Varying structural motifs in oxyanions (NO3−, CO32−) and phenoxyacetate (PhOAc−) bridged coordination polymers derived from alkoxo-bridged dicopper building blocks with {Cu2O2} core†
Shaikh M.
Mobin
abc,
Ashwini K.
Srivastava
*c,
Pradeep
Mathur
*cbd and
Goutam Kumar
Lahiri
*ab
aNational Single Crystal X-ray Diffraction Facility, IIT Bombay, Powai, Mumbai, 400076, India
bDepartment of Chemistry, IIT Bombay, Powai, Mumbai, 400076, India
cDepartment of Chemistry, University of Mumbai, Vidyanagari, Mumbai, 400098, India
dSchool of Sciences, Indian Institute of Technology Indore, Khandwa Road, Indore, 452017, India
First published on 12th September 2011
Abstract
The oxyanions (NO3−, CO32−) and phenoxyacetate (PhOAc−) bridged three 1D-coordination polymeric chains, {[Cu2(μ-hep)2(μ-NO3)]2}n (1), {[Cu2(μ-hep)2(H2O)2]·2H2O(μ-CO3)}n (3) and {[Cu2(μ-hep)2(μ-PhOAc−)]2}n (2) (hep-H = 2-(2-hydroxyethyl)pyridine) have been synthesized. In 1–3 the alkoxide bridged dicopper building units, [Cu(μ-hep)]2 with Cu2O2 core, are linked via the respective anions. Detailed structural analysis reveals that in 1 or 2, two units of NO3− (1) or PhOAc− (2), respectively, bind with the four copper ions in two adjacent alkoxide bridged dimeric units in head-to-head and tail-to-tail fashion and the same binding mode continues along the polymeric chain. This in effect yields a 12-membered metallacyclic ring in between two dimeric core units. However, in 3 only one CO32−group bridges the two copper centres associated with the two neighbouring alkoxide bridged dimeric units in head-to-tail mode which in turn forms a zig-zag polymeric chain. Two coordinated and two lattice water molecules from two adjacent polymeric layers in the structure of 3 form water tetramers. Furthermore, the interaction of water tetramer with the uncoordinated –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group of the bridging CO32− develops an additional zig-zag chain which is being trapped between the two outer zig-zag coordination polymeric chains in 3. The polymeric chains in 1–3 further develop a 2D-network pattern via an extensive non-covalent hydrogen bonding as well as C–H⋯π and π⋯π interactions.
O group of the bridging CO32− develops an additional zig-zag chain which is being trapped between the two outer zig-zag coordination polymeric chains in 3. The polymeric chains in 1–3 further develop a 2D-network pattern via an extensive non-covalent hydrogen bonding as well as C–H⋯π and π⋯π interactions.
Introduction
A convenient practice for fabrication of metal complex derived coordination polymeric materials is to use suitable bridging ligands or spacers such as pyrazine,1bis(4-pyridyl) derivatives: 4,4′-bipyridyl,2bis-(4-pyridyl)ethylene,3bis(4-pyridyl)ethane,4trans-4,4′-azo-pyridine5 and carboxylate groups.6 Previous investigations have also established that the oxyanions, such as sulfate (SO42−), phosphate (PO43−), vanadate (VO43−) and molybdate (MoO42−), can effectively interconnect the primary building units in coordination polymeric chains.7Oxyanions such as nitrate (NO3−) and carbonate (CO32−) are known to be versatile in nature. At the discrete molecular level they can function either as charge compensating counter anions8 in metal complexes, or can bind to the metal ion as simple monodentate (I)9/bidentate (II) ligands10 or can hold two (III) or three metal ions (IV).11 On the other hand, phenoxyacetate (PhOAc−) can only bind with one or two metal ions as shown in I and III.9e,f,11g,h
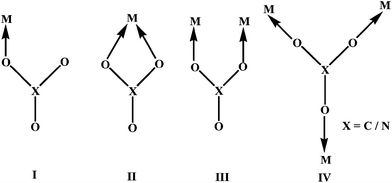
The use of NO3−,12PhOAc−,13CO3,2–14 as bridging units in the construction of extended networks based on copper complexes is also attractive because of the built in flexibility of such anions and this indeed leads to unique molecular frameworks which are established to be useful as novel materials for catalytic15 and magnetic applications.16
The present work originates from our interest in designing newer classes of coordination polymeric frameworks of copper by using selective anions: NO3−, PhOAc− or CO32−, as effective interconnectors between the rather rare alkoxide-bridged dicopper based building units [Cu(μ-hep−)]2 (A) (hep-H = 2-(2-hydroxyethyl)pyridine).17 This approach has led to the formation of varying coordination polymeric motifs in 1, 2 and 3, where the bridging anions link to the same dimeric building blocks, A, in different fashions along the polymeric chain.

It should be noted that to the best of our knowledge no report is available so far on the PhOAc− and CO32− bridged Cu2O2 building block derived coordination polymeric chains, for example in A. Only two examples, [Cu4(L)(OMe)3(NO3)2(H2O)]n (L = [1,3-bis(3-oxo-3-phenylpropionyl)-2-hydroxy-5-methylbenzene])18 and [Cu(ap)NO3]2 (ap = 3-amino-1-propanol)19 are available in the literature where the Cu2O2 building units are bridged by the NO3−group yielding the coordination polymeric chain.
Results and discussion
Synthesis and characterisation
The nitrate (NO3−) bridged coordination polymer (1) has been synthesized by the reactions of 2-(2-hydroxyethyl)pyridine) (hep-H) with a methanolic solution of Cu(NO3)·3H2O at 298 K (Scheme 1). The phenoxyacetate (Ph–O–CH2COO− = PhOAc−) bridged polymeric 2 has been synthesized from a methanolic solution of a mixture of copper acetate dihydrate, hep-H and phenoxyacetic acid at 298 K by the replacement of the acetate group attached to the precursor copper complex by the bulkier phenoxyacetate group (Scheme 1). However, the similar reaction of copper acetate dihydrate with the hep-H ligand in MeOH at 298 K but in the absence of phenoxyacetic acid was reported to result in a discrete dimeric complex [(AcO−)Cu(μ-hep−)]2 (AcO− = acetate) (4) (Scheme 1) instead of a polymeric material as in 2. Thus, a drastic change in molecular framework, dimeric form in 4 to polymeric form in 2, is observed on simple switching from the lighter acetate group to the bulkier phenoxyacetate (PhOAc−) group. In 1 and 2, hep− bridged parent dimeric copper units [Cu(μ-hep)]2 are interconnected by the respective anions along the polymeric chain in head-to-head and tail-to-tail modes (Scheme 1). The hep− derived dimeric building unit in 1 or 2 is stabilised by the central Cu2O2 core as in the discrete dimeric structure of 4. In contrast, the reaction of commercially available copper carbonate hydroxide (Cu2(CO3)·2OH) and hep-H ligand or copper acetate dihydrate, hep-H ligand and sodium carbonate failed to yield any isolable product. Therefore, an alternative synthetic route was explored to synthesize the corresponding carbonate bridged polymeric species. Since in 1 and 2 the dimeric [Cu(μ-hep)]2 units are essentially interconnected by anions (Scheme 1), therefore the recently reported hep−-bridged dimeric complex, [(OAc)Cu(μ-hep)2Cu(OAc)]·2H2O (4), was reacted with Na2CO3 in methanol at 298 K. This indeed resulted in the desired carbonate bridged coordination polymeric complex 3 where the same dimeric building units, [Cu(μ-hep)]2, are linked through the CO32−groups. Though in 1 or 2 the adjacent two dimeric copper units, [Cu(μ-hep)]2, are connected by two units of NO3− or PhOAc− in head-to-head and tail-to-tail modes, in 3 only one CO32−group bridges between the neighbouring dimeric building blocks in head-to-tail fashion (Scheme 1). Moreover, unlike 1 and 2, in 3 one water molecule is bonded to each copper ion in the dimeric unit and there are two lattice water molecules (see later). However, the reaction of 4 with the Ln(NO3)3 or NaNO3 and phenoxyacetic acid results in NO3− and PhOAc− bridged polymeric frameworks as in 1 and 2, respectively, instead of the framework analogous to 3 (Scheme 1).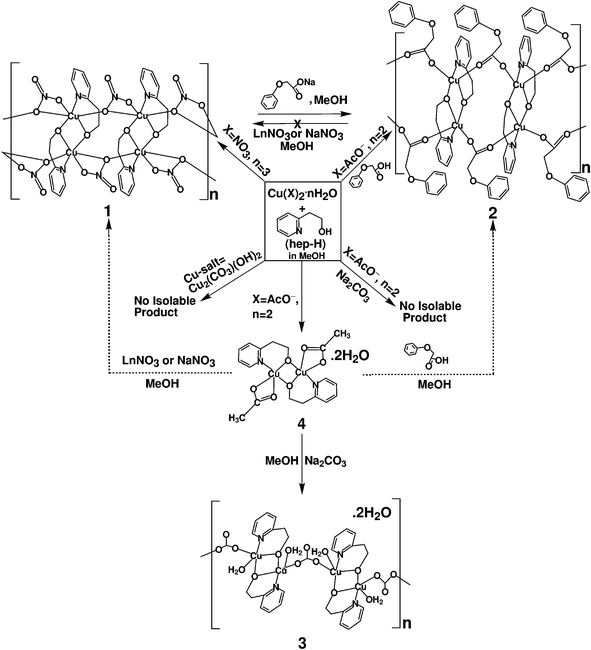 | ||
| Scheme 1 | ||
In addition, 1 can readily be transformed to 2 in the presence of the sodium salt of phenoxyacetate, however, the reverse process, 2 → 1 in the presence of Ln(NO3)3 or NaNO3 surprisingly failed to occur altogether. On the other hand the interconversion of 3 → 1 or 2 by using Ln(NO3)3/NaNO3 or the sodium salt of phenoxyacetate, respectively, and the reverse process of the synthesis of 3 from 1 or 2 by using Na2CO3, failed.
The detailed structural characterisations of 1–3 reveal that the complexes exhibit 1D-coordination polymeric features but with different modes of connectivity along the extended polymeric frameworks based on the bridging anions (see later).
The complexes exhibit satisfactory microanalytical data. The IR spectra of the complexes show characteristic vibrations. The presence of lattice and coordinated water molecules in 3 has been evidenced by strong IR bands near 3400 cm−1 (see Experimental section).
Structural aspects
The coordination polymeric structures of 1–3 have been established by their single crystal X-ray structures. Selective crystallographic parameters and bond distances/angles are listed in Table 1 and Table 2, respectively.| 1 | 2 | 3 | |
|---|---|---|---|
| Empirical formula | C7H8N2O4Cu | C15H15NO4Cu | C15H24N2O9Cu2 |
| Formula weight | 247.69 | 336.82 | 503.44 |
| Crystal symmetry | Monoclinic | Monoclinic | Monoclinic |
| Space group | P21/n | P21/n | C2/c |
| a/Å | 13.5980(5) | 4.794(5) | 18.0701(7) |
| b/Å | 4.7885(2) | 24.697(5) | 6.6051(3) |
| c/Å | 14.5449(8) | 11.500(5) | 15.6456(7) |
| α/° | 90 | 90 | 90 |
| β/° | 112.389(5) | 93.631(5) | 104.807(4) |
| γ/° | 90 | 90 | 90 |
| V/Å3 | 875.69(7) | 1358.8(16) | 1805.37(13) |
| Z | 4 | 4 | 4 |
| μ/mm−1 | 2.482 | 1.622 | 2.410 |
| D c/g cm−3 | 1.879 | 1.646 | 1.852 |
| F(000) | 500 | 692 | 1032 |
| Θ range/° | 3.03 to 24.98 | 3.30 to 25.00 | 3.08 to 25.00 |
| Reflections collected/unique | 5299/1539 [R(int) = 0.0160] | 4222/4222 [R(int) = 0.0000] | 7817/1595 [R(int) = 0.0478] |
| Data/restraints/parameters | 1539/0/127 | 4222/0/196 | 1595/0/144 |
| R 1, wR2 [I > 2σ(I)] | 0.0199, 0.0529 | 0.0450, 0.1118 | 0.0412, 0.0913 |
| R 1, wR2 (all data) | 0.0226, 0.0539 | 0.0601, 0.1192 | 0.0546, 0.0972 |
| GOF | 1.090 | 1.098 | 0.985 |
| largest diff. peak and hole/e Å−3 | 0.319 and −0.265 | 0.425 and −0.711 | 2.615 and −0.462 |
| Bond length (Å)/Bond angle (°) | 1 | 2 | 3 |
|---|---|---|---|
| a Cu–O111 in the case of 3. | |||
| Cu1–Cu1#1 | 2.9650(5) | 3.0057(15) | 3.060(6) |
| Cu1–O1 | 1.9166(14) | 1.940(3) | 1.918(3) |
| Cu1–O1# | 1.9283(13) | 1.930(3) | 1.941(3) |
| Cu1–O2 | 2.0123(14) | 1.988(3) | 1.954(3) |
| Cu1–O3/O111a | 2.3953(14) | 2.349(3) | 2.449(1) |
| Cu1–N1 | 1.9771(17) | 1.997(3) | 2.020(3) |
| Cu(1)–O(1)–Cu(1)#1 | 100.91(6) | 101.88(13) | 104.94(12) |
| O(1)–Cu(1)–O(1)#1 | 79.09(6) | 78.12(13) | 75.06(12) |
| O(1)–Cu(1)–N(1) | 91.91(6) | 90.45(13) | 93.01(12) |
| O(1)#1–Cu(1)–N(1) | 170.79(6) | 168.11(12) | 167.23(13) |
| O(3)–C(8)–O(2) | — | 124.7(4) | — |
| O–X–O | 117.63(16) | 116.3(5) |
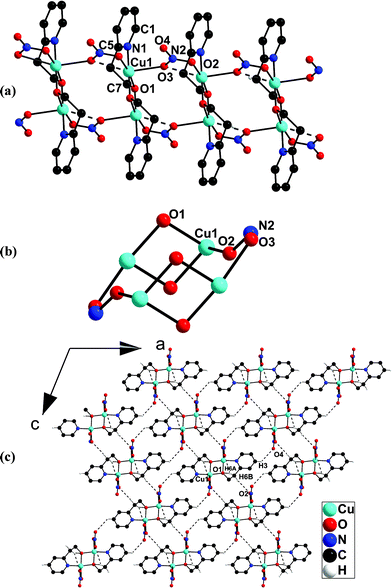 | ||
| Fig. 1 (a) Perspective view of the 1D-coordination polymeric chain in 1. (b) The formation of a 12-membered metallacyclic ring in between the nitrate bridged neighbouring dimeric building units which appears like a chair with two handles. (c) Packing diagram of the hydrogen bonded 2D-network in 1 along the b-axis. | ||
The Cu(1)⋯Cu(1A)# distance and the Cu(1)–O(1)–Cu(1A)# angle of the central Cu2O2 core in the dimeric building unit {[Cu(μ-hep−)]2} in 1 are 2.9650(5) Å and 100.91(6)°, respectively. The other distances associated with the copper-hep− unit in the dimeric building block i.e. Cu–N and Cu–O(bridging hep−) distances are 1.9771(17) and 1.9166(14)/1.9283(13) Å, respectively, which match fairly well with the earlier reported hep−-derived discrete dimeric complex {[Cu(μ-hep−)(OAc−)]2} (4).17b
Each Cu(II) ion along the chain is in distorted octahedral arrangement with CuNO5 configuration, where the basal plane is composed of one nitrogen and two bridging oxygen donors from the bidentate hep− ligands and one monodentate oxygen atom of the NO3−group and the axial positions are occupied by the two bridging oxygen atoms from two NO3−groups. The Cu–O axial distances along the polymeric chain in 1 are alternately long and short, 2.616(12) and 2.3953(14) Å, respectively. The Cu–O distances involving the NO3−group, Cu–O (bridging) and Cu–O (monodentate) are 2.3953(14)/2.616(12) and 2.0123(14) Å, respectively.
If the longer Cu–O(3) (NO3−) distance of 2.616(12) Å in 1 is ignored then a 12-membered ring which apparently looks like a chair with two arms can be considered to form between the nitrate bridged neighbouring dimeric building units (Fig. 1b).
The packing diagram of the 1D polymeric chain of 1 exhibits intramolecular and intermolecular C–H⋯O hydrogen bonds (Fig. 1c, Table 3). The intramolecular C–H⋯O hydrogen bonding involves an α-CH2 group of the hep− ligand and the alkoxo bridged oxygen atom O1 associated with the dimeric building unit and the same continues along the polymeric chain. The intermolecular C–H⋯O hydrogen bond reveals that each layer is connected to two adjacent layers via two different types of hydrogen bonds involving: (i) non-coordinated oxygen atom O(4) of the bridging NO3− and the hydrogen atom H(3) of the coordinated pyridine ring of hep− ligand and (ii) α-CH2 hydrogen atom and monodentate oxygen atom of the bridging NO3−group leading to the formation of a hydrogen bonded 2D network. The distance between the two adjacent layers in the 2D network is 9.26 Å.
| D–H⋯A | H⋯A | D⋯A | D–H⋯A |
|---|---|---|---|
| a Symmetry transformations used to generate equivalent atoms: For 1: #1 −x + 1/2 + 1, +y + 1/2, −z + 1/2. For 2: #1 −x + 1, −y, −z + 2. For 3: #1 x, y, z; #2 −x + 1, −y + 1, −z; #3 x, +y + 1, +z. | |||
| 1 | |||
| C3–H3⋯O4#1 | 2.675(2) | 3.341(3) | 127.60(5) |
| 2 | |||
| C3–H3⋯O4#1 | 2.525(3) | 3.342(5) | 144.10(3) |
| 3 | |||
| O111–H444⋯O222#1 | 2.342(1) | 2.869(5) | 122.42(12) |
| O222–H111⋯O2#1 | 2.003(1) | 2.771(4) | 166.44(21) |
| O222–H111⋯O111#1 | 2.784(6) | 2.869(5) | 88.19(80) |
| O111–H333⋯O222#2 | 1.928(3) | 2.774(5) | 169.62(40) |
| O222–H222⋯O3#3 | 1.789(3) | 2.749(5) | 166.60(87) |
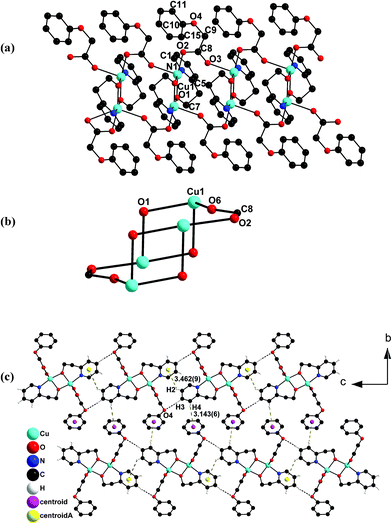 | ||
| Fig. 2 (a) Perspective view of the 1D-coordination polymeric chain in 2. (b) The formation of a 12-membered metallacyclic ring between the phenoxyacetate bridged neighbouring dimeric building units which appears like a chair with two handles. (c) Packing diagram showing the formation of a 2D network in 2 along the a-axis. | ||
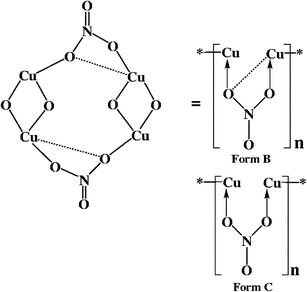
Unlike the bridging NO3−group in 1, the bridging PhOAc− function in 3 binds with the two Cu ions associated with the adjacent dimeric blocks exclusively as a monodentate ligand, Form C. However, the Form B bridging mode of PhOAc− has been reported in one hexameric copper complex, hexakis((μ2-phenoxyacetato-O,O′,O′′)-(μ2-phenoxyacetato-O,O′)-copper(II)).23 Each CuII ion in 2 is therefore exhibiting the penta-coordinated CuO4N configuration with square pyramidal geometry, where the basal plane is composed of one nitrogen and two bridging oxygen donors from the bidentate hep− ligands and one monodentate oxygen atom of the PhOAc−group and the axial position is occupied by the one bridging oxygen atom of another PhOAc−group. The separation between the two copper atoms in the central basic dimeric unit Cu(1)⋯Cu(1A)# of 3.005(2) Å and the Cu(1)–O(1)–Cu(1A) angle of 101.88(3)° are within the range reported for the analogous compounds containing a Cu2O2 core.14a,17
The PhOAc− linked two adjacent basic hep− bridged dimeric units form a 12-membered metallacyclic ring which resembles a chair with two arms (Fig. 2b).
The back and leg-rest fragments in the chair form of the 12-membered metallacyclic part in 1 and 2 show slightly different angular arrangements (Fig. 1b and 2b). The torsion angle in 1, consisting of Cu1 O3 N2 O2, of 164° is in semi-bent conformation whereas that in 2 is almost in linear form: Cu1 O3 C8 O2 of 175°.
It should be stated that the formation of such a large ring (12-membered, Fig. 1b and 2b) due to the head-to-head and tail-to-tail binding modes of the bridging NO3− or PhOAc−group with the adjacent dicopper building units in 1 or 2 has not been reported earlier in other NO3− or PhOAc− bridged polymeric copper complexes.
The packing diagram of 2 reveals the existence of intermolecular strong hydrogen bonding between the adjacent layers of the polymeric unit of 2 involving C(3)–H(3) of the pyridine ring of the coordinated hep− in one layer and the oxygen atom (O4) of the bridging phenoxyacetate group in the next layer with H(3)⋯(O4) = 2.525 Å and C(3)–H(3)⋯(O4) = 144.10° (Fig. 2c, Table 3). Unlike 1, the packing diagram of 2 shows C(2)–H(2)⋯π supramolecular interaction between pyridine rings of hep− ligands in two adjacent layers with edge to face orientation of d = 3.462(9) Å, A = 104.97(4)° and C(4)–H(4)⋯π interaction between the pyridine ring of hep− ligand in one layer and the benzene ring of the phenoxyacetate group in the adjacent layer with an edge to face orientation of d = 3.143(6) Å, A = 166.24(4)°. These indeed result in a 2D network (Fig. 2c) and the distance between the adjacent layers in the 2D network is 8.608 Å.
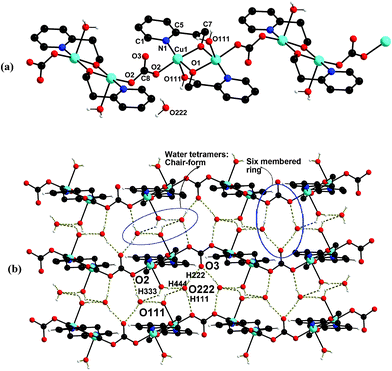 | ||
| Fig. 3 (a) Perspective view of the 1D-coordination polymeric chain in 3. (b) The formation of hydrogen bonded water tetramers and six-membered rings. | ||
3 crystallises in the monoclinic C2/c space group with a crystallographically imposed inversion centre (Tables 1 and 2). Each dimeric unit in the carbonate bridged polymeric 3i.e. {[Cu2(μ-hep)2(H2O)2]·2H2O(μ-CO3)}n consists of two coordinated water molecules trans to each other, and also two lattice water molecules. Each Cu(II) ion in the dimeric unit is in a penta-coordinated (CuO4N) environment with the equatorial plane occupied by two μ-alkoxide oxygen atoms, one pyridine nitrogen of the bidentate hep− ligand and one oxygen donor of the bridging carbonate group. The axial position is occupied by the water molecule resulting in a distorted square pyramidal geometry with the built-in Jahn–Teller distortion (Table 2).
As in 1 and 2 the dimeric core structure {[Cu(μ-hep−)]2} in the polymeric array in 3 is essentially stabilised by the presence of a central four-membered planar Cu2O2 ring with Cu⋯Cu distance of 3.060(6) Å. The carbonate group in 3 bridges between the two copper centres, each in adjacent dimeric building units, {Cu2(μ-hep−)2(H2O)2} in head-to-tail mode through the two oxygen atoms of the carbonate groups leaving the third oxygen atom free as in Form A which however involves strong hydrogen bonding interactions with the lattice water molecules (see later).
The packing diagram of polymeric complex 3 reveals the presence of inter-molecular O–H⋯O and C–H⋯O hydrogen bonds as well C–H⋯π interactions. The hydrogen atoms associated with the two lattice water molecules and two coordinating water molecules from two adjacent layers of the zig-zag coordination polymer of 3 yield a water tetramer (uudd) along the b-axis (Fig. 3b, Table 3). The water tetramer along with the associated two hydrogen atoms in turn forms a six-membered chair like conformation. The water tetramers are further linked with the coordinated and uncoordinated ‘O’ atoms of the bridging CO32−groups in two adjacent layers which eventually forms a six-membered ring (Fig. 3b).
Remarkably, the two lattice water molecules (O222), alternate coordinated water molecules (O111) and uncoordinated ‘O’ atoms (O3) of the bridging CO32−group collectively generate an additional 1D zig-zag chain which is being trapped within the two outer zig-zag coordination polymeric chains of 3 (Fig. 4a,b).
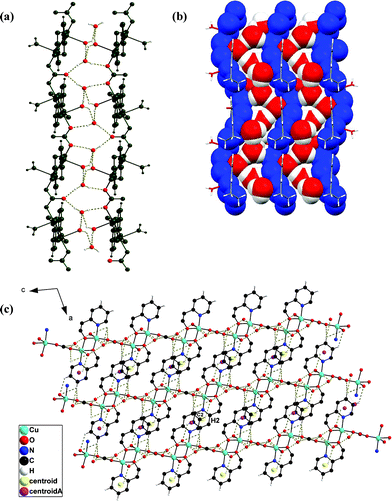 | ||
| Fig. 4 (a) The hydrogen bonded zig-zag chain of oxygen atoms of water tetramers and uncoordinated CO32−groups (red) in between two zig-zag chains of the outer coordination polymers (green). (b) Space filling model of (a). (c) Packing diagram shows the formation of a 2D network in 3 along the b-axis. | ||
Moreover, along the a-axis each of those layers (stated above) is further extended through C–H⋯O and C–H⋯π interactions involving the hydrogen atom H2 of the pyridine ring of hep− from one layer to the coordinated water molecule in another layer (C–H2⋯O111) and the same hydrogen atom (H2) ![[scr I, script letter I]](https://www.rsc.org/images/entities/char_e143.gif) s also simultaneously involved in the C–H⋯π interaction leading to the formation of a 2D network (Fig. 4c).
s also simultaneously involved in the C–H⋯π interaction leading to the formation of a 2D network (Fig. 4c).
Thermal analysis
The DTA (differential thermal analysis) of compound 3 exhibits three absorption peaks at 61 °C, 316 °C, and 453 °C. The first weight loss of 3.8% (calcd: 3.57%) in TGA (thermogravimetric analysis) takes place between 35 and 48 °C corresponding to the loss of one lattice water molecule per formula unit, {[Cu2(μ-hep)2(H2O)2]·2H2O(μ-CO3)}n. The second weight loss of 17.13% (calcd: 17.46%) in the range of 48 to 82 °C accounts for the loss of two CO2 per formula unit and the weight loss of 53.40% (calcd: 52.49%) from 82 to 650 °C corresponds to the loss of one coordinated water molecule and two hep−. Further weight loss at higher temperature matches with the total decomposition.Comparative structural motifs in 1–3
The following striking structural similarities and differences have been recognised in 1–3. (i) Oxyanions (NO3−, CO32−) and phenoxyacetate (PhOAc−) bridged 1–3 exhibit 1D-coordination polymeric features where the dicopper based building blocks, {[Cu(μ-hep−)]2}, stabilise via the presence of a central Cu2O2 core. (ii) 1–3 also form 2D-extended networks through a wide variety of non-covalent interactions. (iii) Two NO3− or PhOAc−groups in 1 or 2 bridge between the dicopper based two adjacent building units ([Cu(μ-hep−)]2) in head-to-head and tail-to-tail modes leading to the formation of a new 12-membered chair-like structure. (iv) In 1 each NO3− function bridges the two copper centres associated with the two adjacent building units as in Form B, whereas in 2 the PhOAc−group bridges between the two adjacent Cu ions as in Form C. (v) Unlike 1 and 2, in 3 only one CO32−group bridges between the two copper centres in two adjacent building units in head-to-tail form which indeed prevents the formation of any large internal ring like in 1 or 2. However, the head-to-tail connecting mode of the CO32−group extends the zig-zag pattern of 3. (vi) The coordinated and lattice water molecules and the non-coordinated oxygen of CO32− in 3 collectively form an additional zig-zag layer between the two outer zig-zag layers of the coordination polymers. (vii) The non-bonded Cu⋯Cu, O⋯O distances and Cu–O–Cu angles in 1, 2 and 3 of 2.97 Å, 2.45 Å, 100.91°; 3.01 Å, 2.44 Å, 101.88° and 3.06 Å, 2.35 Å, 104.94°, respectively, are close to the reported polymeric14a or discrete24copper complexes.Magnetic properties of 1–3
The polymeric complexes 1–3 encompassing building units with a central Cu2O2 core exhibit similar magnetic properties in the temperature range of 300–100 K. The solid state magnetic moments of 1.25, 1.42 and 1.27 B.M. at 300 K for 1, 2 and 3, respectively, sharply decrease to close to zero at around 100 K (Fig. S1, ESI†) which implies that the Cu(II) centres in the complexes are antiferromagnetically coupled.25 The magnetic simulation using the julX program26 extends the J/cm−1 values of −240, −260 and −275 for 1, 2 and 3, respectively.1–3 exhibit axial EPR spectra with g∥ > g⊥ (Fig. S1, inset† and Table 4), consistent with the tetragonally elongated square pyramidal geometry around the copper(II) ion.27 The <g> value in the range of 2.102–2.198 matches fairly well with that obtained from the magnetic simulation of 2.10.
Conclusion
The present article demonstrates the design of oxyanions (NO3−, CO32−) and phenoxyacetate (PhOAc−) bridged varying structural motifs in polymeric copper complexes (1–3) based on alkoxide derived dicopper building units [Cu(μ-hep)]2 with a central Cu2O2 core (Hhep = 2-(2-hydroxyethyl)pyridine). The simultaneous head-to-head and tail-to-tail binding modes of NO3− or PhOAc−groups with the adjacent Cu2O2 units in 1 or 2, respectively, result in unprecedented twelve-membered metallacyclic rings. On the other hand, head-to-tail cross-linking of the CO32−group with the adjacent Cu2O2 units in 3 develops a unique zig-zag coordination polymeric chain via a self-assembly process and two such adjacent chains in turn extend the support to hold the new zig-zag chain of water tetramers inside the outer layers via extensive hydrogen bonding.Experimental
Materials and physical measurements
The commercially available starting materials, dihydrated copper acetate, trihydrate copper nitrate, 2-(2-hydroxyethyl)pyridine (hep-H), phenoxyacetic acid (Ph–O–CH2–COOH), sodium carbonate and reagent grade methanol were used as received. Elemental analyses were carried out with a Perkin-Elmer 240C elemental analyser. FT-IR spectra of complexes as KBr pellets and DTA/TGA experiments were done on a Nicolet spectrophotometer and Perkin Elmer instrument, respectively. The complex [Cu(μ2-hep)(OAc)]2·2H2O (4) was prepared by following the reported methods.17 The magnetic measurements were performed in a commercial PPMS magnetometer (Quantum Design, 6000 Lusk Boulevard, San Diego, CA) equipped with a superconducting 14 T magnet. The program julX written by E. Bill was used for the simulation and analysis of magnetic susceptibility data.26 The EPR measurements were made with a Varian model 109C E-line X-band spectrometer fitted with a quartz Dewar for 77 K. Thermogravimetric analyses for complexes 1–3 were measured under a flow of nitrogen gas from room temperature to 850 °C at a heating rate of 10 °C min−1.Synthesis of 1–3
O)CH3)2·2H2O (199 mg, 1 mmol) in methanol (25 cm3). Phenoxyacetic acid (Ph–O–CH2–COOH, 152 mg, 1 mmol) was added to the above reaction mixture and the resultant solution was stirred for 6 h at room temperature. The solution was then passed through filter paper (Whatman filter paper, 70 mm) in order to remove any unreacted materials. The filtrate was allowed to stand at room temperature for crystallisation. Dark blue single crystals of 2 were obtained within 3 days by slow evaporation of the solvent. M.P.: 249–251 °C. Yield: 290 mg (82%). Anal. Calcd for C15H15CuNO4 (Mr = 336.82): C, 53.57; H, 4.50; N, 4.17. Found: C, 53.60; H, 4.64; N, 4.27. IR (KBr, cm−1): 3066(w), 3030(vw), 2943(w), 2893(w), 2816(m), 1573(vs), 1491(vs), 1386(vs), 1301(s), 1257(s), 1217(vs), 1144(s), 972(w), 913(m), 867(m), 773(s), 750(s), 726(s), 680(s), 604(s), 563(w), 505(w).
Similarly, 2 was prepared by the reaction of methanolic solution of phenoxyacetic acid (Ph–O–CH2–COOH, 75 mg, 0.5 mmol) and preformed [Cu(μ2-hep)(OAc)]2·2H2O (4) (280 mg, 0.5 mmol) in methanol (25 cm3) under magnetic stirring for 4 h at room temperature. The reaction mixture was filtered using Whatman filter paper, 70 mm, and the filtrate was allowed to stand at room temperature for crystallisation. Dark blue single crystals of 2 were obtained within a week by slow evaporation of the solvent. M.P.: 250–252 °C. Yield: 290 mg (82%). Anal. Calcd for C15H15CuNO4 (Mr = 336.82): C, 53.57; H, 4.50; N, 4.17. Found: C, 53.62; H, 4.61; N, 4.23.
2 can also be prepared from 1via the reaction sodium salt of phenoxyacetic acid (Ph–O–CH2–COONa, 75 mg, 0.5 mmol) in methanol (25 cm3) with a methanolic solution of 1 (250 mg, 0.5 mmol) under magnetic stirring for 6 h at 298 K. The reaction mixture was filtered using Whatman filter paper, 70 mm, filter paper and the filtrate was allowed to stand at room temperature for crystallisation. Dark blue single crystals of 2 were obtained within a 10 days by slow evaporation of the solvent. M.P.: 250–252 °C. Yield: 290 mg (82%). Anal. Calcd for C15H15CuNO4 (Mr = 336.82): C, 53.57; H, 4.50; N, 4.17. Found: C, 53.51; H, 4.54; N, 4.13.
Crystal structure determination
Single crystal X-ray structural studies of 1, 2 and 3 were performed on a CCD Oxford Diffraction XCALIBUR-S diffractometer equipped with an Oxford Instruments low-temperature attachment. Data were collected at 150(2) K using graphite-monochromated Mo-Kα radiation (λα = 0.71073 Å). The strategy for the data collection was evaluated by using the CrysAlisPro CCD software. The data were collected by the standard phi-omega scan techniques, and were scaled and reduced using CrysAlisPro RED software. The structures were solved by direct methods using SHELXS-97 and refined by full matrix least squares with SHELXL-97, refining on F.2,282 appears to be a non-merohedral twin, and the problem was tackled using Rotax, which is a program found in WinGX, by using BASF values of 0.23869 0.15492 0.18016 0.03584 0.08242.The positions of all the atoms were obtained by direct methods. All non-hydrogen atoms were refined anisotropically. The water molecules (both coordinated and lattice) in 3 were generated by Fourier maps. The remaining hydrogen atoms were placed in geometrically constrained positions and refined with isotropic temperature factors, generally 1.2 × Ueq of their parent atoms. All the H-bonding interactions, mean plane analyses, and molecular drawings were obtained using the program Diamond (version 3.1d). The crystal and refinement data are summarized in Table 1, and selected bond distances and bond angles are shown in Tables 2 and 3.
Acknowledgements
Financial support received from the Department of Science and Technology (New Delhi, India) is gratefully acknowledged.References
- (a) T. D. Keene, H. R. Ogilvie, M. B. Hursthouse and D. J. Price, Eur. J. Inorg. Chem., 2004, 1007 CrossRef CAS; (b) J-H. Yu, J. Lu, X. Zhang, L. Ye, Q. Hou and J-Q. Xu, Inorg. Chem. Commun., 2006, 9, 415 CrossRef CAS; (c) J-H. Yu, L. Ye, M-H. Bi, Q. Hou, X. Zhang and J-Q. Xu, Inorg. Chim. Acta, 2007, 360, 1987 CrossRef CAS; (d) D-T. Nguyen, E. Chew, Q. Zhang, A. Choi and X. Bu, Inorg. Chem., 2006, 45, 10722 CrossRef CAS; (e) C. Ma, W. Wang, X. Zhang, C. Chen, Q. Liu, H. Zhu, D. Liao and L. Li, Eur. J. Inorg. Chem., 2004, 3522 CrossRef CAS; (f) L. Brammer, M. D. Burgard, C. S. Rodger, J. K. Swearingen and N. P. Rath, Chem. Commun., 2001, 2468 RSC; (g) S. Takamizawa, E. Nakata, T. Akatsuka, C. Kachi-Terajima and R. Miyake, J. Am. Chem. Soc., 2008, 130, 17882 CrossRef CAS; (h) J-X. Yuan, M. L. Hu, Y. Q. Cheng, L. C. Chen and S. W. Ng, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2002, 58, m270 CrossRef.
- (a) C. Hu and U. Englert, Angew. Chem., Int. Ed., 2006, 45, 3457 CrossRef CAS; (b) S. Noro, R. Kitaura, M. Kondo, S. Kitagawa, T. Ishii, H. Matsuzaka and M. Yamashita, J. Am. Chem. Soc., 2002, 124, 2568 CrossRef CAS; (c) T. Gadzikwa, B. S. Zeng, J. T. Hupp and S. T. Nguyen, Chem. Commun., 2008, 3672 RSC; (d) J. W. Johnson, A. J. Jacobson, S. M. Rich and J. F. Brody, J. Am. Chem. Soc., 1981, 103, 5246 CrossRef CAS; (e) R. Fu, S. Hu and X. Wu, Inorg. Chem., 2007, 46, 9630 CrossRef CAS; (f) D. Tanaka, K. Nakagawa, M. Higuchi, S. Horike, Y. Kubota, T. C. Kobayashi, M. Takata and S. Kitagawa, Angew. Chem., Int. Ed., 2008, 47, 3914 CrossRef CAS; (g) G. J. Halder and C. J. Kepert, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2005, 61, m113 Search PubMed; (h) S. Mukhopadhyay, P. B. Chatterjee, D. Mandal, G. Mostafa, A. Caneschi, J. van Slageren, T. J. R. Weakley and M. Chaudhury, Inorg. Chem., 2004, 43, 3413 CrossRef CAS; (i) K. Biradha, K. V. Domasevitch, B. Moulton, C. Seward and M. J. Zaworotko, Chem. Commun., 1999, 1327 RSC; (j) J. T. Sampanthar and J. J. Vittal, J. Chem. Soc., Dalton Trans., 1999, 1993 RSC; (k) S. Shimomura, S. Horike, R. Matsuda and S. Kitagawa, J. Am. Chem. Soc., 2007, 129, 10990 CrossRef CAS.
- (a) X. Li, R. Cao, D. Sun, W. Bi, Y. Wang, X. Li and M. Hong, Cryst. Growth Des., 2004, 4, 775 CrossRef CAS; (b) M. Schweiger, S. R. Seidel, A. M. Arif and P. J. Stang, Inorg. Chem., 2002, 41, 2556 CrossRef CAS; (c) S. W. Lee, H. J. Kim, Y. K. Lee, K. Park, J-H. Son and Y-U. Kwon, Inorg. Chim. Acta, 2003, 353, 151 CrossRef CAS; (d) C. S. Lai and E. R. T. Tiekink, CrystEngComm, 2004, 6, 593 RSC; (e) G. S. Papaefstathiou, I. G. Georgiev, T. Friscic and L. R. MacGillivray, Chem. Commun., 2005, 3974 RSC; (f) E-Y. Choi, K. Park, C-M. Yang, H. Kim, J-H. Son, S. W. Lee, Y. H. Lee, D. Min and Y-U. Kwon, Chem.–Eur. J., 2004, 10, 5535 CrossRef CAS; (g) B. Chen, S. Ma, F. Zapata, F. R. Fronczek, E. B. Lobkovsky and H-C. Zhou, Inorg. Chem., 2007, 46, 1233 CrossRef CAS; (h) T. K. Maji, K. Uemura, H. C. Chang, R. Matsuda and S. Kitagawa, Angew. Chem., Int. Ed., 2004, 43, 326 Search PubMed.
- (a) A. K. Ghosh, D. Ghoshal, E. Zangrando, J. Ribas and N. R. Chaudhuri, Dalton Trans., 2006, 1554 RSC; (b) S. Konar, E. Zangrando, M. G. B. Drew, T. Mallah, J. Ribas and N. R. Chaudhuri, Inorg. Chem., 2003, 42, 5966 CrossRef CAS; (c) J-C. Dai, X-T. Wu, S-M. Hu, Z-Y. Fu, J-J. Zhang, W-X. Du, H-H. Zhang and R-Q. Sun, Eur. J. Inorg. Chem., 2004, 2096 CrossRef CAS; (d) Q-X. Jia, Y-Q. Wang, Q. Yue, Q-L. Wang and E-Q. Gao, Chem. Commun., 2008, 4894 RSC; (e) L-F. Ma, L-Y. Wang, Y-Y. Wang, S. R. Batten and J-G. Wang, Inorg. Chem., 2009, 48, 915 CrossRef CAS; (f) G. J. Halder, K. W. Chapman, S. M. Neville, B. Moubaraki, K. S. Murray, J. F. Letard and C. J. Kepert, J. Am. Chem. Soc., 2008, 130, 17552 CrossRef CAS; (g) B. Rather and M. J. Zaworotko, Chem. Commun., 2003, 830 RSC.
- (a) B. Li, L. Zhu, S. Wang, J. Lang and Y. Zhang, J. Coord. Chem., 2003, 56, 933 CrossRef CAS; (b) M. Kondo, M. Shimamura, S. Noro, T. Yoshitomi, S. Minakoshi and S. Kitagawa, Chem. Lett., 1999, 285 CrossRef CAS; (c) S. Noro, M. Kondo, T. Ishii, S. Kitagawa and H. Matsuzaka, J. Chem. Soc., Dalton Trans., 1999, 1569 RSC; (d) G. J. Halder, C. J. Kepert, B. Moubaraki, K. S. Murray and J. D. Cashion, Science, 2002, 298, 1762 CrossRef CAS; (e) J. P. Barrio, J. N. Rebilly, B. Carter, D. Bradshaw, J. Bacsa, A. Y. Ganin, H. Park, A. Trewin, R. Vaidhyanathan, A. I. Cooper, J. E. Warren and M. J. Rosseinsky, Chem.–Eur. J., 2008, 14, 4521 CrossRef CAS; (f) B. Li, B. Z. Xu, Z. Huang and Q. Wu, J. Coord. Chem., 2002, 55, 1183 CrossRef CAS; (g) S. Noro, S. Kitagawa, T. Nakamura and T. Wada, Inorg. Chem., 2005, 44, 3960 CrossRef CAS; (h) S. Noro, R. Kitaura, S. Kitagawa, T. Akutagawa and T. Nakamura, Inorg. Chem., 2006, 45, 8990 CrossRef CAS.
- (a) E. Colacio, M. Ghazi, R. Kivekäs and J. M. Moreno, Inorg. Chem., 2000, 39, 2882 CrossRef CAS; (b) S. J. Rettig, R. C. Thompson, J. Trotter and S. Xia, Inorg. Chem., 1999, 38, 1360 CrossRef CAS; (c) V. Tangoulis, G. Psomas, C. Dendrinou-Samara, C. P. Raptopoulou, A. Terzis and D. P. Kessissoglou, Inorg. Chem., 1996, 35, 7655 CrossRef CAS; (d) A. D. Burrows, C. G. Frost, M. F. Mahon and C. Richardson, Angew. Chem., Int. Ed., 2008, 47, 8482 CrossRef CAS.
- (a) D. Hagrman, R. P. Hammond, R. Haushalter and J. Zubieta, Chem. Mater., 1998, 10, 2091 CrossRef CAS; (b) L. Carlucci, G. Ciani, D. M. Proserpio and S. Rizzato, CrystEngComm, 2003, 5, 190 RSC; (c) D. Hagrman, R. C. Haushalter and J. Zubieta, Chem. Mater., 1998, 10, 361 CrossRef CAS; (d) R. C. Finn and J. Zubieta, J. Chem. Soc., Dalton Trans., 2002, 856 RSC; (e) L. Carlucci, G. Ciani, D. M. Proserpio and S. Rizzato, J. Chem. Soc., Dalton Trans., 2000, 3821 RSC; (f) K. Abu-Shandi, C. Janiak and B. Kersting, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2001, 57, 1261 CrossRef CAS; (g) Z. Shi, S. Feng, S. Gao, L. Zhang, G. Yang and J. Hua, Angew. Chem., Int. Ed., 2000, 39, 2325 CrossRef CAS.
- (a) I. Viera, M. H. Torre, O. E. Piro, E. E. Castellano and E. J. Baran, J. Inorg. Biochem., 2005, 99, 1250 CrossRef CAS; (b) Y. Zhang, L. K. Thompson, J. N. Bridson and M. Bubenik, Inorg. Chem., 1995, 34, 5870 CrossRef CAS; (c) H. M. J. Hendriks, P. J. M. W. L. Birker, J. van Rijn, G. C. Verschoor and J. Reedijk, J. Am. Chem. Soc., 1982, 104, 3607 CrossRef CAS; (d) L. K. Thompson, A. W. Hanson and B. S. Ramaswamy, Inorg. Chem., 1984, 23, 2459 CrossRef CAS; (e) T. Gajda, Y. Dupre, I. Torok, J. Harmer, A. Schweiger, J. Sander, D. Kuppert and K. Hegetschweiler, Inorg. Chem., 2001, 40, 4918 CrossRef CAS; (f) J-P. Lang, H. Kawaguchi and K. Tatsumi, J. Organomet. Chem., 1998, 569, 109 CrossRef CAS; (g) L. Coghi, M. Lanfranchi, G. Pelizzi and P. Tarasconi, Transition Met. Chem., 1978, 3, 69 CrossRef CAS; (h) G. Mezei, P. Baran and R. G. Raptis, Angew. Chem., Int. Ed., 2004, 43, 574 CrossRef CAS; (i) R. Custelcean, P. Remy and P. V. Bonnesen, Angew. Chem., Int. Ed., 2008, 47, 1866 CrossRef CAS.
- (a) L. Latos-Grazynski, J. Lisowski, M. M. Olmstead and A. L. Balch, J. Am. Chem. Soc., 1987, 109, 4428 CrossRef CAS; (b) P. de Hoog, P. Gamez, M. Luken, O. Roubeau, B. Krebs and J. Reedijk, Inorg. Chim. Acta, 2004, 357, 213 CrossRef CAS; (c) G. De Munno and G. Denti, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1984, 40, 616 Search PubMed; (d) F. A. Mautner and M. A. S. Goher, Polyhedron, 1993, 12, 2823 CrossRef CAS; (e) C. H. L. Kennard, G. Smith, E. J. O'Reilly and K. E. Brown, Inorg. Chim. Acta, 1981, 52, 55 CrossRef CAS; (f) G. Smith, E. J. O'Reilly and C. H. L. Kennard, Cryst. Struct. Commun., 1981, 10, 1397 CAS; (g) G. Smith, E. J. O'Reilly, C. H. L. Kennard and T. C. W. Mak, Inorg. Chim. Acta, 1982, 65, L219 CrossRef CAS.
- (a) I. A. Koval, M. Huisman, A. F. Stassen, P. Gamez, M. Lutz, A. L. Spek and J. Reedijk, Eur. J. Inorg. Chem., 2004, 591 CrossRef CAS; (b) T. D. Keene, M. B. Hursthouse and D. J. Price, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2004, 60, m381 CrossRef; (c) P. C. Healy, C. H. L. Kennard, G. Smith and A. H. White, Cryst. Struct. Commun., 1981, 10, 883 CAS; (d) M-S. Wang, G-C. Guo, L-Z. Cai, G-W. Zhou and J-S. Huang, Jiegou Huaxue (Chin. J. Struct. Chem.), 2003, 22, 427 CAS.
- (a) M. Fondo, A. M. Garcia-Deibe, N. Ocampo, J. Sanmartin and M. R. Bermejo, Dalton Trans., 2004, 2135 RSC; (b) B. F. Abrahams, M. G. Haywood and R. Robson, Chem. Commun., 2004, 938 RSC; (c) S. Kitagawa, M. Kondo, S. Kawata, S. Wada, M. Maekawa and M. Munakata, Inorg. Chem., 1995, 34, 1455 CrossRef CAS; (d) Y. Zhang, L. K. Thompson, J. N. Bridson and M. Bubenik, Inorg. Chem., 1995, 34, 5870 CrossRef CAS; (e) Y-B. Dong, T. Sun, J-P. Ma, X-X. Zhao and R-Q. Huang, Inorg. Chem., 2006, 45, 10613 CrossRef CAS; (f) M-L. Tong, M. Monfort, J. M. C. Juan, X-M. Chen, X-H. Bu, M. Ohba and S. Kitagawa, Chem. Commun., 2005, 233 RSC; (g) K. L. V. Mann, J. C. Jeffery, J. A. McCleverty and M. D. Ward, J. Chem. Soc., Dalton Trans., 1998, 3029 RSC; (h) C. H. L. Kennard, E. J. O'Reilly and G. Smith, Polyhedron, 1984, 3, 689 CrossRef CAS; (i) G. Psomas, C. P. Raptopoulou, L. Iordanidis, C. Dendrinou-Samara, V. Tangoulis and D. P. Kessissoglou, Inorg. Chem., 2000, 39, 3042 CrossRef CAS.
- F. B. Hulsbergen, R. W. M. ten Hoedt, G. C. Verschoor, J. Reedijk and A. L. Spek, J. Chem. Soc., Dalton Trans., 1983, 539 RSC.
- G. Smith, E. J. O'Reilly, C. H. L. Kennard and A. H. White, J. Chem. Soc., Dalton Trans., 1985, 243 RSC.
- (a) R. P. Doyle, M. Julve, F. Lloret, M. Nieuwenhuyzen and P. E. Kruger, Dalton Trans., 2006, 2081 RSC; (b) S. Youngme, N. Wannarit, T. Remsungnen, N. Chaichit, G. A. van Albada and J. Reedijk, Inorg. Chem. Commun., 2008, 11, 179 CrossRef CAS; (c) J. Sertucha, A. Luque, O. Castillo, P. Romàn, F. Lloret and M. Julve, Inorg. Chem. Commun., 1999, 2, 14 CAS; (d) J. Y. Lu, A. L. Reynolds and K. A. Runnels, CrystEngComm, 2001, 3, 144 RSC; (e) J. P. Naskar, M. G. B. Drew, A. Hulme, D. A. Tocher and D. Datta, CrystEngComm, 2005, 7, 67 RSC.
- (a) A. Clearfield, Prog. Inorg. Chem., 1998, 47, 371 CrossRef CAS; (b) A. Hu, H. L. Ngo and W. Lin, Angew. Chem., Int. Ed., 2003, 42, 6000 CrossRef CAS.
- (a) O. Kahn, Molecular Magnetism, VCH Publishers Inc., New York, 1993 Search PubMed; (b) J. S. Miller and M. Drillon, Magnetism: Molecules to Materials, Wiley-VCH, Weinheim, 2001 Search PubMed; (c) C. J. O'Connor, Research Frontiers in Magnetochemistry, World Scientific, Singapore, 1993 Search PubMed.
- (a) M. M. Shaikh, A. K. Srivastava, P. Mathur and G. K. Lahiri, Inorg. Chem., 2009, 48, 4652 CrossRef; (b) M. M. Shaikh, A. K. Srivastava, P. Mathur and G. K. Lahiri, Dalton Trans., 2010, 39, 1447 RSC.
- G. Aromi, J. Ribas, P. Gamez, O. Roubeau, H. Kooijman, A. L. Spek, S. Teat, E. MacLean, H. Stoeckli-Evans and J. Reedijk, Chem.–Eur. J., 2004, 10, 6476 CrossRef CAS.
- T. Lindgren, R. Sillanpaa, K. Rissanen, L. K. Thompson, C. J. O'Connor, G. A. van Albada and J. Reedijk, Inorg. Chim. Acta, 1990, 171, 95 CrossRef CAS.
- (a) B. Duffin, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1968, 24, 396 CrossRef CAS; (b) B. Duffin and S. C.Wallwork, Acta Crystallogr., 1966, 20, 210 CrossRef CAS; (c) Xiang-Jian Kong, Yan-Ping Ren, La-Sheng Long, Rong-Bin Huang and Lan-Sun Zhang, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2005, 61, m2346 Search PubMed; (d) R. Allmann, M. Krestl, C. Bolos, G. Manoussakis and G. S. Nikolov, Inorg. Chim. Acta, 1990, 175, 255 CrossRef CAS.
- (a) K. Sakai, Y. Yamada, T. Tsubomura, M. Yabuki and M. Yamaguchi, Inorg. Chem., 1996, 35, 542 CrossRef CAS; (b) M. Jagoda, S. Warzeska, H. Pritzkow, H. Wadepohl, P. Imhof, J. C. Smith and R. Kramer, J. Am. Chem. Soc., 2005, 127, 15061 CrossRef CAS; (c) J. B. Fontecha, S. Goetz and V. McKee, Angew. Chem., Int. Ed., 2002, 41, 4553 CrossRef CAS.
- A. M. Thomas, A. Mukherjee, M. K. Saha, M. Nethaji and A. R. Chakravarty, Indian J. Chem., Sect. A: Inorg., Bio-inorg., Phys., Theor. Anal. Chem., 2004, 43, 1626 Search PubMed.
- J. R. Carruthers, K. Prout and F. J. C. Rossotti, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1975, 31, 2044 CrossRef.
- V. H. Crawford, H. W. Richardson, J. R. Wasson, D. J. Hodgson and W. E. Hatfield, Inorg. Chem., 1976, 15, 2107 CrossRef CAS.
- J. W. Hall, W. E. Marsh, R. R. Weller and W. E. Hatfield, Inorg. Chem., 1981, 20, 1033 CrossRef CAS.
- http://ewww.mpi-muelheim.mpg.de/bac/logins/bill/julX_en.php .
- A. Banerjee, S. Sarkar, D. Chopra, E. Colacio and K. K. Rajak, Inorg. Chem., 2008, 47, 4023 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, A64, 112 CrossRef CAS . Program for Crystal Structure Solution and Refinement, University of Goettingen, Germany, 1997.
Footnote |
| † Electronic Supplementary Information (ESI) available. See DOI: 10.1039/c1ra00155h/ |
| This journal is © The Royal Society of Chemistry 2011 |