The electrogenerated chemiluminescence detection of IS6110 of Mycobacterium tuberculosis based on a luminol functionalized gold nanoprobe
Jie
Jiang
,
Ying
Chai
and
Hua
Cui
*
CAS Key Laboratory of Soft Matter Chemistry, Department of Chemistry, University of Science & Technology of China, Hefei, Anhui, 230026, People's Republic of China. E-mail: hcui@ustc.edu.cn; Fax: +86-551-3600730; Tel: +86-551-3606645
First published on 2nd August 2011
Abstract
A novel electrogenerated chemiluminescence (ECL) DNA sensor was developed for a fast test for Mycobacterium tuberculosis (MTB) without using the polymerase chain reaction amplification, and was based on luminol functionalized gold nanoprobes with excellent signal amplification functionality. This nanoprobe was formed by the conjugation of luminol functionalized gold nanoparticles (lum–AuNPs) with signal DNA probes. An 81bp segment derived from the Mycobacterium tuberculosis specific insertion sequence IS6110 was chosen as the target strand and corresponding probes were designed to specifically hybridize with it. Biotinylated capture probes can be effectively immobilized directly on a streptavidin coated AuNP modified indium tin oxide electrode. After catching the TB target strand, signal probes tagged with lum–AuNPs were attached to the assembled electrode surface to form a sandwich-type TB sensor. Treating the resulting electrode surface with a carbonate buffer solution containing 1.0 mmol L−1 of H2O2 and applying a double-step potential to the electrode, the ECL response was generated which realized the detection of the TB target strand. The detection limit was estimated to be 6.7 × 10−15 mol L−1 (equal to approximately 1.7 × 10−10 g L−1) of the synthetic TB target strand, which is superior to other genetic methodologies for TB tests based on gold nanoprobes. Genomic DNA from other pathogenic bacterias (Escherichia coli, Pseudomonas aeruginosa, and Staphylococcus aureus) had a negligible effect on its detection, which guaranteed the good selectivity of the TB sensor. The efficacy of the TB sensor was also evaluated for genomic DNA extracted from cultured M. tuberculosis. The TB sensor is sensitive, highly selective, convenient and cost-effective. The simplicity of the assay and the lack of a requirement for sophisticated equipment render the TB sensor a promising candidate as a rapid molecular test for the detection ofM. tuberculosis.
Introduction
Tuberculosis (TB) is an infectious bacterial disease caused by Mycobacterium tuberculosis (MTB). It is one of the major causes of global morbidity and mortality and constitutes a serious public health problem worldwide. Approximately two million deaths annually are attributed to the disease, mainly in developing countries.1 Accurate and rapid diagnosis is the first priority in controlling the growing epidemic. Conventional smear microscopy and culture methods are widely used in the diagnosis of TB. The former is insensitive2 and the latter takes up to 6–8 weeks to provide a result, limiting the value of these methods in aiding diagnosis and immediate decisions on treatment. Major advances in molecular biology and the availability of new information generated after deciphering the complete genome sequence of M. tuberculosis3–4 stimulated the development of new tools for the rapid diagnosis of tuberculosis, differentiation of M. tuberculosis from nontuberculous mycobacteria, and for the rapid detection of drug resistance.5–7 Using the insertion sequence IS6110 and/or 16S ribosomal DNA (16S rDNA) as the detection targets, many in-house and commercially available nucleic acid amplification (NAA) based detection systems have been developed as rapid tests for the direct identification of the M. tuberculosis complex in clinical specimens.8–10 The procedures based on NAA techniques mostly use the polymerase chain reaction (PCR) to amplify the target sequences, followed by a detection step performed on agarose or acrylamide gel, or hybridization in various formats. The sensitivity and specificity have been confirmed in many publications,11–14 but in most cases the reliable application of these diagnostic methods requires highly trained personnel and very often, dedicated equipment that can be of considerable cost. Therefore, additional efforts should be made to develop a new diagnostic assay that would be easily applicable, and would allow specific and sensitive detection and identification of M. tuberculosis from clinical samples without the need for high-cost sophisticated equipment.Nanodiagnostics, i.e. the use of nanotechnology based strategies for diagnostic purposes, has provided researchers with more and improved techniques for clinical diagnostics with increased sensitivity at lower costs. Recently, many gold-nanoprobe-based sensing schemes have been described for detection of specific DNA sequences of M. tuberculosis. Baptista and coworkers developed a colorimetric detection method for DNA identification in homogenous solution based on the aggregation of gold nanoprobes. The method relies on a spectroscopic change of the solution before and after salt induced gold nanoprobe aggregation: hybridization of complementary target prevents aggregation and the solution remains red; noncomplementary/mismatched targets do not prevent gold nanoprobe aggregation, resulting in a visible change of color from red to blue. They have already applied the non-cross-linking method for the identification of M. tuberculosis in clinical samples based on the ropB locus,15–16 and optimized the method for detection of point mutations.17–18 Soo and coworkers reported a simple assay for the identification of M. tuberculosis using two gold nanoprobes with covalently bound oligonucleotides complementary to the IS6110 target DNA.19 When encountering target strands, these gold nanoprobes are polymerized and form network structures, accompanied by a red to blue color change. However, the measurements become more difficult at low concentrations of target strands because of the decrease in the surface plasmon resonance (SPR) shift effect. Visual detection of IS6110 target DNA by sandwiching it between DNA–latex beads as the separator and red DNA-gold nanoparticles as the reporter has been demonstrated.20 By adding silver enhancement solution to gold nanoparticles retained on filters, Ag ions are deposited on their surfaces to form Au–Ag core shell structures, leading to changes in the color of the spot from red to black. However, the main limitation of these above approaches is their low sensitivity. It is still a critical demand on simple, rapid, sensitive, and low-cost detection technologies for the identification and detection ofM. tuberculosis.
Electrogenerated chemiluminescence (ECL), involving a light emission process during a redox reaction of electrogenerated reactants, combines electrochemical and luminescent techniques. By integrating the high-affinity hybridization of oligonucleotides, ECL DNA sensors have become a powerful analytical tool due to their high sensitivity, wide dynamic range and simply instrumentation.21–22 To the best of our knowledge, nanoprobe-based ECL detection for DNA of M. tuberculosis has not been reported.
In our previous work, luminol functionalized gold nanoparticles (lum–AuNPs) were synthesized by a one-pot method via the direct reduction of chloroauric acid (HAuCl4) by luminol in aqueous solution,23 and thousands of luminol molecules acting as stabilizers were coated on the surface of the AuNPs through the weak covalent interactions between the gold and the nitrogen atoms in their amino groups,24 enabling the AuNPs be chemiluminescent.23 Based on the lum–AuNPs labeling, a general-purpose concept for ultrasensitive DNA ECL detection was demonstrated by using a model DNA sequence.25 In the present study, effort was made to develop a rapid test for M. tuberculosis without PCR amplification based on the previously proposed ultrasensitive, simple and low-cost strategy. IS6110, an insertion sequence present exclusively in the MTB complex genome, is the most widely used target gene for detection ofM. tuberculosis.26–29 An 81bp segment derived from the Mycobacterium tuberculosis specific insertion sequence IS6110 was chosen as the DNA target in this study. An indium tin oxide (ITO)-coated glass slide was utilized as an assembly substrate owing to its excellent compatibility with microfabrication and miniaturization; it was reversibly bound with a hollow polydimethylsiloxane (PDMS) layer on its surface to confine the area of ITO electrode. Using the luminol functionalized gold nanoprobe, a single lum–AuNP conjugated with a signal probe could bind thousands of luminol molecules. Besides, another kind of AuNP functionalized with streptavidin (SA) was introduced into the TB sensor as the immobilization matrix for further improvement of ECL intensity. By the combination of smart sensor fabrication, luminol functionalized gold nanoprobes, the amplification of the AuNP and biotin-SA system, a novel ECL DNA sensor was proposed for sensitive, highly selective, convenient and cost-effective detection ofM. tuberculosis. Electrochemical impedance techniques were used to characterize and confirm the fabrication of the ECL TB sensor. The conditions for ECL detection were optimized. The analytical performance and selectivity of this assay were examined. Finally, the sensor was evaluated for the assay of genomic DNA extracted from cultured M. tuberculosis.
Experimental
Chemicals and materials
The oligonucletides used in the present study were synthesized and HPLC-purified by Sangon Inc. (Shanghai, China). The target sequence comprised nucleotides 786–866 of the IS6110 sequence30 (GenBank accession no.M29899). The two nucleotide probes were complementary to each end of the target strand. Their exact sequences and modification are listed in Table 1. Standard strains of Mycobacterium tuberculosisH37Rv (ATCC27294) were obtained from Anhui Provincial TB Institute. 3-Mercaptopropyltrimethoxysilane (MPTMS) and luminol standard powder were purchased from Sigma-Aldrich. Bovine serum albumins (BSA), streptavidin (SA) were obtained from Solarbio (Beijing, China). A HAuCl4 stock solution (2‰ HAuCl4, w/w) was prepared by dissolving 1.0 g of HAuCl4·4H2O (Shanghai Reagent, China) in 412 ml of purified water and stored at 4 °C. A luminol stock solution (10 mmol L−1) was prepared by dissolving luminol in 0.1 mol L−1sodium hydroxide solution and stored in the dark. The indium tin oxide (ITO)-coated glass (1.1 mm thick and <10 Ω cm−2 resistant) was purchased from CSG Holding Co., Ltd. (Shenzhen, China). Sylgard 184 silicone elastomer and curing agent were obtained from Dow Corning (Midland, MI). All other reagents were of analytical grade. Ultra-pure water was prepared by a Millipore Milli-Q system and used throughout.| DNA | Sequence(5′ → 3′) |
|---|---|
| TB target strand | CCAGCACCTA ACCGGCTGTG GGTAGCAGAC CTCACCTATG TGTCGACCTG GGCAGGGTTC GCCTACGTGG CCTTTGTCAC C |
| GP1(TB capture probe) | GGTGAGGTCT GCTACCCAC-Biotin |
| GP2(TB signal probe) | Biotin-GGCCACGTAG GCGAACC |
Preparation of lum–AuNPs labeled TB signal probe
The lum–AuNPs with an average diameter of 20 nm were synthesized by the method of reduction of HAuCl4 with luminol.23 Briefly, 100 mL of 0.02% (w/w) HAuCl4 solution was boiled with vigorous stirring, and then, 1.8 mL of 1.0 × 10−2 mol L−1luminol stock solution were rapidly added to the boiling solution. The solution was maintained at the boiling point for 30 min. The color of the solution turned from yellow to black to purple before a wine-red color was reached, indicating the formation of lum–AuNPs. After cooling down the resulting colloidal suspension to room temperature with stirring, the colloidal lum–AuNPs were obtained. The unreacted reagents and the by-products (small molecules) were removed via six sets of 2 days of dialysis with ultrapure water under stirring using of a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 molecular weight cutoff dialysis membrane to obtain lum–AuNPs. 1 mL pH-adjusted (pH 8.0) lum–AuNPs solution was added to 25 μL of SA (1.0 mg mL−1), after being incubated at room temperature for 0.5 h, to the solution was further added 5% BSA solution to reach a final concentration of 1% BSA, and was then stirred for 5 min. The conjugate of lum–AuNPs with SA was centrifuged at 12500 rpm for 20 min (Universal 320, Hettich, Germany), and the red precipitates were dispersed with 0.05 mol L−1 pH 8.0 Tris-HCl containing 0.3 mol L−1NaCl. A biotinylated TB signal probe (4.0 × 10−7 mol L−1) was added to the red solution and incubated at 37 °C for 1 h. The as-prepared mixture was centrifuged at 12500 rpm for 10 min, and the red precipitates were suspended with 0.05 mol L−1 pH 8.0 Tris-HCl containing 0.3 mol L−1NaCl to obtain the lum–AuNPs labeled TB signal probe.
500 molecular weight cutoff dialysis membrane to obtain lum–AuNPs. 1 mL pH-adjusted (pH 8.0) lum–AuNPs solution was added to 25 μL of SA (1.0 mg mL−1), after being incubated at room temperature for 0.5 h, to the solution was further added 5% BSA solution to reach a final concentration of 1% BSA, and was then stirred for 5 min. The conjugate of lum–AuNPs with SA was centrifuged at 12500 rpm for 20 min (Universal 320, Hettich, Germany), and the red precipitates were dispersed with 0.05 mol L−1 pH 8.0 Tris-HCl containing 0.3 mol L−1NaCl. A biotinylated TB signal probe (4.0 × 10−7 mol L−1) was added to the red solution and incubated at 37 °C for 1 h. The as-prepared mixture was centrifuged at 12500 rpm for 10 min, and the red precipitates were suspended with 0.05 mol L−1 pH 8.0 Tris-HCl containing 0.3 mol L−1NaCl to obtain the lum–AuNPs labeled TB signal probe.
Fabrication of the TB sensor
ITO-coated glass slides were used as an assembly electrode owing to their stability, easy miniaturization and compatibility with microfabrication.31 An ITO glass slide with a dimension of 6 cm × 1.5 cm was sonicated alternately with acetone, ultra-pure water and ethanol, each for 10 min to get a clean ITO surface. After rinsing with ethanol and drying with a N2 stream, 1/3 of the ITO electrode was immersed in 5% MPTMS ethanol solution overnight. Through the bifunctional linker, MPTMS, the ITO surface was functionalized with an SH-rich self-assembled layer. After exhaustive rinsing with ethanol, the ITO electrode was heated to 80 °C to remove loosely bound MPTMS molecules (loosely bound MPTMS can lead to gold nanoparticle aggregates attaching the ITO electrode with the SA coated AuNPs32). Then the modified part was reversibly bound to a punched polydimethylsiloxane (PDMS) layer, which was prepared by curing the mixture of PDMS monomer and the curing agent (10:1) in an oven at 75 °C for 1.5 h. The effective electrode area was confined by the punched round hole with a diameter of 5 mm, which was used as a reservoir for subsequent assembly. 50 μL of SA coated AuNPs was introduced into the reservoir of the ITO electrode, followed by a 10 h self-assembly at room temperature. After rinsing with excess ultrapure water, the AuNP-modified electrode was ready for further experiments.
To immobilization of the capture probe, 50 μL of the biotinylated TB capture probe (1.0 × 10−6 mol L−1) was dropped on the above AuNP-modified ITO electrode for 1 h at 37 °C. The modified electrode was subsequently rinsed thoroughly with washing buffer (7.0 mmol L−1 pH 8.0 Tris-HCl containing 0.17 mol L−1NaCl) to remove any weakly absorbed DNA capture probe. A 50-μL portion of 0.05 mol L−1 Tris-HCl (pH 8.0) containing 1% (w/w) BSA was used to saturate the possible bare AuNPs for 30 min at 37 °C and flushed with washing buffer. After that, TB target strand sample (50 μL) with different concentrations was dropped on the electrode, incubated 1 h at 37 °C, and subsequently washed with copious amounts of washing buffer. After the binding of TB target strand, 50 μL of the lum–AuNPs labeled TB signal probe was dropped on the electrode. Again, incubated at 37 °C for 1 h and rinsed with the similar washing steps as above.
ECL detection
ECL measurements were carried out with a homemade ECL system consisting of a model CHI760B workstation (Chenhua, Shanghai, China), a model RFL-1 luminometer (Remax, Xi'an, China), an H-type electrochemical cell (self-designed) and a computer. The generation of ECL response comprises electrochemical reactions of the ECL reagent luminol on the surface of the AuNPs and H2O2 with a conventional three-electrode setup and a light emission process from the excited state to the ground state of the oxidation products of luminol. The modified part of the ITO electrode was immersed in 1.0 mmol L−1H2O2/0.02 mol L−1 pH 9.8 carbonate buffer solution (CBS) as working electrode. When a double-step potential (30 s pulse period, 0.1 s pulse time, 1.0 V pulse potential and 0 V initial pulse potential) was applied to the working electrode, an ECL signal was generated and recorded.Preparation of genomic DNA
Genomic DNA from cultured M. tuberculosis (standard strain H37Rv) was extracted by MYCOBACTERIUM DNA EXTRACTION KIT (MT81813, from Sangon) and dissolved in Tris-EDTA (10 mmol L−1 Tris, 1.0 mmol L−1EDTA, pH 8.0) buffer, and the yield was estimated by measuring the absorbance at 260 nm and 280 nm. Before hybridization with the capture probe, the genomic double-stranded DNA was denatured to single-stranded DNA by keeping the tube at 95 °C for 10 min and then in a freezing mixture consisting NaCl and ice for 1 min.Results and discussion
Principle of the TB sensor protocol
The schematic diagram of the principle of the present TB sensor protocol was depicted in Fig. 1. The MPTMS-functionalized ITO electrode was prepared to immobilize streptavidin coated AuNPs through an Au–S bond. A streptavidin coated AuNP modified ITO electrode was then bound with a large number of biotinylated TB capture probes through biotin–SA specific interaction. The resulting TB capture probe linked electrode was hybridized with the TB target strand by the double-stranded assembly between capture probe strand and the 19-mer portion of the TB target strand sequences. When treated with lum–AuNPs labeled TB signal probe, a “sandwich” configuration of capture probe/TB target strand/luminol–AuNPs labeled signal probe was formed on the modified ITO electrode. i.e.TB sensor. As the TB sensor was immersed in a H2O2-containing electrolyte solution, ECL response emerged under an applied double-step potential.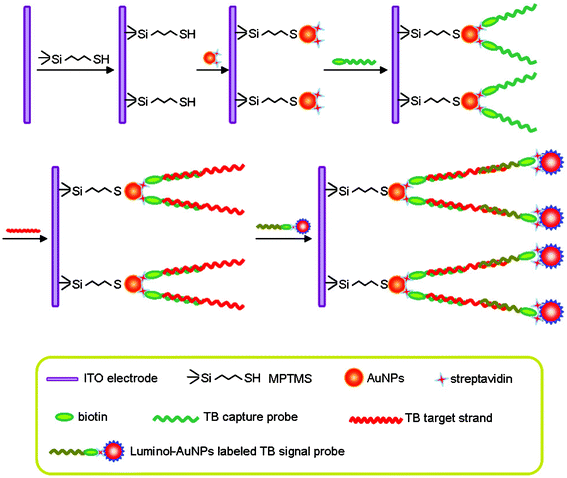 | ||
| Fig. 1 Schematic illustration of the proposed TB sensor. | ||
ECL behavior
The ECL property of the TB sensor was studied with a double-step potential (1.0 V pulse potential, 0.1 s pulse time, 30 s pulse period, 0 V initial pulse potential) in 0.02 mol L−1 pH 9.8 carbonate buffer solution (CBS) containing 1.0 mmol L−1H2O2. To gain a better understanding of the ECL signal generation, the ECL signal of the modified electrodes during different stages was examined. The stepwise ECL measurements of the modified ITO electrode at different modification steps were carried out and recorded as shown in Fig. 2. Curves a–e show the ECL responses on the bare ITO electrode, MPTMS self-assembled ITO electrode, SA coated AuNPs/MPTMS/ITO electrode, TB target strand (1.0 × 10−13 mol L−1)/TB capture probe/SA coated AuNPs/MPTMS/ITO electrode, lum–AuNPs labeled TB signal probe/TB target strand (1.0 × 10−13 mol L−1)/TB capture probe/SA coated AuNPs/MPTMS/ITO electrode, respectively. The results indicated that only the electrode linked to the luminol–AuNP labeled TB signal probe could produce ECL signals. Therefore, the ECL signals were from the luminol radicals electro-oxidized by luminol with H2O2 under the catalysis of AuNPs.33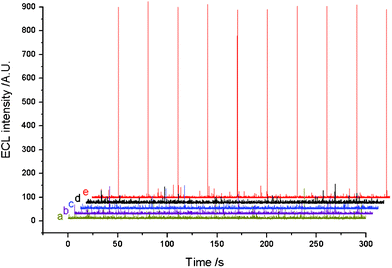 | ||
| Fig. 2 ECL signals under pulse potential. Initial potential, 0 V; pulse period, 30 s; pulse time, 0.1 s; pulse potential, 0.8 V. ECL signals were obtained (a) on a bare ITO electrode, (b) on a MPTMS/ITO electrode, (c) on a SA-coated AuNPs/MPTMS/ITO electrode, (d) on a TB target strand (1.0 × 10−13 mol L−1)/TB capture probe/SA-coated AuNPs/MPTMS/ITO electrode, (e) on a lum–AuNPs labeled TB signal probe/TB target strand (1.0 × 10−13 mol L−1)/TB capture probe/SA-coated AuNPs/MPTMS/ITO electrode. All ECL signals were measured in 0.02 mol L−1 CBS (pH 9.95) solution containing 1.0 mmol L−1H2O2. | ||
EIS monitoring of the fabrication of the TB sensor
Electrochemical impedance spectra (EIS) is an effective tool for probing the features of surface-modified and biomaterial-functionalized electrodes. The stepwise EIS measurements of the modified ITO electrode at different modification steps were carried out in the PDMS reservoir as this ensures a constant electrode area. When the stepwise modification finished, the reservoir was washed, dried and filled with redox probe solution (0.1 mol L−1, PBS (pH 7.4) containing 1.0 mmol L−1[Fe(CN)6]3−/4−) for the EIS experiment. The results presented as Nyquist plot are illustrated in Fig. 3. Nyquist plot of the EIS measurement showed a semicircle in the high-frequency region while showed a straight line in the low-frequency region, demonstrating that the electrode process was controlled by electron transfer at high frequency and by diffusion at low frequency. In EIS, the value of Ret equals the semicircle diameter. Significant differences in the EIS spectra were observed during the step-by-step modification. It can be seen that the bare ITO electrode exhibited a very small Ret value (Fig. 3a). Silanization of the ITO surface with MPTMS generated an insulating layer on the electrode surface, which resulted in a remarkable increased electron transfer resistance (Fig. 3b). The Ret value decreased obviously when streptavidin–AuNPs were further modified on the MPTMS/ITO electrode (Fig. 3c). This might be because the AuNPs could facilitate electron-transfer. It was reported that the assembly of AuNPs on an ITO electrode behaved as an array of randomly distributed nanoparticles.32 The fast electrode charge transfer kinetics on an AuNPs modified ITO electrode originated from the enlarged surface area of the AuNPs monolayer. Subsequently, when the TB capture probe and the TB target strand were assembled successively on the modified electrode above, as expected, the electron transfer resistance was greatly elevated as shown in Fig. 3d. Generally, negatively charged oligonucleotides repel the negatively charged redox probe [Fe(CN)6]3−/4− and thus the interfacial electron transfer resistance will increase. Finally, when the electrode was associated with luminol functionalized AuNP-labeled signal probes, the Ret value decreased again (Fig. 3e). This indicated the excellent conductive performance of luminol functionalized AuNPs, which facilitate the electron transfer exceeding the repulsion interactions between the negatively charged oligonucleotides and the probe ions and leading to a net decrease of electron transfer resistance. The entire EIS data indicated that the electrode was modified as expected, and the results were consistent with other published work.34![Nyquist plot for the Faradaic impedance measurements of the modified ITO electrode by layer-by-layer self-assembly. (a) Bare ITO surface, (b) MPTMS/ITO surface, (c) SA-coated AuNPs/MPTMS/ITO surface, (d) TB target strand (1.0 × 10−13 mol L−1)/TB capture probe/SA-coated AuNPs/MPTMS/ITO surface, (e) Lum–AuNPs labeled TB signal probe/TB target strand (1.0 × 10−13 mol L−1)/TB capture probe/SA-coated AuNPs/MPTMS/ITO surface. The spectra were recorded in 0.1 mol L−1PBS (PH 7.4) containing 1 mmol L−1[Fe(CN)6]3−/4− as the redox probe, using a frequency of 10 kHz to 0.1 Hz under an oscillation potential of 5 mV.](/image/article/2011/RA/c1ra00206f/c1ra00206f-f3.gif) | ||
| Fig. 3 Nyquist plot for the Faradaic impedance measurements of the modified ITO electrode by layer-by-layer self-assembly. (a) Bare ITO surface, (b) MPTMS/ITO surface, (c) SA-coated AuNPs/MPTMS/ITO surface, (d) TB target strand (1.0 × 10−13 mol L−1)/TB capture probe/SA-coated AuNPs/MPTMS/ITO surface, (e) Lum–AuNPs labeled TB signal probe/TB target strand (1.0 × 10−13 mol L−1)/TB capture probe/SA-coated AuNPs/MPTMS/ITO surface. The spectra were recorded in 0.1 mol L−1PBS (PH 7.4) containing 1 mmol L−1[Fe(CN)6]3−/4− as the redox probe, using a frequency of 10 kHz to 0.1 Hz under an oscillation potential of 5 mV. | ||
Optimization of the ECL detection conditions
An electropulse signal double-step potential was used to initiate the ECL of the TB sensor. The pulse ECL intensity reached a stable value in the second to tenth periods in every experiment. The average intensity of nine pulse ECL signals (periods two to ten ) was used as an analytical signal for detection. To obtain the maximum ECL intensity, some important experimental parameters influencing the ECL intensity, including pH value, H2O2 concentration, pulse period, pulse time, pulse potential and initial potential, were optimized as shown in Fig. 4a–f.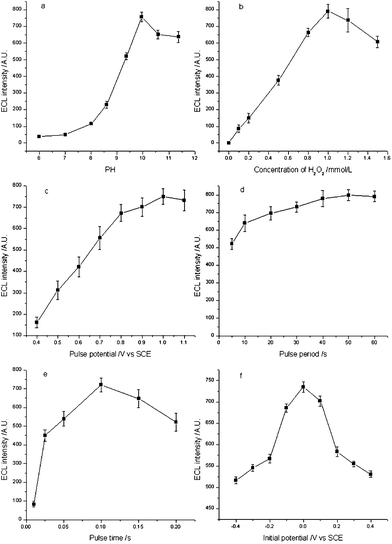 | ||
| Fig. 4 Effect of (a) pH value, (b) H2O2 concentration, (c) pulse potential, (d) pulse period, (e) pulse time, and (f) initial pulse potential on ECL intensity. | ||
It is well known that the ECL performance of luminol was dependent greatly on the pH of the solution. The influence of pH in the range of 6.0–8.0 (PBS, 0.02 mol L−1) and 8.6–11.3 (CBS, 0.02 mol L−1) on the ECL intensity was examined. As shown in Fig. 4a, the maximum ECL intensity was obtained at pH 9.95. Therefore, pH 9.95 (CBS, 0.02 mol L−1) was chosen as the optimum pH condition for the following experiments.
The ECL intensity was also dependent on the concentration of H2O2. As shown in Fig. 4b, the ECL intensity increased when the concentration of H2O2 increased from 0.1 mmol L−1 to 1.0 mmol L−1 and reached a maximum at 1.0 mmol L−1, which might be attributed to the co-oxidation function of H2O2. When the concentration of H2O2 was higher than 1.0 mmol L−1, the ECL intensity decreased. Therefore, the optimized concentration of H2O2 was 1.0 mmol L−1 for the following experiments.
The electrochemical parameters played very important roles since the ECL reaction was initiated by an electrochemical reaction at the electrode surface. A pulse wave was used as the work waveform in the present work, because a pulse wave probably produces more photons in the same time than other waveforms.
The effect of pulse potential over the range 0.4–1.1 V (versusSCE) in CBS solution (0.1 mol L−1) containing 1.0 mmol L−1H2O2 at pH 9.95 was studied (see Fig. 4c). In the view of high intensity and good stability, a pulse potential of 1.0 V was employed for the following experiments.
The effect of pulse period on the ECL intensity was investigated in the range of 5–60 s (see Fig. 4d). The ECL signal showed a positive correlation with the pulse period. This phenomenon was attributed to the more effective diffusion of H2O2 in the longer pulse period. To make a compromise between higher ECL intensity and shorter analytical time, a pulse period of 30 s was chosen in the following experiments.
The effect of pulse time was examined in the range of 0.01–0.2 s. As shown in Fig. 4e, the ECL intensity increased when the pulse time increased from 0.01 to 0.1 s and reached a maximum at 0.1 s. However, when the pulse time was longer than 0.1 s, the ECL intensity decreased rapidly at the second period, and could not reach a stable ECL signal in the following period. This is because the diffusion layer on the surface of the electrode became thicker in a relatively long pulse time, and was difficult to recover in the next pulse. Therefore, the optimized pulse time of 0.1 s was used for the following experiments.
The effect of initial potential on the ECL intensity was also tested in the range from −0.4 V to 0.4 V, as shown in Fig. 4f. The maximum ECL intensity was achieved at the initial potential of 0.0 V. Therefore, this initial potential was used in the following experiments.
Ultimately, the optimized conditions for the electrochemical parameters were optimized as follows: 1.0 V pulse potential, 30 s pulse period, 0.1 s pulse time and 0 V initial pulse potential.
Analytical performance
The principle of the TB sensor was demonstrated initially with a synthetic TB target strand. A series of dilutions containing 1.0 × 10−14–1.0 × 10−12 mol L−1 of target was made. Under the optimized conditions, the ECL intensity of the TB sensor was linear with the logarithm of the concentration of the synthetic TB target strand in the range from 1.0 × 10−14–1.0 × 10−12 mol L−1 (Fig. 5). The regression equation was I = 7716.45 + 535.50 logC with a regression coefficient of 0.9966, where I is ECL intensity and C is the concentration of the synthetic TB target strand. The limit of detection (LOD), at a signal of noise ratio of three (S/N = 3), was 6.7 × 10−15 mol L−1 (which approximately equals 1.7 × 10−10 g L−1). The reproducibility of the TB sensor was evaluated by detecting the synthetic TB target strand five times. The relative standard deviations (RSDs) for 2.0 × 10−13 mol L−1 synthetic TB target strand was 6.86%, indicating a good analytical performance of the present TB DNA-assay method in terms of reproducibility.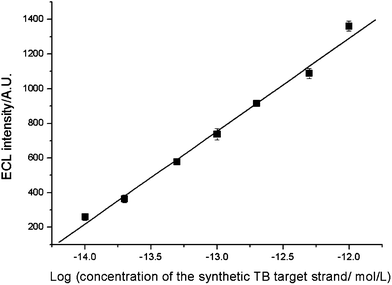 | ||
| Fig. 5 Calibration curve of ECL intensity versus concentration of the synthetic TB target strand. Conditions: initial potential, 0 V; pulse period, 30 s; pulse time, 0.1 s; pulse potential, 1.0 V; H2O2, 1.0 mmol L−1; CBS, 0.02 mmol L−1 (pH 9.95). | ||
Detection selectivity
In order to verify the selectivity of the proposed TB sensor for the detection of its TB target strand, we performed a series of contrast experiments using genomic DNA from Escherichia coli (ATCC25922), Pseudomonas aeruginosa (ATCC27853), and Staphylococcus aureus (ATCC25923) as negative controls. 12.5 pg mL−1 (eguals 5.0 × 10−13 mol L−1) of the TB target strand was detected, even in the presence of 500 ng mL−1 genomic E. coliDNA, 500 ng mL−1 genomic P. aeruginosaDNA and 500 ng mL−1 genomic S. aureusDNA. All samples were denatured by thermal denaturation before hybridization. As shown in Fig. 6, no remarkable change of ECL signal was observed as compared to that of the TB target strand only. On the other hand, a much weaker response was observed by replacing the TB target strand with genomic DNA from E. coli, P. aeruginosa and S. aureus in the solution. The results suggest that the as-proposed TB sensor responded to its target with good selectivity while genomic DNA from other pathogenic types of bacteria have almost negligible effect on the detection of the TB target strand. Moreover, it remains sufficiently sensitive to detect the TB target sequence among a large pool of nucleotides.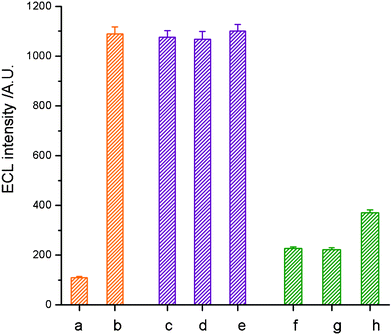 | ||
| Fig. 6 Investigation of the selectivity of the TB sensor to the synthetic TB target strand by comparison with E. coli, S. aureus and P. aeruginosa as negative controls. The ECL responses of the TB sensor to (a) blank, (b) 12.5 pg mL−1TB target strand, (c) 12.5 pg mL−1TB target strand in 500 ng mL−1 genomic E. coliDNA, (d) 12.5 pg mL−1TB target strand in 500 ng mL−1 genomic P. aeruginosaDNA, (e) 12.5 pg mL−1TB target strand in 500 ng mL−1 genomic S. aureusDNA, (f) 500 ng mL−1 genomic E. coliDNA, (g) 500 ng mL−1 genomic P. aeruginosaDNA, and (h) 500 ng mL−1 genomic S. aureusDNA. Conditions: initial potential, 0 V; pulse period, 30 s; pulse time, 0.1 s; pulse potential, 1.0 V; H2O2, 1.0 mmol L−1; CBS, 0.02 mmol L−1 (pH 9.95). | ||
Evaluation of the TB sensor in cultured M. tuberculosis
The proposed TB sensor was evaluated for genomic DNA extracted from cultured M. tuberculosis. The amount of genomic DNA extracted from standard strains of M. tuberculosisH37Rv (ATCC27294) was quantified and serially diluted from 500 ng mL−1 to 5 ng mL−1. Under the optimized conditions, the curve for the determination of H37Rv genomic DNA is shown in Fig. 7. The lower detection limit of the TB sensor is 5 ng mL−1 of H37Rv genomic DNA. This detection limit corresponds to 106cells mL−1 as there are 16 copies of IS6110 present in strain H37Rv,35 and a single genome contains 5 fg of DNA. Therefore, the present TB sensor demonstrates a strong applicability in the diagnosis of M. tuberculosis directly from cultured M. tuberculosis samples.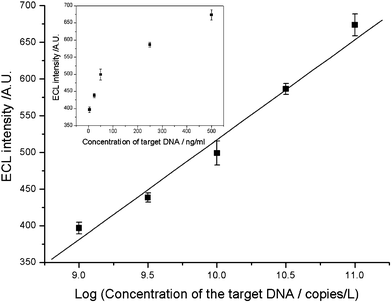 | ||
| Fig. 7 Calibration curve of ECL intensity versus concentration of genomic DNA extracted from standard strains M. tuberculosisH37Rv (ATCC27294). Conditions: initial potential, 0 V; pulse period, 30 s; pulse time, 0.1 s; pulse potential, 1.0 V; H2O2, 1.0 mmol L−1; CBS, 0.02 mmol L−1 (pH 9.95). | ||
Conclusion
A novel ECL DNA sensor was proposed for the detection of tuberculosis by using luminol functionalized gold nanoprobes with excellent signal amplification functionality. An 81bp segment derived from the Mycobacterium tuberculosis specific insertion sequence IS6110 of Mycobacterium tuberculosis was chosen as the target strand and corresponding probes were designed to specifically hybridize with it. A biotinylated capture probe can be effectively immobilized directly on a streptavidin coated AuNP modified ITO electrode. After catching the TB target strand, signal probes tagged with luminol functionalized AuNPs were attached to the assembled electrode surface to form sandwich-type TB sensor. By treating the resulting electrode surface with carbonate buffer solution containing 1.0 mmol L−1 of H2O2 and applying a double-step potential to the electrode, an ECL response was generated, which realized the detection of the TB target strand. The detection limit was estimated to be 6.7 × 10−15 mol L−1 (equal to approximately 1.7 × 10−10 g L−1) of the synthetic TB target strand, which is superior to other genetic methodologies for TB tests based on gold nanoprobes. Genomic DNA from other types of pathogenic bacteria (E. coli, P. aeruginosa and S. aureus) had a negligible effect on its detection, which guaranteed the good selectivity of the TB sensor. The TB sensor was successfully applied for genomic DNA extracted from cultured M. tuberculosis. This sensor is sensitive, highly selective, convenient and cost-effective. The simplicity of the assay and the lack of requirement for sophisticated equipment render the TB sensor a promising candidate as a rapid molecular test for the detection ofM. tuberculosis.Acknowledgements
The support of this research by the National Natural Science Foundation of P.R. China (Grant Nos. 20625517, 20573101 and 21075115) and the Overseas Outstanding Young Scientist Program of Chinese Academy of Sciences are gratefully acknowledged.References
- M. Arentz and T. R. Hawn, Drug Discovery Today Dis. Mech., 2007, 4, 231–236 CrossRef.
- M. Caws, S. M. Wilson, C. Clough and F. Drobniewski, J. Clin. Microbiol., 2000, 38, 3150–3155 CAS.
- J. C. Camus, M. J. Pryor, C. Medigue and S. T. Cole, Microbiol., 2002, 148, 2967–2973 CAS.
- S. T. Cole, R. Brosch, J. Parkhill, T. Garnier and C. Churcher, Nature, 1998, 393, 537–544 CrossRef CAS.
- M. Pai, S. Kalantri and K. Dheda, Expert Rev. Mol. Diagn., 2006, 6, 423–432 CrossRef CAS.
- J. C. Palomino, Eur. Respir. J., 2005, 26, 339–350 CrossRef CAS.
- I. C. Shamputa, L. Rigouts and F. Portaels, APMIS, 2004, 112, 728–752 CrossRef CAS.
- D. F. Gardiner and K. G. Beavis, Semin. Respir. Infect., 2000, 15, 132–143 CrossRef CAS.
- H. Soini and J. M. Musser, Clin. Chem., 2001, 47, 809–814 CAS.
- G. L. Woods, Arch. Pathol. Lab. Med., 1999, 123, 1002–1006 CAS.
- M. Bogard, J. Vincelette, R. Antinozzi, R. Alonso and T. Fenner, Eur. J. Clin. Microbiol. Infect. Dis., 2001, 20, 724–731 CAS.
- W. H. F. Goessens, P. de Man, J. G. M. Koeleman, A. Luijendijk, R. te Witt, H. P. Endtz and A. van Belkum, J. Clin. Microbiol., 2005, 43, 2563–2566 CrossRef CAS.
- N. Lemaìtre, S. Armand, A. Vache'e, O. Capilliez, C. Dumoulin and R. J. Courcol, J. Clin. Microbiol., 2004, 42, 4307–4309 CrossRef.
- S. Levidiotou, G. Vrioni, E. Galanakis, E. Gesouli, C. Pappa and D. Stefanou, Eur. J. Clin. Microbiol. Infect. Dis., 2003, 22, 349–356 CrossRef CAS.
- P. V. Baptista, M. Koziol-Montewka, J. Paluch-Oles, G. Doria and R. Franco, Clin. Chem., 2006, 52, 1433–1434 CAS.
- P. Costa, A. Amaro, A. Botelho, J. Inacio and P. V. Baptista, Clin. Microbiol. Infect., 2010, 16, 1464–1469 CrossRef CAS.
- G. Doria, B. G. Baumgartner, R. Franco and P. V. Baptista, Colloids Surf., B, 2010, 77, 122–124 CrossRef CAS.
- B. Veigas, D. Machado, J. Perdigao, I. Portugal, I. Couto, M. Viveiros and P. V. Baptista, Nanotechnology, 2010, 21, 415101–415115 CrossRef.
- P. C. Soo, Y. T. Horng, K. C. Chang, J. Y. Wang, P. R. Hsueh, C. Y. Chuang, C. C. Lu and H. C. Lai, Mol. Cell. Probes, 2009, 23, 240–246 CrossRef CAS.
- P. Upadhyay, M. Hanif and S. Bhaskar, Clin. Microbiol. Infect., 2006, 12, 1118–1122 CrossRef CAS.
- P. Bertoncello and R. J. Forster, Biosens. Bioelectron., 2009, 24, 3191–3200 CrossRef CAS.
- M. M. Richter, Chem. Rev., 2004, 104, 3003–3036 CrossRef CAS.
- H. Cui, W. Wang, C. F. Duan, Y. P. Dong and J. Z. Guo, Chem.–Eur. J., 2007, 13, 6975–6984 CrossRef CAS.
- W. Wang, T. Xiong and H. Cui, Langmuir, 2008, 24, 2826–2833 CrossRef CAS.
- Y. Chai, D. Y. Tian, W. Wang and H. Cui, Chem. Commun., 2010, 46, 7560–7562 RSC.
- J. Almeda, A. Garcia, J. Gonzalez, L. Quinto, P. J. Ventura and R. Vidal, Eur. J. Clin. Microbiol. Infect. Dis., 2000, 19, 859–867 CrossRef CAS.
- V. C. C. Cheng, W. C. Yam, I. F. N. Hung, P. C. Y. Woo, S. K. P. Lau, B. S. F. Tang and K. Y. Yuen, J. Clin. Pathol., 2004, 57, 281–285 CrossRef CAS.
- R. Lazraq, J. El Baghdadi, J. L. Guesdon and A. Benslimane, Pathol. Biol., 1999, 47, 790–796 CAS.
- D. Thierry, A. Brisson-Noël, V. Vincent-Lévy-Frébault, S. Nguyen, J. Guesdon and B. Gicquel, J. Clin. Microbiol., 1990, 28, 2668–2673 CAS.
- D. Thierry, M. D. Cave, K. D. Eisenach, J. T. Crawford, J. H. Bates, B. Gicquel and J. L. Guesdon, Nucleic Acids Res., 1990, 18, 188–192 CrossRef CAS.
- T. M. H. Lee, L. L. Li and I. M. Hsing, Langmuir, 2003, 19, 4338–4343 CrossRef CAS.
- W. L. Cheng, S. J. Dong and E. K. Wang, Anal. Chem., 2002, 74, 3599–3604 CrossRef CAS.
- H. Cui, Y. Xu and Z. Zhang, Anal. Chem., 2004, 76, 4002–4010 CrossRef CAS.
- L. Y. Fang, Z. Z. Lu, H. Wei and E. Wang, Anal. Chim. Acta, 2008, 628, 80–86 CrossRef CAS.
- Y. C. Manabe, A. M. Dannenberg and W. R. Bishai, Int. J. Tuberc. Lung Dis., 2000, 4, S18–23 CAS.
| This journal is © The Royal Society of Chemistry 2011 |