The synergistic effects of stimuli-responsive polymers with nano- structured surfaces: wettability and protein adsorption†
Qian
Yu
ab,
Xin
Li
a,
Yanxia
Zhang
ab,
Lin
Yuan
*a,
Tieliang
Zhao
ab and
Hong
Chen
*a
aCollege of Chemistry, Chemical Engineering and Materials Science, Soochow University, Suzhou, 215123, China. E-mail: chenh@suda.edu.cn; yuanl@suda.edu.cn; Fax: +86-512-65880827; Tel: +86-512-65880827
bSchool of Materials Science and Engineering, Wuhan University of Technology, Wuhan, 430070, China
First published on 2nd August 2011
Abstract
Surface modification with stimuli-responsive polymers leads to switchable wettability and bioadhesion that varies in response to environmental stimuli. The introduction of nanoscale structure onto surfaces also results in changes to the surface properties. However, the synergistic effects of stimuli-responsive polymers with nanoscale structures are unclear. In this work, two typical stimuli-responsive polymers, thermo-responsive poly(N-isopropylacrylamide) (poly(NIPAAm)) and pH-responsive poly(methacrylic acid) (poly(MAA)), were grafted from initiator-immobilized silicon nanowire arrays (SiNWAs) with nanoscale topography via surface-initiated atom transfer radical polymerization. Because of the synergistic effects of the stimuli-responsive conformation transition of polymer chains and the nano-effects of three-dimensional nanostructured SiNWAs, these new platforms possess several unique properties. Compared with their corresponding modified flat silicon surfaces, the introduction of nanoscale roughness enhanced the thermo-responsive wettability of SiNWAs-poly(NIPAAm) but weakened the pH-responsive wettability of SiNWAs-poly(MAA). More importantly, these surfaces exhibited special protein-adsorption behavior. The SiNWAs-poly(NIPAAm) surface showed good non-specific protein resistance regardless of temperature, suggesting a weakened thermo-responsivity to protein adsorption. The SiNWAs-poly(MAA) surface showed obvious enhancement of pH-dependent protein adsorption behavior.
Introduction
In recent years, nanomaterials have been widely used for various biomedical and biotechnology applications, including biosensors, drug delivery, and diagnostics.1,2 Understanding and controlling the interactions of nanomaterials with biological molecules, such as proteins is not only of great theoretical interest but also of crucial practical importance. Properties inherent to the nanomaterial, such as size, curvature and surface chemistry strongly influence the quality, secondary structure and activity of adsorbed/conjugated proteins, all of which influence biofunctionality.3–5 On the other hand, conjugation of nanomaterials with appropriate proteins offers potential opportunities for biomolecule delivery, targeted therapy and the detection of small molecules.6–9Silicon nanowires are widely used as a typical one-dimensional nanomaterial for field-effect transistors,10 biosensors11,12 and thermoelectric materials.13,14Silicon nanowire arrays (SiNWAs) have attracted increased attention because of their novel electronic properties and surface activity; they are good candidate substrates for surface-enhanced Raman scattering15 and solar cells.16 In recent years, some groups have investigated the biocompatibility of SiNWAs and broadened their application to the biomedical and biomaterial fields.17–21 It was found that the nano-topography of SiNWAs enhanced the interaction between cells and surfaces and therefore benefits cell adhesion.22,23 With proper modification, SiNWAs acquired unique wettability that enabled resistance to the adhesion of platelets24 and bacteria.25 Moreover, SiNWAs exhibited efficient tumor cell capture19 and controlled drug release20 because of their three-dimensional (3D) nanostructure. However, to the best of our knowledge, there are few reports concentrating on protein adsorption on SiNWAs.26
The interfaces that control protein adsorption and desorption through the modulation of environmental stimuli are important for many biomedical and biotechnological applications, including controlled drug delivery, separation science and biosensors.27–31 Persistent efforts have been made towards the design of such smart interfaces, and many reports concern surface modification with stimuli-responsive or “smart” polymers.32–35poly(N-isopropylacrylamide) (poly(NIPAAm)) is the best known thermo-responsive polymer,36 and numerous reports indicate that poly(NIPAAm)-modified surfaces can switch their wettability, topography and bioadhesion in response to environmental temperature in the vicinity of its lower critical solution temperature (LCST).37–40 This novel property has attracted considerable attention and can be applied to the fabrication of surfaces with switchable wettability,41 the separation of proteins and other biomolecules,42,43 and the regulation of the adhesion/detachment of cells,44platelets45 and bacteria.46Poly(methacrylic acid) (poly(MAA)) is another type of smart polymer that exhibits an extended/collapsed conformational transition as pH changes; its swelling behavior has been extensively studied.47–51 When grafted onto a solid surface, this transition results in pH-responsive changes in surface wettability and charge, which in turn influences protein-surface interactions because of the change in hydrophobic and electrostatic interactions.52
Because previous reports have shown that the stimuli-responsivity of surface wettability can be greatly enhanced by nanoscale roughness,41,53,54 it is natural to explore whether protein adsorption can also be enhanced when nanoscale topography is introduced on these smart surfaces. However, the influence of nanoscale structure on the interactions of smart surfaces with biomolecules or cells is not consistent. Chen et al. found that the introduction of nanoscale topography onto poly(NIPAAm) modified SiNWAs surfaces resulted in the disappearance of thermo-responsive platelet adhesion.24 In contrast, our recent research indicates that the poly(MAA)-modified SiNWAs surfaces exhibit significantly pH-responsive lysozyme adsorption as compared with poly(MAA)-modified flat silicon surfaces.26 These results suggest that it is important to investigate the synergistic effects of both stimuli-responsive polymers and nanoscale structures on “smart” surface properties (wettability and bioadhesion), which are not only of great theoretical interest but also of crucial importance for designing and fabricating novel devices in biomaterial and biomedical applications.
In the present study, two nanoscale surfaces of SiNWAs modified with typical smart polymers, namely thermo-responsive poly(NIPAAm) and pH-responsive poly(MAA) were prepared via surface-initiated atom-transfer radical polymerization (SI-ATRP). We investigated the synergistic effects of stimuli-responsive properties and nanoscale roughness on the surface wettability and protein adsorption. It is interesting that the introduction of nanostructure results in some novel properties, which may be favorable for biomaterial and biomedical applications. The poly(NIPAAm)-modified SiNWAs exhibit high resistance to non-specific proteins regardless of temperature, whereas the poly(MAA)-modified SiNWAs show an extremely high capacity for binding protein at low pH. Most of the adsorbed protein can be released by simply increasing the pH.
Experimental section
Materials
N-Isopropylacrylamide (NIPAAm, Acros, 99%) was recrystallized from toluene/hexane solution (50%, v/v) and dried under vacuum prior to use. tert-Butyl methacrylate (tBMA, Aldrich, 98%) was passed through a column of activated basic alumina prior to use. Copper(I) bromide (CuBr, Fluka) and Copper(II) bromide (CuBr2, Aldrich) were recrystallized before use. 3-Aminopropyltriethoxysilane (APTES, Aldrich), bromoisobutyryl bromide (BIBB, Fluka) and 1,1,4,7,7-pentamethyldiethylenetriamine (PMDETA, Aldrich) were used as received. All other solvents were purchased from Shanghai Chemical Reagent Co. and purified according to standard methods before use. Silicon wafers were purchased from Guangzhou Semiconductor Materials (Guangzhou, China). The as-received silicon wafers were cut into square chips about 0.5 cm × 0.5 cm. Deionized water was purified by a Millipore water purification system to give a minimum resistivity of 18.2 MΩ·cm and used in all experiments. Fibrinogen (MW = 341 kDa) was purchased from Calbiochem (La Jolla, CA).Preparation of polymer-grafted silicon nanowire arrays
The SiNWAs were prepared by chemical etching of crystalline silicon in HF/AgNO3 aqueous solution (the detailed procedure is given in ESI†). Then, the initiator was immobilized on the SiNWAs following the procedures reported previously.40 SI-ATRP grafting of NIPAAm was carried out in a glovebox purged with argon. NIPAAm (6.25 g, 55.23 mmol), PMDETA (0.7 mL, 3.35 mmol) and CuBr (0.16 g, 1.12 mmol) were dissolved in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of methanol and water (25 mL). The reaction solution was sonicated for 2 min and then added to a glass vessel, into which the initiator-functionalized SiNWAs were also placed. The polymerization was carried out at room temperature for 2 h. The obtained SiNWAs-poly(NIPAAm) surfaces were rinsed with deionized water and dried under an argon flow. SI-ATRP grafting of tBMA was carried out as follows. tBMA (4 mL, 25 mmol), CuBr2 (3.3 mg, 0.015 mmol), CuBr (7.15 mg, 0.05 mmol), PMDETA (0.031 mL, 0.15 mmol) and 3 mL acetone were mixed in a flask. The heterogeneous reaction solution was degassed with three freeze–pump–thaw cycles and then stirred at 60 °C for 20 min until it became clear and homogeneous. The solution was then transferred to a Schlenk flask containing the initiator-functionalized SiNWAs via syringe. The polymerization was allowed to proceed at 60 °C for 4 h. The obtained SiNWAs-poly(tBMA) surfaces were rinsed thoroughly with acetone, dried under an argon flow, and placed in a flask containing a mixture of 1,4-dioxane (20 mL) and concentrated HCl (37%, 3 mL). The reaction was carried out at 80 °C for 2 h to hydrolyze poly(tBMA) to poly(MAA). The resulting surfaces were thoroughly rinsed with acetone and ethanol and dried under an argon flow. For comparison, polymer-grafted smooth silicon surfaces (Si-poly(NIPAAm) and Si-poly(MAA)) were also prepared following the same procedures mentioned above.
:1 mixture of methanol and water (25 mL). The reaction solution was sonicated for 2 min and then added to a glass vessel, into which the initiator-functionalized SiNWAs were also placed. The polymerization was carried out at room temperature for 2 h. The obtained SiNWAs-poly(NIPAAm) surfaces were rinsed with deionized water and dried under an argon flow. SI-ATRP grafting of tBMA was carried out as follows. tBMA (4 mL, 25 mmol), CuBr2 (3.3 mg, 0.015 mmol), CuBr (7.15 mg, 0.05 mmol), PMDETA (0.031 mL, 0.15 mmol) and 3 mL acetone were mixed in a flask. The heterogeneous reaction solution was degassed with three freeze–pump–thaw cycles and then stirred at 60 °C for 20 min until it became clear and homogeneous. The solution was then transferred to a Schlenk flask containing the initiator-functionalized SiNWAs via syringe. The polymerization was allowed to proceed at 60 °C for 4 h. The obtained SiNWAs-poly(tBMA) surfaces were rinsed thoroughly with acetone, dried under an argon flow, and placed in a flask containing a mixture of 1,4-dioxane (20 mL) and concentrated HCl (37%, 3 mL). The reaction was carried out at 80 °C for 2 h to hydrolyze poly(tBMA) to poly(MAA). The resulting surfaces were thoroughly rinsed with acetone and ethanol and dried under an argon flow. For comparison, polymer-grafted smooth silicon surfaces (Si-poly(NIPAAm) and Si-poly(MAA)) were also prepared following the same procedures mentioned above.
Surface characterization
The chemical composition of the modified silicon surfaces was determined with an ESCALAB MK II X-ray photoelectron spectrometer (XPS) (VG Scientific Ltd.). All XPS data were analyzed using XPS Peak 4.1 software. The thicknesses of the polymer grafts on the smooth silicon substrate were measured by an M-88 spectroscopic ellipsometer (J. A. Woollam Co., Inc.). The nanostructures and surface morphology of pristine and modified SiNWAs were observed using scanning electron microscopy (SEM, S-4800, Japan).Contact angle measurements
Contact angle (CA) measurements were performed using an SL-200C optical contact angle meter (Solon Information Technology Co., Ltd.) with a heating element placed on the sample stage to control the temperature of the sample surfaces. The static water contact angles were measured using the sessile drop method in the dry state. For poly(MAA)-grafted surfaces, the samples were first immersed in phosphate buffered saline (PBS) at a specified pH for 30 min and dried immediately in an argon flow. The pH of the PBS was pre-adjusted by adding aqueous NaOH or HCl solution until the desired values were reached. To further investigate the wettability of the surfaces in the wet state, the oil (dichloromethane, CH2Cl2) contact angles and captive bubble contact angles in PBS under different temperatures (for poly(NIPAAm)-grafted surfaces) or at different pH values (for poly(MAA)-grafted surfaces) were also measured.Protein adsorption
Fibrinogen adsorption from PBS solution (1 mg mL−1) was determined by radiolabeling with 125I using a Wizard 3′′1480 Automatic Gamma Counter (Perkin-Elmer Life Sciences).40PBS at different pH levels was used to prepare protein solutions. Adsorption was allowed to proceed for 3 h under static conditions. For PNIPAAm-grafted surfaces, the adsorption temperatures were chosen at either room temperature (23 °C) or body temperature (37 °C) at pH 7.4; for PMAA-grafted surfaces, the pH of protein solutions were 4 or 9 at 23 °C. Because it is very difficult to calculate the absolute surface area of SiNWAs, the amount of protein adsorption is expressed as μg/disc (for one disc, the “apparent” surface area is 0.5 cm2).Results and discussion
Characterization of SiNWAs modified with polymers
The general process for the formation of poly(NIPAAm) or poly(MAA) brushes on SiNWAs (SiNWAs-poly(NIPAAm) or SiNWAs-poly(MAA)) is illustrated in Scheme 1. First, the SiNWAs were prepared by a chemical etching method, as previously reported.55 The length and diameter of the resulting SiNWAs depended on the etching time; homogeneous SiNWAs were prepared with an optimized etching time (Fig. S1, ESI†). The initiator was immobilized, followed by SI-ATRP of NIPAAm or tBMA using these surfaces as substrates. The poly(tBMA) chains were further hydrolyzed in acidic solution to obtain the corresponding poly(MAA) chains. For comparison, polymers grafted onto smooth silicon surfaces (Si-poly(NIPAAm) and Si-poly(MAA)) were also prepared following the same procedures. The resulting poly(NIPAAm)- and poly(MAA)-grafted layers had a thickness of ∼39.2 nm and ∼18.5 nm, respectively, as determined by ellipsometry. These values are consistent with the values reported elsewhere.40,56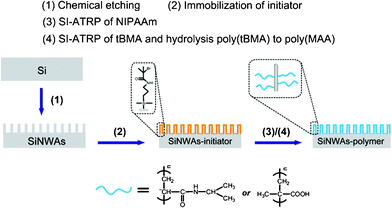 | ||
| Scheme 1 Process for grafting stimuli-responsive polymer (poly(NIPAAm) or poly(MAA)) from SiNWAs. | ||
Changes in the chemical composition of SiNWAs after the grafting of the polymers were determined from XPS data. The survey spectra of the poly(NIPAAm)- and poly(MAA)-grafted surfaces are shown in Fig. 1, and the corresponding chemical composition of these surfaces is summarized in Table 1. The experimental atomic ratios for the resulting surfaces were close to the theoretical values, suggesting a successful modification process. The nanostructures and surface morphology of unmodified and modified SiNWAs were observed using SEM. The top view and cross-sectional view are shown in Fig. 2, indicating a unique nanoscale porous-type structure. All the nanowires were observed to have similar diameters and lengths, and the majority were erect and distributed evenly. The nanowires having diameters of about 100 nm and lengths of about 30 μm, combined together to form nanoclusters. The unique nanoscale topography was preserved after polymer modification.
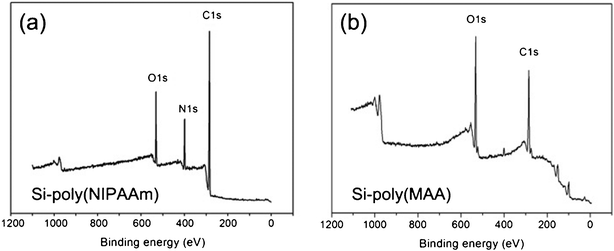 | ||
| Fig. 1 XPS survey spectra of (a) SiNWAs-poly(NIPAAm) and (b) SiNWAs-poly(MAA) surfaces. | ||
 | ||
| Fig. 2 SEM images of (a), (b) unmodified and (c), (d) polymer modified SiNWAs. (a) and (c): top view; (b) and (d): cross-sectional view. | ||
| Surface | C | N | O | C/O |
|---|---|---|---|---|
| SiNWAs-poly(NIPAAm) | 74.6 | 12.2 | 13.2 | 5.65 |
| theory | 75.0 | 12.5 | 12.5 | 6.00 |
| SiNWAs-poly(MAA) | 65.1 | 0.8 | 34.1 | 1.91 |
| theory | 66.7 | — | 33.3 | 2.00 |
Stimuli-responsivity of surface wettability
Stimuli-responsive polymers can display a reversible expanded/collapsed conformational transition in response to environmental stimuli changes.57 When they are immobilized onto a solid surface, the conformational change of chains will definitely influence the surface properties and typically will lead to tunable surface wettability.58,59 Based on many previous reports, the introduction of nanoscale roughness onto a surface will significantly affect surface wettability.60,61 Therefore, it is of interest to investigate the influence of the nanostructure of SiNWAs on the wettability.Fig. 3a shows the water contact angle of Si-poly(NIPAAm) and SiNWAs-poly(NIPAAm) surfaces as a function of temperature. For both surfaces, an obvious jump in the water contact angle occurred between 31–35 °C. The thermo-responsive wettability change was significantly enhanced by the nanotopography of SiNWAs. As indicated in Fig. 3b, as the temperature increased from 23 °C to 37 °C, the difference in water CA value was ∼14° for the Si-poly(NIPAAm) surface and ∼120° for the SiNWAs-poly(NIPAAm) surface. This nano-enhanced thermo-responsive wettability was consistent with another report.24
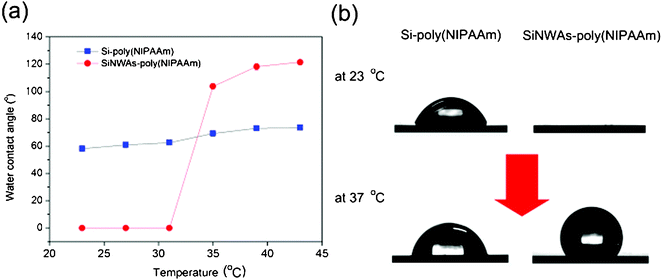 | ||
| Fig. 3 (a) Water contact angle of Si-poly(NIPAAm) and SiNWAs-poly(NIPAAm) surfaces as a function of temperature. The typical contact angle images for these surfaces at both 23 °C and 37 °C are shown in (b). Each value is the average of six parallel measurements. The standard error is less than 2°. | ||
Because further protein adsorption was carried out in PBS solution, it was important to investigate the surface wettability in the liquid phase. In this work, two methods were used: the captive bubble method to measure the water contact angle and the sessile drop method to measure the oil (CH2Cl2) contact angle in PBS. These tests were conducted under various temperatures, and the results are summarized in Table 2. It is suggested that the oil contact angle reflects the surface hydrophilicity in the wetted state because a hydrophilic surface in a water/air/solid system shows oleophobic behavior in an oil/water/solid system.62 In PBS, the two surfaces exhibited different wettability. For the SiNWAs-poly(NIPAAm) surface, the CA value was almost unchanged as the temperature increased, suggesting that the thermo-responsivity weakened or even disappeared. The surface showed superhydrophilic and superoleophobic properties independent of temperature. We believe this phenomenon results from the nanoscale porous structure of SiNWAs, which harbors water molecules.24 From another point of view, it is suggested that in aqueous solution the grafted poly(NIPAAm) chains will swell and exhibit an extended conformation. These swollen chains may “block up” the gaps between the silicon nanowires, resulting in the transition from a rough surface to a smooth one. This transition may also contribute to the specific wettability of the SiNWAs-poly(NIPAAm) surface in PBS.
| contact angle (°) a | Si-poly(NIPAAm) | SiNWAs-poly(NIPAAm)b | |
|---|---|---|---|
| a Each value is the average of six parallel measurements. The standard error is less than 2°. b - indicates that it was difficult to adhere the bubble to the surface. | |||
| at 23 °C | in air | 58.1 | 0 |
| in PBS (bubble) | 39.7 | - | |
| in PBS (CH2Cl2) | 129.2 | 145.6 | |
| at 37 °C | in air | 72.1 | 118.7 |
| in PBS (bubble) | 43.7 | - | |
| in PBS (CH2Cl2) | 110.3 | 151.2 | |
We also investigated the influence of nanostructure on the wettability of poly(MAA) grafted surfaces in response to pH. Poly(MAA) is a typical weak polyacid, and the change in pH will control the extent of ionization and further conformational change of the polymer chains.47 As shown in Fig. 4a, there was a concomitant decrease in the CA value of the smooth Si-poly(MAA) surface as the pH increased from 4 to 6; a further increase in pH led to an obvious drop in CA value from ∼45° to ∼15°. This enhanced hydrophilicity of the poly(MAA) brush in response to increasing pH has been previously reported and can be explained by a conformational transition of the polymer chains. 51 For poly(MAA), the addition of a base will deprotonate the pendant acidic groups, resulting in swollen polymer chains with many ionized COO−, which favors the entrance of water molecules and makes the surface more hydrophilic. However, in the case of the SiNWAs-poly(MAA) surface, the pH-responsive wettability disappeared because the surface remained superhydrophilic (with a CA value of ∼0°). The introduction of nanoscale roughness is believed to make the surface superhydrophilic independent of pH; in the measured pH range, the CA value of the flat Si-poly(MAA) surface was lower than 65°, suggesting a hydrophilic surface.63 The CA values measured in PBS also indicated the superhydrophilicity and superoleophobicity of SiNWAs-poly(MAA) surface (Table 3). Both the air bubble and oil drop easily rolled on the surface and were difficult to adhere (Fig. S2, ESI†).
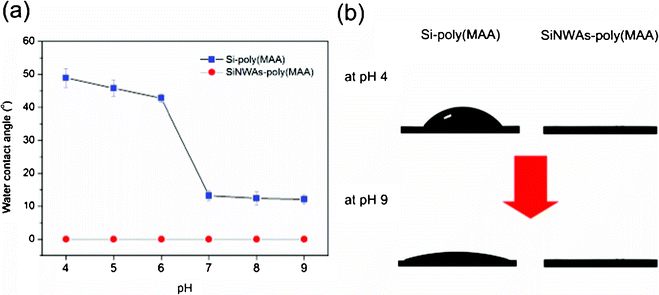 | ||
| Fig. 4 (a) Water contact angle of Si-poly(MAA) and SiNWAs-poly(MAA) surfaces as a function of pH. The typical contact angle images for these surfaces at both pH 4 and pH 9 are shown in (b). Each value is the average of six parallel measurements. | ||
| contact angle (°) a | Si-poly(MAA)b | SiNWAs-poly(MAA)b | |
|---|---|---|---|
| a Each value is the average of six parallel measurements. The standard error is less than 2°. b - indicates that it was difficult to adhere the bubble to the surface. -- means the surface is superoleophobic, with a CA value above 150° | |||
| at pH 4 | in air | 48.9 | 0 |
| in PBS (bubble) | 29.8 | - | |
| in PBS (CH2Cl2) | 136.4 | -- | |
| at pH 9 | in air | 12.1 | 0 |
| in PBS (bubble) | - | - | |
| in PBS (CH2Cl2) | -- | -- | |
Stimuli-responsivity of protein adsorption
The results of the contact angle measurements show that the combination of nanostructure and smart polymers will endow the modified polymer with novel surface wettability in response to environmental stimuli. It is well known that surface wettability is one of the most important factors for protein adsorption on biomaterial surfaces.64,65 Therefore, we further measured protein adsorption on these modified surfaces under different conditions to investigate the effects of surface nanostructure and stimuli-responsive conformational change on polymer chains. Fibrinogen (Fg) was used as a model protein because it is abundant in plasma and plays a major role in blood coagulation.66,67Using poly(NIPAAm)-modified surfaces, adsorption from 1 mg mL−1Fg in PBS solution was measured at both room temperature (23 °C) and body temperature (37 °C). As shown in Fig. 5a, as the temperature increased, the adsorption on the smooth silicon surface increased ∼10%, whereas that of the Si-poly(NIPAAm) surface increased ∼110% because of the enhanced hydrophobicity resulting from the thermo-responsive transition of poly(NIPAAm) chains.40 From another point of view, the Si-poly(NIPAAm) surface exhibits good protein-resistant properties. The adsorption onto one disc (the surface area of one disc is 0.5 cm2) is ∼50 ng even at 37 °C, corresponding to a reduction of ∼90% as compared with the unmodified Si surface. This antifouling property of the poly(NIPAAm) layer is consistent with previous reports.68,69 The introduction of nanoscale roughness onto the smooth samples resulted in a significant increase in surface area, which led to an obvious increase in the amount of adsorption as compared with the unmodified Si surface. However, it is interesting that there was no distinct difference in protein adsorption between the Si-poly(NIPAAm) and SiNWAs-poly(NIPAAm) surfaces. Compared with the SiNWAs surface, the SiNWAs-poly(NIPAAm) reduced more than ∼99% of Fg adsorption, regardless of temperature (Fig. 5b). It is suggested that this high protein resistance resulted from the introduction of this nanoscale structure. More water molecules were trapped in the interstices of the nanowire arrays and formed a strong hydration layer, which prevents intimate molecular contact between proteins and the surface. This water-trapping effect was clear from the contact angle (Table 2) and adhesive force measurements24 in aqueous solution. Recently, Chen et al. found that SiNWAs-poly(NIPAAm) surfaces showed largely reduced platelet adhesion in vitro both below and above the LCST.24 This phenomenon can be explained by the reduction of Fg adsorption on the surface because it is well accepted that Fg plays a crucial role in platelet adhesion and activation.66 To investigate this antifouling aspect further, we measured the adsorption of two other typical proteins, lysozyme and human serum albumin (HSA), with very different charge and size70,71 (Fig. S3, ESI†). These results show that the SiNWAs-poly(NIPAAm) surface exhibited effective nonspecific protein resistance. Therefore, this novel surface is a promising candidate for biomaterial and biomedical applications when nonfouling properties are required.
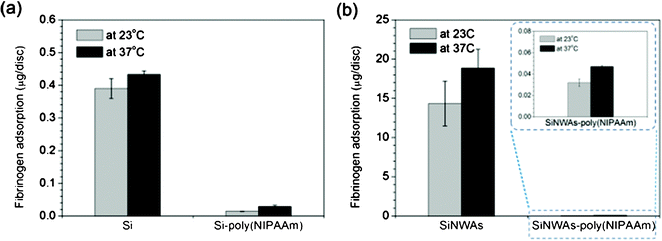 | ||
| Fig. 5 Adsorption of 1 mg mL−1fibrinogen from PBS solution over a 3 h period on (a) pristine and poly(NIPAAm)-modified silicon surfaces and (b) pristine and poly(NIPAAm)-modified SiNWAs surfaces at 23 °C and 37 °C. For SiNWAs samples, the “apparent” surface area of one disc is 0.5 cm2. Data consist of the mean ± standard error (n=3). | ||
The possible pH-responsive protein adsorption on poly(MAA)-modified surfaces was also investigated. It was shown that Fg adsorption on smooth silicon surfaces at pH 9 was slightly lower than at pH 4, whereas after modification with poly(MAA), the effect of pH was greater. The adsorption at pH 4 was ∼38 times higher than that at pH 9, but the amount of adsorbed Fg is still lower than 4 μg/disc (Fig. 6a). The introduction of nanoscale roughness onto the smooth Si substrate led to an obvious increase in the amount of adsorption as a result of the enlarged surface area, but the pH-responsiveness was not enhanced. Interestingly, the combination of pH-sensitivity of poly(MAA) chains and the nano-effects of 3D nanostructured SiNWAs gave the SiNWAs-poly(MAA) material an extremely high capacity for binding Fg at pH 4, with adsorption greater than 70 μg per disc, i.e., a more than 70-fold increase compared with smooth silicon surface and a ∼20-fold increase compared with smooth Si-poly(MAA) (Fig. 6b). As shown in Fig. 6c, the differences in adsorbed protein and the ratio of adsorbed quality between pH 4 and pH 9 on SiNWAs-poly(MAA) surface were greatest. This significant pH-sensitive protein adsorption may be attributed to the synergistic effect between the conformational transition of poly(MAA) chains and nano-enhanced effects of SiNWAs.19,72 At pH 4, the poly(MAA) chains are collapsed and more hydrophobic, and hydrogen bonds might be formed between the -COOH groups on poly(MAA) chains and -CONH groups on proteins, resulting in an increase in protein adsorption. In addition, the collapsed poly(MAA) chains may favor access of Fg to the interstices of the nanowire arrays. At pH 9, the -COOH groups are ionized to -COO−groups, resulting in extended hydrophilic chains with negative charges. Because Fg also carries negative charges, a reduction in hydrophobic interaction and enhanced electrostatic repulsion lead to reduced protein adsorption at pH 9. On the other hand, it is assumed that interactions between Fg and poly(MAA) are significantly enhanced by the nanostructure of SiNWAs. Water molecules are favored to be trapped in the interstices of the nanowire arrays, giving a high degree of hydration and reducing protein adsorption, especially under basic conditions at pH 9. This result is consistent with our previous report,26 suggesting that the introduction of nanoscale structure onto poly(MAA)-modified surfaces leads to significantly enhanced pH-responsivity of protein adsorption, although the pH-responsive wettability is weakened.
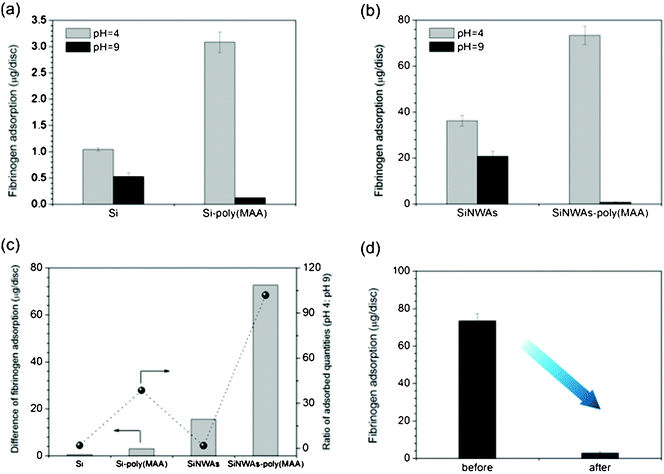 | ||
| Fig. 6 Adsorption of 1 mg mL−1fibrinogen from PBS solution over a 3 h period on sample surfaces at pH 4 and pH 9. (a) Comparison of pristine and poly(MAA)-modified silicon surfaces; (b) comparison of pristine and poly(MAA)-modified SiNWAs surfaces; (c) the difference in adsorption and ratio of adsorbed quantities between pH 4 and pH 9; (d) the remaining adsorbed protein at pH 4 before and after incubation in PBS at pH 9 for 3 h. For SiNWAs samples, the “apparent” surface area of one disc is 0.5 cm2. Data consist of the mean ± standard error (n=3). | ||
The significant adsorption difference motivated us to question whether the Fg adsorbed at pH 4 can be desorbed by increasing the pH. To that end, SiNWAs-poly(MAA) samples were allowed to adsorb Fg at pH 4. They were then incubated in PBS at pH 9 for 3 h, and the quantity of the remaining adsorbed Fg was measured. For one disc, more than 70 μg (corresponding to ∼96%) adsorbed Fg was released from substrate into solution by simply increasing the pH (Fig. 6d). Such pH-controlled binding of proteins is of great importance for many biomedical applications, including protein delivery and bioseparations.73,74 The potentially reversible process of adsorption and release by cycled pH change will be tested in a future study.
Conclusion
In summary, two stimuli-responsive polymers, poly(NIPAAm) and poly(MAA) were grafted from initiator-immobilized SiNWAs with nanoscale topography. The two resulting polymer-coated surfaces exhibited different wettability and protein adsorption in response to environmental stimuli. For the SiNWAs-poly(NIPAAm) surface, changes in temperature from 23 °C to 37 °C lead to a significant change in wettability in the dry state (from superhydrophilic (water CA ∼0°) to strongly hydrophobic (water CA ∼120°)), but no obvious difference in protein adsorption. This surface exhibited good non-specific protein resistance regardless of temperature, probably because the nanoscale topography bound many water molecules to form a strong hydration layer. In contrast, for SiNWAs-poly(MAA) surface, the introduction of nanoscale roughness resulted in a superhydrophilic surface in the treatment pH range (4 to 9), suggesting the disappearance of pH-responsive wettability. However, the pH-responsivity of protein adsorption was enlarged. These results indicated that the introduction of the same nanoscale structure onto surfaces modified with different stimuli-responsive polymers leads to different effects on surface properties. Moreover, for surfaces with nanotopography, surface wettability and bioadhesion may not show consistent trends. This surface showed an extremely high capacity for binding protein at pH 4, and most of the adsorbed protein can be released by simply increasing pH. Based on our results and taking into account the good biocompatibility of SiNWAs, we believe that these surfaces will find applications in biomedical devices, protein delivery and biosensors. In addition, the work presented here also offers a new strategy for designing and fabricating other nanomaterials with special properties.Acknowledgements
This work was supported by the National Natural Science Foundation (21074083, 20974086, and 20920102035). We are grateful to Professor Hongwei Ma's group for assistance with SEM measurements.References
- P. Asuri, S. S. Bale, S. S. Karajanagi and R. S. Kane, Curr. Opin. Biotechnol., 2006, 17, 562–568 CrossRef CAS.
- R. S. Kane and A. D. Stroock, Biotechnol. Prog., 2007, 23, 316–319 CrossRef CAS.
- D. D. Deligianni, N. Katsala, S. Ladas, D. Sotiropoulou, J. Amedee and Y. F. Missirlis, Biomaterials, 2001, 22, 1241–1251 CrossRef CAS.
- K. Rechendorff, M. B. Hovgaard, M. Foss, V. P. Zhdanov and F. Besenbacher, Langmuir, 2006, 22, 10885–10888 CrossRef CAS.
- P. Roach, D. Farrar and C. C. Perry, J. Am. Chem. Soc., 2006, 128, 3939–3945 CrossRef CAS.
- N. Kam, T. Jessop, P. Wender and H. Dai, J. Am. Chem. Soc., 2004, 126, 6850–6851 CrossRef CAS.
- I. Medintz, H. Uyeda, E. Goldman and H. Mattoussi, Nat. Mater., 2005, 4, 435–446 CrossRef CAS.
- D. Pantarotto, J. Briand, M. Prato and A. Bianco, Chem. Commun., 2004, 16–17 RSC.
- F. Patolsky and C. Lieber, Mater. Today, 2005, 8, 20–28 CrossRef CAS.
- Y. Cui and C. M. Lieber, Science, 2001, 291, 851–853 CrossRef CAS.
- Y. Cui, Q. Q. Wei, H. K. Park and C. M. Lieber, Science, 2001, 293, 1289–1292 CrossRef CAS.
- F. Patolsky, B. P. Timko, G. H. Yu, Y. Fang, A. B. Greytak, G. F. Zheng and C. M. Lieber, Science, 2006, 313, 1100–1104 CrossRef CAS.
- A. I. Boukai, Y. Bunimovich, J. Tahir-Kheli, J. K. Yu, W. A. Goddard and J. R. Heath, Nature, 2008, 451, 168–171 CrossRef CAS.
- A. I. Hochbaum, R. K. Chen, R. D. Delgado, W. J. Liang, E. C. Garnett, M. Najarian, A. Majumdar and P. D. Yang, Nature, 2008, 451, 163 CrossRef CAS.
- M. Becker, V. Sivakov, U. Gosele, T. Stelzner, G. Andra, H. J. Reich, S. Hoffmann, J. Michler and S. H. Christiansen, Small, 2008, 4, 398–404 CrossRef CAS.
- K. Q. Peng, X. Wang, X. L. Wu and S. T. Lee, Nano Lett., 2009, 9, 3704–3709 CrossRef CAS.
- H. Wang, L. Wang, P. Zhang, L. Yuan, Q. Yu and H. Chen, Colloids Surf., B, 2011, 83, 355–359 CrossRef CAS.
- L. Yuan, H. Wang, Q. Yu, Z. Wu, J. L. Brash and H. Chen, J. Mater. Chem., 2011, 21, 6148–6151 RSC.
- S. T. Wang, H. Wang, J. Jiao, K. J. Chen, G. E. Owens, K. I. Kamei, J. Sun, D. J. Sherman, C. P. Behrenbruch, H. Wu and H. R. Tseng, Angew. Chem., Int. Ed., 2009, 48, 8970–8973 CrossRef CAS.
- K. S. Brammer, C. Choi, S. Oh, C. J. Cobb, L. S. Connelly, M. Loya, S. D. Kong and S. Jin, Nano Lett., 2009, 9, 3570–3574 CrossRef CAS.
- S. J. Qi, C. Q. Yi, S. L. Ji, C. C. Fong and M. S. Yang, ACS Appl. Mater. Interfaces, 2009, 1, 30–34 CAS.
- Z. Li, J. Song, G. Mantini, M. Y. Lu, H. Fang, C. Falconi, L. J. Chen and Z. L. Wang, Nano Lett., 2009, 9, 3575–3580 CrossRef CAS.
- S. J. Qi, C. Q. Yi, W. W. Chen, C. C. Fong, S. T. Lee and M. S. Yang, ChemBioChem, 2007, 8, 1115–1118 CrossRef CAS.
- L. Chen, M. Liu, H. Bai, P. Chen, F. Xia, D. Han and L. Jiang, J. Am. Chem. Soc., 2009, 131, 10467–10472 CrossRef CAS.
- E. Galopin, G. Piret, S. Szunerits, Y. Lequette, C. Faille and R. Boukherroub, Langmuir, 2010, 26, 3479–3484 CrossRef CAS.
- Q. Yu, H. Chen, Y. Zhang, L. Yuan, T. Zhao, X. Li and H. Wang, Langmuir, 2010, 26, 17812–17815 CrossRef CAS.
- C. D. H. Alarcon, S. Pennadam and C. Alexander, Chem. Soc. Rev., 2005, 34, 276–285 RSC.
- M. Yamato, Y. Akiyama, J. Kobayashi, J. Yang, A. Kikuchi and T. Okano, Prog. Polym. Sci., 2007, 32, 1123–1133 CrossRef CAS.
- J. F. Mano, Adv. Eng. Mater., 2008, 10, 515–527 CrossRef CAS.
- H. Nandivada, A. M. Ross and J. Lahann, Prog. Polym. Sci., 2010, 35, 141–154 CrossRef CAS.
- M. Motornov, Y. Roiter, I. Tokarev and S. Minko, Prog. Polym. Sci., 2010, 35, 174–211 CrossRef CAS.
- P. M. Mendes, Chem. Soc. Rev., 2008, 37, 2512–2529 RSC.
- F. Xia, Y. Zhu, L. Feng and L. Jiang, Soft Matter, 2009, 5, 275–281 RSC.
- M. A. Cole, N. H. Voelcker, H. Thissen and H. J. Griesser, Biomaterials, 2009, 30, 1827–1850 CrossRef CAS.
- M. A. C. Stuart, W. T. S. Huck, J. Genzer, M. Muller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban, F. Winnik, S. Zauscher, I. Luzinov and S. Minko, Nat. Mater., 2010, 9, 101–113 CrossRef.
- H. G. Schild, Prog. Polym. Sci., 1992, 17, 163–249 CrossRef CAS.
- D. Cunliffe, C. D. Alarcon, V. Peters, J. R. Smith and C. Alexander, Langmuir, 2003, 19, 2888–2899 CrossRef CAS.
- X. H. Cheng, H. E. Canavan, M. J. Stein, J. R. Hull, S. J. Kweskin, M. S. Wagner, G. A. Somorjai, D. G. Castner and B. D. Ratner, Langmuir, 2005, 21, 7833–7841 CrossRef CAS.
- E. C. Cho, D.-H. Kim and K. Cho, Langmuir, 2008, 24, 9974–9978 CrossRef CAS.
- Q. Yu, Y. Zhang, H. Chen, Z. Wu, H. Huang and C. Cheng, Colloids Surf., B, 2010, 76, 468–474 CrossRef CAS.
- T. L. Sun, G. J. Wang, L. Feng, B. Q. Liu, Y. M. Ma, L. Jiang and D. B. Zhu, Angew. Chem., Int. Ed., 2004, 43, 357–360 CrossRef CAS.
- D. L. Huber, R. P. Manginell, M. A. Samara, B.-I. Kim and B. C. Bunker, Science, 2003, 301, 352–354 CrossRef CAS.
- K. Nagase, J. Kobayashi, A. Kikuchi, Y. Akiyama, H. Kanazawa and T. Okano, Langmuir, 2007, 23, 9409–9415 CrossRef CAS.
- N. Matsuda, T. Shimizu, M. Yamato and T. Okano, Adv. Mater., 2007, 19, 3089–3099 CrossRef CAS.
- K. Uchida, K. Sakai, E. Ito, O. Hyeong Kwon, A. Kikuchi, M. Yamato and T. Okano, Biomaterials, 2000, 21, 923–929 CrossRef CAS.
- C. D. H. Alarcon, T. Farhan, V. L. Osborne, W. T. S. Huck and C. Alexander, J. Mater. Chem., 2005, 15, 2089–2094 RSC.
- M. Biesalski, D. Johannsmann and J. Ruhe, J. Chem. Phys., 2002, 117, 4988–4994 CrossRef CAS.
- R. Konradi and J. Ruhe, Macromolecules, 2005, 38, 4345–4354 CrossRef CAS.
- H. Zhang and J. Ruhe, Macromolecules, 2005, 38, 4855–4860 CrossRef CAS.
- R. Heeb, R. M. Bielecki, S. Lee and N. D. Spencer, Macromolecules, 2009, 42, 9124–9132 CrossRef CAS.
- A. J. Parnell, S. J. Martin, C. C. Dang, M. Geoghegan, R. A. L. Jones, C. J. Crook, J. R. Howse and A. J. Ryan, Polymer, 2009, 50, 1005–1014 CrossRef CAS.
- H. Chen, L. Yuan, W. Song, Z. Wu and D. Li, Prog. Polym. Sci., 2008, 33, 1059–1087 CrossRef CAS.
- F. Xia, H. Ge, Y. Hou, T. L. Sun, L. Chen, G. Z. Zhang and L. Jiang, Adv. Mater., 2007, 19, 2520–2524 CrossRef CAS.
- F. Xia, L. Feng, S. T. Wang, T. L. Sun, W. L. Song, W. H. Jiang and L. Jiang, Adv. Mater., 2006, 18, 432–436 CrossRef CAS.
- K. Q. Peng, Z. P. Huang and J. Zhu, Adv. Mater., 2004, 16, 73–76 CrossRef CAS.
- N. Ayres, S. G. Boyes and W. J. Brittain, Langmuir, 2007, 23, 182–189 CrossRef CAS.
- E. S. Gil and S. A. Hudson, Prog. Polym. Sci., 2004, 29, 1173–1222 CrossRef CAS.
- F. Xia and L. Jiang, Adv. Mater., 2008, 20, 2842–2858 CrossRef CAS.
- T. Chen, R. Ferris, J. Zhang, R. Ducker and S. Zauscher, Prog. Polym. Sci., 2010, 35, 94–112 CrossRef CAS.
- D. Qurere, Annu. Rev. Mater. Res., 2008, 38, 71–99 CrossRef.
- M. Liu, Y. Zheng, J. Zhai and L. Jiang, Acc. Chem. Res., 2010, 43, 368–377 CrossRef CAS.
- M. J. Liu, S. T. Wang, Z. X. Wei, Y. L. Song and L. Jiang, Adv. Mater., 2009, 21, 665–669 CrossRef CAS.
- E. A. Vogler, Adv. Colloid Interface Sci., 1998, 74, 69–117 CrossRef CAS.
- A. Sethuraman, M. Han, R. S. Kane and G. Belfort, Langmuir, 2004, 20, 7779–7788 CrossRef CAS.
- K. L. Prime and G. M. Whitesides, J. Am. Chem. Soc., 1993, 115, 10714–10721 CrossRef CAS.
- D. Kwak, Y. G. Wu and T. A. Horbett, J. Biomed. Mater. Res., Part A, 2005, 74A, 69–83 CrossRef CAS.
- M. C. Shen, M. S. Wagner, D. G. Castner, B. D. Ratner and T. A. Horbett, Langmuir, 2003, 19, 1692–1699 CrossRef CAS.
- Q. Yu, Y. Zhang, H. Chen, F. Zhou, Z. Wu, H. Huang and J. L. Brash, Langmuir, 2010, 26, 8582–8588 CrossRef CAS.
- S. Burkert, E. Bittrich, M. Kuntzsch, M. Muller, K.-J. Eichhorn, C. Bellmann, P. Uhlmann and M. Stamm, Langmuir, 2010, 26, 1786–1795 CrossRef CAS.
- A. M. Brzozowska, B. Hofs, A. de Keizer, R. Fokkink, M. A. Cohen Stuart and W. Norde, Colloids Surf., A, 2009, 347, 146–155 CrossRef CAS.
- J. Kim and G. A. Somorjai, J. Am. Chem. Soc., 2003, 125, 3150–3158 CrossRef CAS.
- W. M. de Vos, P. M. Biesheuvel, A. de Keizer, J. M. Kleijn and M. A. Cohen Stuart, Langmuir, 2008, 24, 6575–6584 CrossRef CAS.
- B. K. Nfor, P. D. E. M. Verhaert, L. A. M. van der Wielen, J. Hubbuch and M. Ottens, Trends Biotechnol., 2009, 27, 673–679 CrossRef CAS.
- D. Schmaljohann, Adv. Drug Delivery Rev., 2006, 58, 1655–1670 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available: Detailed procedures of preparation and optimization of SiNWAs, contact angle images of oil drop and bubble in PBS, and HSA and lysozyme adsorption on SiNWAs and SiNWAs-poly(NIPAAm) surfaces. See DOI:10.1039/c1ra00201e/ |
| This journal is © The Royal Society of Chemistry 2011 |