 s and the acquisition time was 1.01 s. The spectral width was 80 ppm with the number of data points 32 k. The number of acquired transients was 16. 31P spectra of CS/β-GP solutions in the presence of urea (or isobutanol) were acquired in situ at the interval of 2 min at 34 °C. The magnetic field was locked with D2O. Sample temperature was controlled by a Bruker BVT3000 temperature control device.Fluorescence measurements
Fluorescence measurement was detected at 34 °C using a FLS-920 fluorescence spectrophotometer (Edinburgh Instruments, U.K.) with a 1.0 cm path length quartz cell. The kinetics of the formation of hydrophobic domain during gelation of CS/β-GP in the presence of urea (or isobutanol) was monitored by anilinonaphthalene-8-sulfonic acid ammonium salt (ANS) binding. The concentration of ANS was 200 μM. The samples were excited at 390 nm, and the fluorescence emission spectra were recorded between 400 and 700 nm. Stock solutions of ANS were prepared in deionized water and diluted to the final concentration.Atomic force microscopy (AFM) observations
CS/β-GP solution was incubated at 34 °C and diluted 2,000 times with de-ionized water. 20 μL of the diluted solution was dipped onto freshly cleaved mica attached to a magnetic steel disc (serving as sample holder). The samples were allowed to dry in air overnight at room temperature. Imaging was recorded on a Nanoscope IV Digital Instruments atomic force microscope (Veeco Metrology Group, USA) in a tapping mode, using a Si3N4 cantilever with a spring constant of 50 N m−1 and a resonance frequency of 340 kHz.Dynamic light scattering (DLS) measurements
The CS/β-GP solution was diluted 100 fold with de-ionized water, and then the size of the clusters was measured immediately by DLS using a Zeta-sizer Nano (Malvern Instruments Ltd., Malvern, U.K.) at 34 °C. Laser light at 632.8 nm was used to measure the fluctuation in intensity of light scattered by clusters. Data were collected for 1 min for each sample.Results and discussion
Rheological measurements
To investigate the possible mechanism of CS/β-GP gelation, dynamic rheological measurements were employed to monitor the change of viscoelastic properties during the hydrogel formation process at different conditions. Frequency sweeps for the CS/β-GP system over a period of 0 to 180 min at 34 °C were performed. Fig. 1 shows the shear storage modulus (G′) and loss modulus (G′′) as a function of angular frequency of CS/β-GP solution at fixed interval (the curves of CS/β-GP/urea and CS/β-GP/isobutanol solutions are not shown here because they are similar to that of CS/β-GP solution). Both G′ and G′′ decrease with the decreases of frequency and show a crossover point in the first 60 min. Then from 90 to 180 min, G′ becomes larger than G′′ at lower frequency and presents a plateau-like behavior (which is almost independent of frequency), indicating the formation of a solid but tenuous network.34 Interestingly, the appearance of the frequency-independent plateau in G′ at low frequencies is common in suspensions of colloidal particles,34–36 implying the existence of particles or clusters in these CS/β-GP solutions regardless of the presence of urea (or isobutanol) or not. Such frequency-independent plateaus enlarge significantly with the increase of the time. In addition, the height of the plateau in G′ also increases with the time, corresponding to the growth of the network with stronger association among molecular chains due to the increase of inter-molecular interactions. From these rheological results (G′ is almost independent of frequency and larger than G′′ within the entire frequency range in our experiments), we may conclude that CS/β-GP forms hydrogel after 60 min.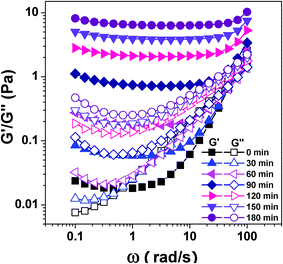 | ||
| Fig. 1 Storage modulus G′ and loss modulus G′′ as a function of angular frequency ω for CS/β-GP solution at different times at 34 °C (cCS = 1 wt%, cβ-GP = 7 wt%). | ||
The effects of urea and isobutanol on the evolution of the plateau moduli in G′ for CS/β-GP systems, as captured by the frequency sweeps during the gelation process, are shown in Fig. 2. For pristine CS/β-GP sample, G′ increases rapidly with time during the first 90 min, and then followed by a relatively slow increase when the gelation happens. Different from pristine CS/β-GP, the sample with urea shows three regions: a slow increase of G′ in the first 75 min; then a fast increase of G′ during the gelation process; and finally a smooth growth of G′ after 120 min, indicating the retarding effect of urea on the early stage of the sol–gel transition. The effect of isobutanol on the gelation process is contrary to that of urea. Isobutanol accelerates the gelation process at initial stage, leading G′ to a sharp increase in the first 15 min, and then to a slow growth region. Moreover, the final storage modulus of CS/β-GP/isobutanol hydrogel is less than CS/β-GP hydrogel, but is much higher than CS/β-GP/urea hydrogel (Fig. 3). The dissimilar behavior of G′ evolution with time and the difference of final strength of the hydrogels suggest that urea and isobutanol have a significant influence on the gelation behavior of CS/β-GP system. We should point out here that the difference of G′ in Fig. 2 and Fig. 3 results from the incubative discrepancy of the hydrogels. The G′ shown in Fig. 3 was measured after the hydrogel was fully formed (in total 48 h of incubation), however, G′ shown in Fig. 2 was monitored under the shearing force from rheometer, which caused the breakage of hydrogel network and the significant decrease of G′ values.
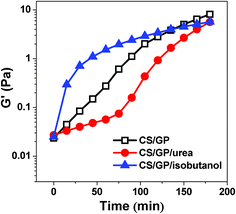 | ||
| Fig. 2 Plateau modulus of G′ as a function of time at 34 °C for CS/β-GP, CS/β-GP/urea and CS/β-GP/isobutanol solutions, respectively (cCS = 1 wt%, cβ-GP = 7 wt%, curea = 5 wt% and cisobutanol = 5 wt%). | ||
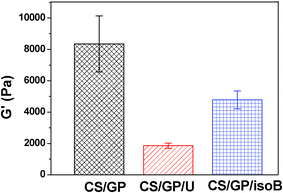 | ||
| Fig. 3 Final storage modulus G′ for CS/β-GP, CS/β-GP/U (urea), and CS/β-GP/isoB (isobutanol) hydrogels after incubated at 34 °C for 48 h (cCS = 1 wt%, cβ-GP = 7 wt%, curea = 5 wt%, and cisobutanol = 5 wt%). | ||
The gelation time and temperature were also measured in the presence of urea and isobutanol as plotted in Fig. 4. The gelation time of the CS/β-GP increases from 38 min to 85 min when the urea concentration increases from 0 to 7 wt%. On the contrary, higher isobutanol concentration shortens the gelation time. It is from 38 min to 15 min as the isobutanol concentration increases from 0 to 5 wt%. This indicates that urea slows down the gelation process, while isobutanol accelerates it, which is consistent with the results of frequency sweep experiments shown above. As expected, the gelation temperature of the CS/β-GP system increases from 42 to 48 °C with the increase of urea concentration from 0 to 7 wt%, but drops from 42 to 38 °C with the increase of isobutanol concentration from 0 to 5 wt%.
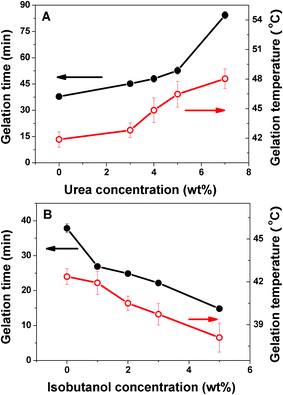 | ||
| Fig. 4 Gelation time and temperature as a function of urea (A) and isobutanol (B) concentration in the CS/β-GP system (cCS = 1 wt%, cβ-GP = 7 wt%). | ||
31P NMR analysis
To further understand the gelation mechanism of the CS/β-GP system, 31P NMR measurements were performed. 31P NMR provides the possibility to investigate the interactions related to the gelation by using β-GP as a powerful probe. It is well-known that chemical shift is sensitive to the chemical nature of the related nucleus. The increase of magnetic shielding of a nucleus, which is determined mainly by the shell of electrons, induces an upfield shift.37 As shown in Fig. 5, the significant downfield shift is experienced by the 31P nuclei of β-GP in β-GP/isobutanol solution (line c) compared with the signal in pure β-GP solution (line a). This reflects the existence of interactions between β-GP and isobutanol that causes the low shielding values of 31P nuclei. However, the chemical shift of 31P nuclei of β-GP in β-GP/urea solution (line b) is almost the same as that in pure β-GP solution (line a), suggesting that the local environment of the β-GP molecules is nearly unchanged. In the pristine CS/β-GP solution (line d), the chemical shift shows an upfield shift of more than 0.5 ppm, and the line width of the signal also increases. This observation demonstrates the decrease of the mobility of the phosphate group caused by the interaction between CS and β-GP. After 1 h incubation of CS/β-GP solution, the chemical shifts of 31P nuclei is found to move towards further upfield side (line g), indicating the increase of the interactions between CS and β-GP. Moreover, the signal becomes even broader than that in the original CS/β-GP solution (line d) due to the further restriction of the motion of 31P nuclei, which reflects the formation of hydrogel.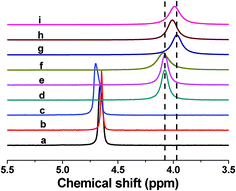 | ||
| Fig. 5 31P NMR spectra of pristine β-GP (a), β-GP/urea (b), β-GP/isobutanol (c), original CS/β-GP (d), original CS/β-GP/urea (e), original CS/β-GP/isobutanol (f), and CS/β-GP (g), CS/β-GP/urea (h), CS/β-GP/isobutanol (i) after incubation for 1 h at 34 °C. | ||
The final state of chemical shifts of those NMR spectra after 1 h incubation (line g, h, and i) show the similar upfield change, but the real-time chemical shift changes of the 31P nuclei of β-GP in CS/β-GP, CS/β-GP/urea and CS/β-GP/isobutanol systems during the gelation process are different (Fig. 6). The chemical shifts of 31P nuclei decrease immediately after CS is mixed with β-GP, probably due to the intermolecular interactions between CS and β-GP. It's interesting to find that in the CS/β-GP/urea system, the value of the chemical shifts of 31P nuclei remains the same in the initial stage (about 8 min), and then decreases with time. The chemical shift of 31P nuclei in CS/β-GP/isobutanol shows another trend, i.e., increases slightly in the first 6 min and then decreases sharply. The chemical shifts of all three systems approach a constant value on the timescale of the experiment, indicating they all form the hydrogel. However, the different behavior of chemical shift at the earlier stage implies that urea and isobutanol have diverse effects on the interactions which are responsible for the gelation of CS/β-GP, as with the rheological results shown above.
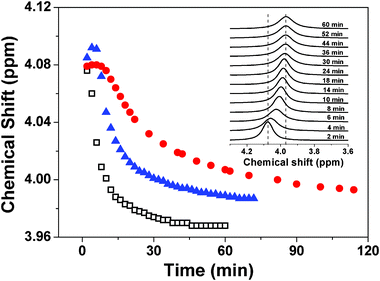 | ||
| Fig. 6 Time-dependent chemical shift of 31P nuclei at 34 °C of CS/β-GP (□), CS/β-GP/urea (●) and CS/β-GP/isobutanol (▲), respectively. The insert figure shows the real-time 31P NMR spectra of CS/β-GP at 34 °C (cCS = 1 wt%, cβ-GP = 7 wt%, curea = 5 wt% and cisobutanol = 5 wt%). | ||
Fluorescence analysis
The environmental sensitivity of ANS fluorescence makes it an excellent subsidiary tool to probe the change of hydrophobicity in various systems, such as protein aggregation38–40 and gelation kinetics.41–43 Fluorescence of ANS is largely quenched in aqueous media due to the dynamic collisional quenching, but increases when trapped in hydrophobic cavities/clefts. In the meantime, a shift of the emission maximum also supports the change in the local environment of ANS. Thus, if it shows both the increase in the intensity and the blue shift of the emission maximum of ANS fluorescence in the course of gelation, it implies the existence of new accessible hydrophobic sites to the probe. Although the microviscosity of the system surrounding the probe may change with gelation and has some effects on fluorescence properties, it is mainly reflected by fluorescence lifetime from the charge transfer state of ANS rather than its intensity.42The fluorescence spectrum of pure ANS exhibits a wide band with low fluorescence emission intensity and an emission maximum of 535 nm in polar environments (Fig. 7). After binding to the hydrophobic region of CS, the fluorescence intensity of ANS increases and its emission maximum shows a blue shift to 505 nm. The addition of β-GP to CS solution results in an increase in ANS fluorescence intensity which indicates that the ANS molecules are in the more hydrophobic environment. As shown in Fig. 7, the fluorescence intensity of ANS increases significantly during the gelation process of CS/β-GP, accompanied by a blue shift of the emission maximum from 505 to 493 nm, indicating that the hydrophobic interactions are strengthened. The emission maxima of CS/β-GP/urea and CS/β-GP/isobutanol also show a similar blue shift like that of CS/β-GP, implying a similar enhancement of the hydrophobic interactions with gelation.
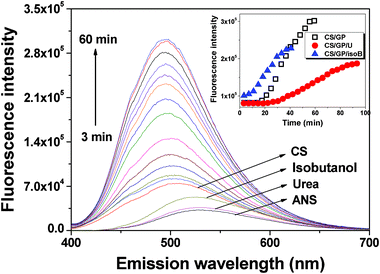 | ||
| Fig. 7 Change in the ANS fluorescence intensity of CS/β-GP with time at 34 °C. The spectra of pristine ANS as well as ANS with pure CS, isobutanol, and urea are pointed separately. Samples were excited at 390 nm. The inserted illustration shows the fluorescence intensity at λmax as a function of time at 34 °C of CS/β-GP, CS/β-GP/U (urea) and CS/β-GP/isoB (isobutanol) solutions, respectively (cCS = 1 wt%, cβ-GP = 7 wt%, curea = 5 wt% and cisobutanol = 5 wt%). | ||
The inset illustration in Fig. 7 shows the fluorescence intensity changes of ANS at the maximum emission wavelength after binding to CS in the gelation process of CS/β-GP, CS/β-GP/urea and CS/β-GP/isobutanol. It should be noted that the fluorescence intensity decreased because of the decline in light transmittance in the final stage when an opaque gel was formed, so we did not show those data here. For CS/β-GP, the fluorescence intensity remains constant in the first 14 min and then increases abruptly with time. Such a plateau extends to about 27 min with the addition of urea, and followed by a slow increase of fluorescence intensity. This indicates that urea has no influence on the hydrophobic region of CS/β-GP in the initial stage but retards the growth of hydrophobic interaction during gelation. In contrast, the fluorescence intensity starts to increase from the very beginning of the measurement with the presence of isobutanol in CS/β-GP solution, and its initial fluorescence intensity is also higher than that of CS/β-GP and CS/β-GP/urea. This may be because the addition of isobutanol decreases of polarity of surrounding environment and thus increases the hydrophobic domains in CS/β-GP system.
Morphology observation
To determine the nature of CS/β-GP hydrogels, we monitored the aggregation state of CS/β-GP in aqueous solutions with AFM, which has been widely applied to obtain surface-dependent information in three dimensions on the nanometre scale.44AFM images show a granular nanostructure of CS/β-GP at the initial stage of gelation (Fig. 8A), revealing that the CS chains self-assembled into aggregates. The DLS results obtained in situ at 34 °C shows that initial size (hydrodynamic diameter) of aggregates is about 200 nm and does not change significantly in the first few minutes (Fig. 8D). Then, the further aggregation of the CS clusters during gelation gives rise to a dramatic increase of particle size, and levels off at about 40 min. The clusters are highly heterogeneous, but final size of these cluster aggregations is about 1–2 μm, which is quite consistent with the result from laser scanning confocal microscopy (LSCM) by Crompton, et al.23AFM observation also gives further information on the shape of those aggregates (Fig. 8B and 8C). That is, a large CS aggregate is formed by the connection of several relatively smaller clusters, which are partially fused from one to another. Aggregation of clusters is observed by both AFM and DLS during gelation, but there is discrepancy between results from different methods, mainly attributing to the difference of sample states (dried particles for AFM, but swollen aggregates for DLS).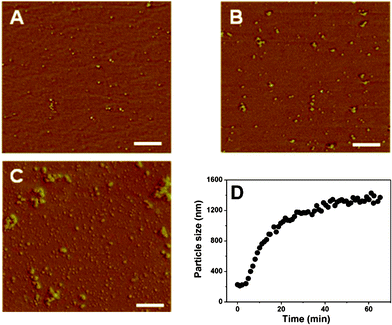 | ||
| Fig. 8 AFM images of the CS/β-GP samples with different incubation times: 0 min (A); 5 min (B); 10 min (C), and the particle size of CS/β-GP aggregates determined by DLS analysis as a function of time at 34 °C (D). All scale bars represent 500 nm (cCS = 1 wt%, cβ-GP = 7 wt%). | ||
Formation mechanism of CS/β-GP hydrogel
According to the investigation of viscoelasticity change of CS/β-GP during gelation, it is found that a solid but tenuous network with junctions is formed by inter-molecular interactions at the initial stage of gelation. The strength of such a network increases with the incubation time. The initial fast growth of G′ is originated from CS aggregates, but the increase of the viscosity of the whole system during the gelation process slows down the further aggregation of CS. Consequently, the increase of G′ slows down and then levels off at about 5 h. During this period, the microstructure of the gels is found to be composed of aggregated clusters that become larger with the time (Fig. 8).45 However, the addition of urea and isobutanol changes the evolution of G′ of CS/β-GP system (Fig. 2). Our work shows that the increase of G′ of CS/β-GP/urea system at the beginning is quite limited, indicating that urea has a retarding effect on the gelation process. The reason is that the urea hinders the formation of intermolecular hydrogen bonds, which is necessary to build the network.29–31 On the contrary, the addition of isobutanol leads to a prompt increase in G′ in the initial stage. It indicates that isobutanol, as a non-solvent for CS, accelerates phase separation by favoring the formation of hydrophobic domains between CS molecules32 and eventually manifests as gelation. This observation is similar to those previous studies on the effect of polyol on CS/β-GP and other related gelation systems.33,46 These results reveal that both hydrogen bonding and the hydrophobic effect are responsible for gel formation in the CS/β-GP system. In addition, the difference among final gel strength also confirms our assumption about the possible interactions existed during the formation of CS/β-GP hydrogel. Urea reduces the crosslinking degree of CS/β-GP hydrogel by disrupting intermolecular hydrogen bonds, so it results in a weak hydrogel (Fig. 3). In the meantime, it brings about the longer gelation time and the higher gelation temperature compared to a pure CS/β-GP system (Fig. 4A), because more energy is needed for the sol–gel transition.21 Since isobutanol strengthens the CS-CS hydrophobic interactions, it shows the shortest gelation time and the lowest gelation temperature (Fig. 4B). However, the restriction of molecular chain motion resulted from the rapid gelation at beginning causes less hydrogen bonding and hydrophobic interactions that serve as crosslinking point in CS/β-GP/isobutanol hydrogel. Thus, the final strength of CS/β-GP/isobutanol hydrogel is less than CS/β-GP hydrogel, but is still much higher than CS/β-GP/urea hydrogel (Fig. 3).31P NMR analysis provides further reliable evidence for the role of inter-molecular interactions in the CS/β-GP gelation process. Our work shows that the chemical shift of 31P in CS/β-GP solution appears upfield compared with that of pristine β-GP solution. Lavertu et al.27 simply attributed it to the change of ionization degree of β-GP with pH value. However, hydrogen bonding is also a reason for the change of chemical shift.47–49 The chemical shift of 31P moves to upfield in CS/β-GP solution, indicating that N–Hδ+⋯Oδ−–P hydrogen bonds may be formed between β-GP and the amino groups in CS. The interaction between CS and β-GP causes the steric repulsion to hinder further aggregation of CS molecules from precipitation. Meanwhile, this interaction may also restrict the mobility of β-GP molecules and eventually give rise to the broadening of line width of 31P NMR spectrum, which is consistent with our previous study with 1H NMR techniques.28
The decrease of 31P chemical shift with time during the gelation process means that more hydrogen bonds form between CS and β-GP. Urea is known as a hydrogen bond-breaking agent,29–31 so the constant chemical shift at the early stage of sol–gel transition process (within 8 min) with the addition of urea indicates an equilibrium between the formation and the destruction of hydrogen bonding. On the other hand, when in the presence of isobutanol, as oxygen has the stronger electro-negativity than nitrogen, the electron cloud density of oxygen adjacent to phosphorus in the case of O–Hδ+⋯Oδ−–P is lower than that of N–Hδ+⋯Oδ−–P, thus causes a downfield shift of 31P chemical shift. This explains the increase of chemical shift of 31P nuclei in the first few minutes (within 6 min) in CS/β-GP/isobutanol system, which attributes to the formation of hydrogen bonds between β-GP and the hydroxyl group of isobutanol.
Hydrophobic interaction is thought to be the main driving force for the gelation of CS/β-GP according to the literature,3,18,19,21 which is also confirmed by the accelerating effect of isobutanol in the current fluorescence study. The increase of fluorescence intensity of ANS with the time is another strong evidence for the gradual enhancement of hydrophobic interaction during the gelation process of CS/β-GP and eventually leads to the formation of the hydrogel.
Polymers with both hydrophobic and hydrophilic segments can self-assemble into distinct structures in aqueous solution, such as micelles, vesicles and tubules.40,50,51 It is largely due to the hydrophobic effect, which drives the non-polar region of each polymer molecule away from water and towards each other. The formation of submicrometric chain aggregates driven by hydrophobic interactions is evidenced in CS hydrogel system.51 Therefore, the formation of CS aggregates in the presence of β-GP indicates that the hydrophobic interactions among the CS molecules are strengthened by the addition of β-GP, leading to the self-assembling of CS. From the morphology studies, it is found that the aggregation of CS clusters takes place after the formation of the hydrogen bonds, but before the enhancement of hydrophobic interaction. According to this finding, we suppose that the initiatory hydrogen bond formation makes the hydrophobic action site become more accessible for intermolecular junction, thus eventually accelerates the formation of the gel network.
Finally, we summarize the results from various measurements we performed here to make it clearer for the formation mechanism of CS/β-GP hydrogel we proposed. From the fluorescence spectra, we find that fluorescence intensity is almost independent of the time at the earlier stage in all three systems, but the duration of the constant intensity plateau is quite different. The fluorescence intensity of ANS for the pure CS/β-GP system keeps constant for almost 14 min, but its 31P chemical shift changes immediately with the time. This further verifies our deduction that the formation of hydrogen bond facilitates the connection of hydrophobic domains. In the case of CS/β-GP/urea, due to the detrimental effect of urea on the inter-molecular hydrogen bonds, duration of the constant fluorescence intensity range is much longer than that of CS/β-GP, so hydrophobic interactions among CS molecules are retarded, which gives the reason for the slow increase of G′ at the initial stage (Fig. 2). In addition, urea damages the CS–CS hydrophobic interaction as well,21 so it can be another reason for the appearance of a longer constant fluorescence intensity plateau as well as the tardy increases of fluorescence intensity which followed. On the other hand, the presence of isobutanol in CS/β-GP decreases the polarity of the environment which favors the formation of hydrophobic domains in CS solution,32 causing the immediate increase of fluorescence intensity. The fast formation of hydrophobic interactions is obviously favorable to the gel formation, which accords very well with the sharp increase of G′ at the initial stage (Fig. 2). This is also the possible reason for the broader line width of CS/β-GP/isobutanol system in 31P NMR spectrum (Fig. 5, line f), which due to the restriction of molecular chain motions from the hydrophobic interaction between CS and isobutanol.
Conclusion
In this work, we proposed a formation mechanism for CS/β-GP hydrogel according to the results from a combination of measurements, i.e., the rheological investigation, NMR analysis, fluorescence measurement, and morphology observation. We found that both hydrogen bonding and hydrophobic interaction among CS molecules or between CS and β-GP molecules had a synergistic effect on the gelation behavior of CS/β-GP system. During the gelation process, hydrogen bonds are formed before the enhancement of hydrophobic interactions, making the hydrophobic site more accessible for the creation of more hydrophobic interactions to finally form a gel network. The effects of urea and isobutanol on the gelation behavior of CS/β-GP system were investigated in order to assess the driving intermolecular interactions during the sol–gel transition. The results show that the gelation process is retarded by the addition of urea because it disrupts the hydrogen bonding and hinders the hydrophobic interactions. On the contrary, the gelation process is accelerated by isobutanol through the strengthening of CS–CS hydrophobic interactions. Since the gelation mechanism is critical to the potential development and application of hydrogel materials, we believe the useful information present in this article enables the further optimization of this unique natural thermosensitive hydrogel for wide and practical applications.Acknowledgements
This work is supported by the National Natural Science Foundation of China (No. 20874018 and 20674011) and the Program for New Century Excellent Talents in University of MOE of China (NCET-06-0354). We thank Dr Zuguang Gong for helpful advice and discussion.References
- M. H. Cho, K. S. Kim, H. H. Ahn, M. S. Kim, S. H. Kim, G. Khang, B. Lee and H. B. Lee, Tissue Eng. A, 2008, 14, 1099 CrossRef CAS
.
- Y. Qiu and K. Park, Adv. Drug Delivery Rev., 2001, 53, 321 CrossRef CAS
- A. Chenite, C. Chaput, D. Wang, C. Combes, M. D. Buschmann, C. D. Hoemann, J. C. Leroux, B. L. Atkinson, F. Binette and A. Selmani, Biomaterials, 2000, 21, 2155 CrossRef CAS
- G. Franz, Adv. Polym. Sci., 1986, 76, 1 Search PubMed
-
J. M. Guenet, in Thermoreversible Gelation of Polymers and Biopolymers, Academic Press, London, 1992 Search PubMed
- M. K. Joo, M. H. Park, B. G. Choi and B. Jeong, J. Mater. Chem., 2009, 19, 5891 RSC
- L. Yu and J. D. Ding, Chem. Soc. Rev., 2008, 37, 1473 RSC
- M. Rinaudo, Prog. Polym. Sci., 2006, 31, 603 CrossRef CAS
- G. Sharma, J. L. Italia, K. Sonaje, K. Tikoo and M. Kumar, J. Controlled Release, 2007, 118, 27 CrossRef CAS
- N. Kashyap, B. Viswanad, G. Sharma, V. Bhardwaj, P. Ramarao and M. Kumar, Biomaterials, 2007, 28, 2051 CrossRef CAS
- E. Ruel-Gariepy, M. Shive, A. Bichara, M. Berrada, D. Le Garrec, A. Chenite and J. C. Leroux, Eur. J. Pharm. Biopharm., 2004, 57, 53 CrossRef CAS
- E. Ruel-Gariepy, A. Chenite, C. Chaput, S. Guirguis and J. C. Leroux, Int. J. Pharm., 2000, 203, 89 CrossRef CAS
- S. M. Richardson, N. Hughes, J. A. Hunt, A. J. Freemont and J. A. Hoyland, Biomaterials, 2008, 29, 85 CrossRef CAS
- C. D. Hoemann, J. Sun, M. D. McKee, A. Chevrier, E. Rossomacha, G. E. Rivard, M. Hurtig and M. D. Buschmann, Osteoarthritis Cartilage, 2007, 15, 78 CrossRef CAS
- A. Chevrier, C. D. Hoemann, J. Sun and M. D. Buschmann, Osteoarthritis Cartilage, 2007, 15, 316 CrossRef CAS
- C. D. Hoemann, J. Sun, A. Legare, M. D. McKee and M. D. Buschmann, Osteoarthritis Cartilage, 2005, 13, 318 CrossRef CAS
- C. D. Hoemann, M. Hurtig, E. Rossomacha, J. Sun, A. Chevrier, M. S. Shive and M. D. Buschmann, J. Bone Jt. Surg., 2005, 87A, 2671 CrossRef
- A. Chenite, M. Buschmann, D. Wang, C. Chaput and N. Kandani, Carbohydr. Polym., 2001, 46, 39 CrossRef CAS
- J. Y. Cho, M. C. Heuzey, A. Begin and P. J. Carreau, Biomacromolecules, 2005, 6, 3267 CrossRef CAS
- K. E. Crompton, R. J. Prankerd, D. M. Paganin, T. F. Scott, M. K. Horne, D. I. Finkelstein, K. A. Gross and J. S. Forsythe, Biophys. Chem., 2005, 117, 47 CrossRef CAS
- J. Cho, M. C. Heuzey, A. Begin and P. J. Carreau, Carbohydr. Polym., 2006, 63, 507 CrossRef CAS
- D. Filion, M. Lavertu and M. D. Buschmann, Biomacromolecules, 2007, 8, 3224 CrossRef CAS
- K. E. Crompton, J. S. Forsythe, M. K. Horne, D. I. Finkelstein and R. B. Knott, Soft Matter, 2009, 5, 4704 RSC
- U. Scheler, Curr. Opin. Colloid Interface Sci., 2009, 14, 212 CrossRef CAS
- M. Reibarkh, T. J. Malia and G. Wagner, J. Am. Chem. Soc., 2006, 128, 2160 CrossRef CAS
- F. Ilmain, T. Tanaka and E. Kokufuta, Nature, 1991, 349, 400 CrossRef CAS
- M. Lavertu, D. Filion and M. D. Buschmann, Biomacromolecules, 2008, 9, 640 CrossRef CAS
- R. Zeng, Z. C. Feng, R. Smith, Z. Z. Shao, X. Chen and Y. H. Yang, Acta Chim. Sin., 2007, 65, 2459 CAS
- G. G. Hammes and J. C. Swann, Biochemistry, 1967, 6, 1591 CrossRef CAS
- S. Kim, D. Sarathchandra and D. E. Mainwaring, J. Appl. Polym. Sci., 1996, 59, 1979 CrossRef CAS
- A. L. Kjoniksen, M. Hiorth, J. Roots and B. Nystrom, J. Phys. Chem. B, 2003, 107, 6324 CrossRef CAS
- O. E. Philippova, E. V. Volkov, N. L. Sitnikova, A. R. Khokhlov, J. Desbrieres and M. Rinaudo, Biomacromolecules, 2001, 2, 483 CrossRef CAS
- C. Jarry, J.-C. Leroux, J. Haeck and C. Chaput, Chem. Pharm. Bull., 2002, 50, 1335 CrossRef CAS
- V. Trappe and D. A. Weitz, Phys. Rev. Lett., 2000, 85, 449 CrossRef CAS
- P. Sollich, F. Lequeux, P. Hebraud and M. E. Cates, Phys. Rev. Lett., 1997, 78, 2020 CrossRef CAS
- E. K. Hobbie and D. J. Fry, J. Chem. Phys., 2007, 126, 124907 CrossRef CAS
-
H. Friebolin, in Basic One- and Two-Dimensional NMR Spectroscopy, 4th edn, WILEY-VCH, Weinheim, 2005 Search PubMed
- Y. H. Yang, Z. Z. Shao, X. Chen and P. Zhou, Biomacromolecules, 2004, 5, 773 CrossRef CAS
- P. H. Risso, D. M. Borraccetti, C. Araujo, M. E. Hidalgo and C. A. Gatti, Colloid Polym. Sci., 2008, 286, 1369 CAS
- V. Vetri and V. Militello, Biophys. Chem., 2005, 113, 83 CrossRef CAS
- S. Mukhopadhyay, U. Maitra, G. Krishnamoorthy, J. Schmidt and Y. Talmon, J. Am. Chem. Soc., 2004, 126, 15905 CrossRef CAS
- Y. Someya and H. Yui, Anal. Chem., 2010, 82, 5470 CrossRef CAS
- H. D. Wang, L. Y. Chu, X. Q. Yu, R. Xie, M. Yang, D. Xu, J. Zhang and L. Hu, Ind. Eng. Chem. Res., 2007, 46, 1511 CrossRef CAS
- Z. B. Cao, X. Chen, J. R. Yao, L. Huang and Z. Z. Shao, Soft Matter, 2007, 3, 910 RSC
- T. Inoue, G. H. Chen, K. Nakamae and A. S. Hoffman, Polym. Gels Networks, 1997, 5, 561 CrossRef CAS
- K. Gekko, H. Mugishima and S. Koga, Int. J. Biol. Macromol., 1987, 9, 146 CrossRef CAS
- P. Mateus, R. Delgado, P. Brandao and V. Felix, J. Org. Chem., 2009, 74, 8638 CrossRef CAS
- V. Kral, H. Furuta, K. Shreder, V. Lynch and J. L. Sessler, J. Am. Chem. Soc., 1996, 118, 1595 CrossRef CAS
- P. J. Cozzone and O. Jardetzky, Biochemistry, 1976, 15, 4853 CrossRef CAS
- O. Rathore and D. Y. Sogah, J. Am. Chem. Soc., 2001, 123, 5231 CrossRef CAS
- S. P. Nita, P. Alcouffe, C. Rochas, L. David and A. Domard, Biomacromolecules, 2010, 11, 6 CrossRef
| This journal is © The Royal Society of Chemistry 2011 |