Modulation of the photophysical properties of BODIPY dyes by substitution at their meso position.†
Jorge
Bañuelos
*a,
Ismael J.
Arroyo-Córdoba
b,
Ismael
Valois-Escamilla
c,
Alejandro
Alvarez-Hernández
c,
Eduardo
Peña-Cabrera
b,
Rongrong
Hu
d,
Ben
Zhong Tang
d,
Ixone
Esnal
a,
Virginia
Martínez
a and
Iñigo
López Arbeloa
a
aDepartamento Química Física, Universidad del País Vasco, -EHU, Apartado 644, 48080-Bilbao, Spain. E-mail: jorge.banuelos@ehu.es; Fax: +34946013500; Tel: +34946015384
bDepartamento Química. Universidad de Guanajuato. Col. Noria Alta. Guanajuato, Gto. 36050, Mexico. E-mail: eduardop@quijote.ugto.mx
cCentro Investigaciones Químicas, Universidad Autónoma del Estado de Hidalgo. Carretera Pachuca-Tulancingo Km. 4.5. Cd. Universitaria, Mineral de la Reforma, Hidalgo 42076, Mexico. E-mail: alvarez@uaeh.edu.mx
dDepartment of Chemistry, Hong Kong University of Science and Technology, Clear Water Bay, Kowloon, Hong Kong, China. E-mail: tangbenz@ust.hk
First published on 24th August 2011
Abstract
We report the photophysical properties of new BODIPY derivatives monosubstituted at the central position. The presence of different functional groups induced the appearance of new photophysical processes in BODIPY dyes, such as intramolecular charge or energy transfer. These phenomena are sensitive to solvent properties (mainly the polarity) and have a potential use as fluorescent probes. Adequate modifications in their molecular structure or in the environment polarity can modulate the emission region of these fluorophores in the visible spectral region. Specifically, different processes and photophysical behaviors can be achieved depending on the excited chromophore and/or the solvent characteristics in a bichromophoric pyrene-BODIPY system.
Introduction
4,4-difluoro-4-bora-3a,4a-diaza-s-indacene dyes (henceforth abbreviated as BODIPY) are widely known active media of tunable dye lasers, generally in the green-yellow part of the visible spectral region.1BODIPY dyes efficiently absorb electromagnetic radiation and emit sharp fluorescence bands, usually characterized by high fluorescence quantum yields, in some cases close to the unit.2 Due to their quasi-aromatic electron delocalization, the intersystem crossing probability is very low,3 dismissing the triplet–triplet absorption ability in these dyes; probably the main factor which reduces the laser efficiency of many laser dyes. Moreover, BODIPY dyes have excellent chemical and thermal stabilities. All these factors contribute to the important application of BODIPY dyes in photonics and as fluorescent probes in many biological systems.4 The synthesis of new BODIPY derivatives with adequate aromatic substituents can extend the chromophoric π-system and shifts the emission band to the red.5 Thus, photoactive materials with a wide range of the visible spectral region can be achieved with the same laser dye family.Since the discovery of BODIPY at the beginning of the 90s as efficient laser dyes,6BODIPY fluorophores are widely used in light harvesting arrays or antenna systems, as energy injectors or acceptors to collect and transport the light to a reaction centre, or as sensitizers in solar cells.7 However, probably the most successful application is as fluorescence probes or sensors to monitor the evolution of biological processes, to characterize the surrounding environment of biomolecules or to recognize the presence of certain analytes.8 Indeed, there is a huge variety of functionalized BODIPY dyes to study biochemistry processes in biological systems (DNA, proteins, lipids and so on).9 Most BODIPY-probes are based on the possibility of modulating their photophysical behaviour by the incorporation of appropriate functional groups in the molecular structure of the chromophore. Typically, the photophysics of BODIPY dyes are quite insensitive to the solvent properties,2 but the presence of adequate substituents induces new photophysical processes, which can be sensitive to certain environmental characteristics.
The present work is focused in the controlled modification of the photophysical properties of BODIPY dyes by the inclusion of appropriate substituents at the meso position of the BODIPY core. This central position is the most sensitive one to the substituent effect because an important modification in the electronic density localized at this meso position takes place upon excitation. The new BODIPY derivatives considered in this work are depicted in Scheme 1. The aim of the 8-alkyl derivatives (compounds 1–3) was to shift the emission band of the BODIPY chromophore to the blue part of the visible region, a barely exploited region with BODIPY. Additionally, different aromatic groups: p-bromophenyl (4), triphenylaminethiophene (5) and pyrene (6) were included in the search for new photophysical phenomena sensitive to the solvent and to expand the applicability of BODIPY dyes.
 | ||
| Scheme 1 Molecular structure of BODIPY derivatives | ||
Experimental
BODIPY derivatives 4–6 were prepared as previously described.10 The preparation of derivatives 1–3 will be reported elsewhere. The photophysical properties of the new BODIPY derivatives (1–6, Scheme 1) were explored in diluted solutions (2 × 10−6 M) using solvents (spectroscopy grade), from apolar to polar and polar/protic, in 1 cm optical pathway quartz cuvettes. The samples in different media were prepared by adding the corresponding solvent to an adequate amount of a concentrated stock solution (ca. 10−3 M) of the corresponding dye in acetone, after vacuum evaporation of the solvent.UV-Vis absorption and fluorescence spectra were recorded on a Varian model CARY 4E spectrophotometer and a SPEX Fluorolog 3–22 fluorimeter, respectively. The fluorescence quantum yield (φ) was obtained using a methanolic solution of adequate commercial BODIPY dyes as reference. Radiative decay curves were registered with the time correlated single-photon counting technique (Edinburgh Instruments, model FL920) with a time resolution of 30 ps by means of a multichannel plate detector (Hamamatsu MCP C4878). The emission was monitored with a double monochromator at the maximum emission wavelength after excitation at 370 or 470 nm, depending on the dye, by means of pulsed diode lasers (PicoQuant, model LDH370 and 470, respectively) with recorded wide pulses of 150 ps and operating at 20 MHz repetition rate. The measured fluorescence decay curves were deconvoluted from the instrument response function (recorded by a LUDOX scatter) and determined from the slope of the exponential fit by means of iterative software supplied with the instruments. The goodness of the fit was described by the chi-square statistical parameter and by the analysis of the residual distribution. The radiative (kfl) and non-radiative (knr) rate constants were calculated by means of ϕ/τ and (1 − ϕ)/τ, respectively.
Quantum mechanic calculations were performed by Gaussian 09 software. The ground and excited state geometries were fully optimized with the B3LYP (Density Functional Theory, DFT) and ab initioCIS methods respectively, using in both cases the double valence 6-31G basis set. Absorption properties were predicted by the Time Dependent (TD-B3LYP) method and semiempirical ZINDO method as the Franck–Condon transition from the ground states. The solvent effect was considered by the Polarizable Continuum Model (PCM).
Results and Discussion
Alkyl-BODIPY (1–3).
Monosubstituted alkyl BODIPY dyes bearing n-propyl, n-penthyl and 1-phenylethyl groups at the meso position (Scheme 1) showed the typical absorption and emission bands of BODIPY chromophore.11 They were located at around 495 and 505 nm, respectively (Fig. 1), at slightly higher energies with respect to unsubstituted BODIPY (505 nm and 515 nm, respectively). Such hypsochromic shifts can be related to the inductive effect of the alkyl groups. Indeed, quantum mechanical calculations suggest that the electron density at the central 8-carbon of the BODIPY chromophore increases upon excitation (Fig. 1).2 Consequently the inductive effect +I of the alkyl groups will raise up the energy of the LUMO state with regard to the HOMO, slightly increasing the energy gap between both states. The small bathochromic shift (around 5 nm) observed for dye 2 with respect to the other n-alkyl analogs (1 and 3), should be ascribed to the presence of the phenyl group, which reduces the inductive donor character of the substituent, recovering the spectral band position of the parent BODIPY core. In fact, the calculated energy gap slightly increases (0.1 eV) for dye 2.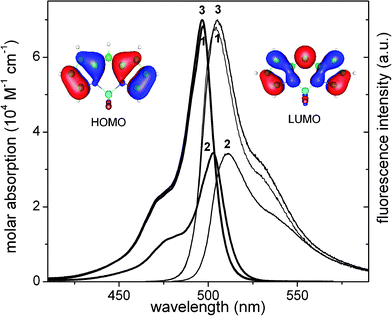 | ||
| Fig. 1 Absorption and fluorescence normalized spectra of dye 1, 2 and 3 in diluted (2 × 10−6 M) solutions of c-hexane. Electronic density in the HOMO and LUMO state of the unsubstituted BODIPY. | ||
The photophysical behaviour of these derivatives (Table 1) were very similar to that of the 8-unsubstituted chromophore,11 with a high fluorescence quantum yield (φ > 0.90) and lifetime (τ > 6 ns), leading to a high radiative rate constant (kfl > 1.4 × 108 s−1) and a low non-radiative deactivation process (knr < 0.1 × 108 s−1), a short Stokes shift (ΔνSt < 400 cm−1) and a high molar absorption coefficient (εmax ∼ 7 × 104 M−1 cm−1). As an exception, dye 2 showed an unusual low molar absorption coefficient.
| BODIPY | λ ab (nm) | ε max (104M−1cm−1) | λ fl (nm) | Δν St (cm−1) | φ | τ (ns) | k fl (108s−1) | k nr (108s−1) |
|---|---|---|---|---|---|---|---|---|
| absorption (λabs) and fluorescence (λflu) wavelength, Stokes shift (ΔνSt), molar absorption coefficient (εmax), fluorescence quantum yield (φ) and lifetime (τ), radiative (kfl) and non-radiative (knr) rate constant. | ||||||||
| BDP | 503.5 | 7.6 | 510.5 | 265 | 0.96 | 6.47 | 1.48 | 0.06 |
| 1 | 497.0 | 6.8 | 505.0 | 315 | 0.95 | 6.23 | 1.52 | 0.08 |
| 2 | 503.0 | 3.4 | 512.0 | 355 | 0.92 | 6.65 | 1.38 | 0.12 |
| 3 | 496.5 | 7.0 | 506.5 | 385 | 0.97 | 6.50 | 1.49 | 0.04 |
The solvent dependence of the photophysical parameters of these derivatives (see Table S1, ESI†) was as commonly observed for an 8-unsubstituted derivative, with short displacement of the absorption and fluorescence bands to lower energies from apolar to polar and protic solvents. The fluorescence capacity did not depend practically on the nature of the solvent.
Consequently, alkylation at the meso position of the BODIPY core did not damage its excellent fluorescence capacity.
p-bromophenyl-BODIPY (4)
The presence of the electron withdrawing p-bromophenyl group at central position of the BODIPY chromophore (Scheme 1) induced important changes in the photophysical behavior. Fig. 2 shows a large bathochromic shift of 4 in the absorption and fluorescence bands (around 80–90 nm) and a decrease in the fluorescence lifetime compared with 1 (Fig. 2). | ||
| Fig. 2 Absorption and fluorescence spectra (A) and decay curve (B) of compound 4 in diluted solutions of c-hexane. The corresponding spectra and curve of dye 1 are also included for comparison. | ||
As mentioned, the meso position was very sensitive to the presence of substituents with electronic acceptor/donor character, since an important increase in the electronic density at this position took place when an electron jumped from the HOMO to the LUMO state, the involved molecular orbitals in the S0 → S1 spectral transition (Fig. 1). Therefore, important spectral shifts are expected by the incorporation of substituents with high electron donor/acceptor ability at this central position. In the present case, the electron withdrawing Br atom stabilized the LUMO state more extensively than the HOMO state, and hence the energy gap between both states decreased.
Table 2 summarizes the photophysical parameters of compound 4 in several solvents, covering the complete apolar, polar and polar/protic range. Although the molar absorption coefficient of this dye remained high (εmax ∼ 7.7 × 104 M−1 cm−1), both the fluorescence quantum yield (φ ∼ 0.4) and the lifetime (τ ∼ 3 ns) decreased with respect to those values of its alkyl partners (Table 1) in not very acid protic solvents. These changes are mainly due to an increase in the non-radiative deactivation rate constant (knr ∼ 2 × 108s−1) rather than to a decrease in the radiative deactivation (kfl ∼ 1.3 × 108 s−1). The increase in the non-radiative deactivation may be due to an augmentation to the internal conversion and/or intersystem crossing processes. For instance, the presence of the Br atom could imply an increase in the intersystem crossing probability by the heavy atom effect. However, the Br atom is attached to the BODIPY chromopore by a phenyl group, and consequently its intramolecular heavy atom effect should be very low.
| Solvent | λ ab (nm) | ε max (104M−1cm−1) | λ fl (nm) | Δν St (cm−1) | φ | τ (ns) | k fl (108s−1) | k nr (108s−1) |
|---|---|---|---|---|---|---|---|---|
| F3-ethanol | 574.0 | 7.5 | 591.5 | 525 | 0.54 | 5.40 | 1.00 | 0.85 |
| methanol | 577.5 | 7.7 | 597.0 | 570 | 0.40 | 3.53 | 1.13 | 1.70 |
| ethanol | 579.0 | 7.5 | 598.5 | 525 | 0.43 | 3.74 | 1.15 | 1.52 |
| acetone | 578.5 | 7.6 | 599.0 | 595 | 0.39 | 3.31 | 1.17 | 1.84 |
| ethyl acetate | 579.0 | 7.9 | 599.0 | 580 | 0.39 | 3.46 | 1.12 | 1.76 |
| c-hexane | 583.0 | 7.5 | 600.0 | 485 | 0.39 | 2.89 | 1.35 | 2.11 |
Therefore, the reduction in the fluorescence capacity should be mainly assigned to an increase in the internal conversion process, probably due to the rotational motion of the phenyl group directly linked to the BODIPY core. Indeed, it has been demonstrated that the free rotation of a phenyl group attached at the central 8-position of BODIPY core drastically reduces the fluorescence capacity of the chromophore,12 but when this rotational motion is reduced (for instance, by the incorporation of methyl groups at the ortho position of the phenyl group and/or at the 1- and 7-position of the BODIPY core) the fluorescence ability of the chromophore is recovered.13
The highest values of the fluorescence quantum yield and lifetime were achieved in polar/protic media due to a decrease in the non radiative rate constant (Table 2). This dependency of the knr value on the solvent characteristics confirms that the non-radiative deactivation from the excited state is due to internal conversion mechanisms rather than intersystem crossing processes.
From these results it can be concluded that dye 4 can operate as an active medium of dye lasers emitting at the red part of the visible region. Furthermore, improvement in the lasing efficiency could be obtained with a more constricted structure, for instance by restricting the intramolecular rotational motion of the p-Br-phenyl group by attaching bulky groups at the ortho positions of the phenyl group and/or at the 1- and 7- positions of the BODIPY ring.
Triphenylaminethiophene-BODIPY (5)
The derivative bearing a thiophene group, α-substituted with a triphenylamine (TPA) unit, at the meso position of the BODIPY core (Scheme 1), presents a clear change in the shape of its absorption spectrum, as shown in Fig. 3. In fact, as well as the typical absorption band of alkyl-BODIPYs, centered at around 520 nm, a new shoulder appeared at a lower energy (around 570 nm in c-hexane). This new band became much more evident and bathochromically shifted in polar media, suggesting the formation of a new absorbing entity characterized by a high dipole moment. Roncali and coworkers have studied the formation of intramolecular charge transfer (ICT) complexes between TPA-thiophene and several π-conjugated systems by UV-Vis spectroscopy.14 The authors point out the high electron donor capacity of such a group and that the intensity and position of the absorbing complexes depends on the electron acceptor properties of the π-system attached to the thiophene group. In compound 5, the same donor unit is present at the meso position of the BODIPY core, which can act as an electron acceptor. The HOMO − 1 and LUMO molecular orbitals are exclusively located in the BODIPY, while the HOMO and LUMO + 1 are located in the TPA-thiophene (Fig. S1, ESI†). Consequently, the new absorption band placed at lower energies should be assigned to an intramolecular charge transfer (ICT) complex formed between the TPA-thiophene partner and the BODIPY core, which already exists in the ground state (i.e., transition from the HOMO in the donor TPA-thiohene to the LUMO in the acceptor BODIPY). Such an ICT complex is characterized by a high dipole moment and is further stabilized in polar media. Theoretical simulation was not able to reproduce such new bathochromic band (Table S2, ESI†), even in polar solvent. | ||
| Fig. 3 Absorption and excitation (dashed line) (A) and fluorescence (B) spectra of compound 5 in c-hexane (a), acetone (b) and F3-ethanol (c). Inset: evolution of the fluorescence spectra in ethyl acetate/c-hexane mixtures with different volume fractions of ethyl acetate: a-0%, b-5%, c-10%, d-25%, e-50%, f-75% and g-100%. | ||
The excitation at the BODIPY absorption band (490 nm) leads to a broad fluorescence band, centered at 600 nm in c-hexane (Fig. 3). Consequently, compound 5 is characterized by a large Stokes shift (2565 cm−1), much higher than the typical one for alkyl-BODIPY dyes (around 500 cm−1). The fluorescence quantum yield (φ = 0.32) and lifetime (τ = 1.36 ns) were reduced by more than 50% compared to the values of the alkyl-BODIPY derivatives as a result of an important increase in the probability of the non-radiative deactivation processes (knr = 5×108s−1).
The shape and position of the emission band did not change with the excitation wavelength, even after exciting the new CT absorption band (around 570 nm). The corresponding excitation spectrum perfectly matched the absorption one (Fig. 3). All this experimental evidence indicates that the fluorescence band is due to the emission from the excited state of the ICT complex, which can be populated either by direct excitation (570 nm) or from the locally excited (LE) state of BODIPY (490 nm) after electron transfer from the donor TPA-thiophene. The fluorescence decay curve after excitation at 470 nm was analyzed as monoexponential, without any grown-in component, suggesting a very fast population (faster than the time resolution of our single photon counter, ~30 ps) of the ICT state.
Polar environments further reduced the fluorescent ability of this derivative and it became nearly non-fluorescent in the most polar media (φ < 0.01 in F3-ethanol, Fig. 3). Just a weak remnant fluorescence band was observed at 550–600 nm, depending on the solvent, after the excitation at the main absorption band. Generally, the ICT states were stabilized in polar media due to their high dipole moment. However, the charge transfer process can be so favored that it leads to a charge separation (CS) ionic state (zwitterionic form), which is not fluorescent and quenches the emission from the CT complex. Thus, in polar media only a remnant emission was observed at shorter wavelengths (for wide excitation and emission slits), which should be ascribed to the weak emission from the LE state.
The CT nature of the emissive species was confirmed by the evolution of the fluorescence band in c-hexane/ethyl acetate mixtures. Indeed, a progressive increase in the solvent polarity by increasing the ethyl acetate content led to a red shift of the emission band and a concomitant decrease of the fluorescence intensity (inset Fig. 3). In pure ethyl acetate the fluorescence band is nearly negligible.
Taking into account that the fluorescence efficiency of this derivative is very sensitive to the solvent polarity, it can be potentially used as a fluorescent probe to monitor environmental polarity, achieving a red fluorescence emission in apolar environments, but nearly no fluorescence signal in polar media.
Pyrene-BODIPY dyad (6)
Finally, we considered the influence of a pyrene chromophore, attached also at the meso position, on the photophysics of the BODIPY (Scheme 1). The UV-Vis absorption spectrum of this dye, depicted in Fig. 4, is characterized by a strong (εmax ∼ 7×104 M−1 cm−1) band in the visible region (centered at around 505 nm), attributed to the BODIPY core, and several UV bands assignable to different S0 → Sn electronic transitions of the pyrene chromophore with their typical and clear vibrational structure. In this UV region (around 370 nm) the BODIPY also presents weak absorption bands (Fig. S2, ESI†). This result suggests that there is no electronic interaction between the two moieties in the ground state. Indeed, the optimized geometry places the pyrene at a 63° with respect to the BODIPY fragment, without altering the BODIPY core planarity. Thus, the π-electronic clouds of both chromophores do not overlap and both chromophoric systems are independent of each other. These trends are theoretically confirmed since similar absorption properties to the BDP chromophore are predicted upon the presence of the pyrene, corroborating the nearly negligible effect of such unit, at least in the ground state. | ||
| Fig. 4 UV-Vis absorption and fluorescence, under λexc = 480 nm (A) and under λexc = 340 nm (B), spectra of compound 6 in c-hexane. | ||
The direct excitation of the S0-S1 absorption band of the BODIPY chromophore at the visible region (480 nm) provides the typical fluorescent emission of the BODIPY core. In c-hexane this emission band is placed at 520 nm (Fig. 4) with a modest fluorescence quantum yield (φ = 0.30) and low lifetime (τ = 1.93 ns), owing to a high non-radiative deactivation rate constant (knr = 3.6 × 108 s−1). Probably, the pyrene group can have some freedom to rotate, like in other 8-aromatic-BODIPY dyes,13 enhancing the non-radiative deactivation processes. Blocking such internal movement, by the substitution at the adjacent positions to the meso, places the chromophore in a more rigid and perpendicular disposition, improving the fluorescent ability.15
On the other hand, the UV excitation (340 nm) at the pyrene moiety gave rise to a weak fluorescent band at around 420 nm, typical of the pyrene chromophore, and an intense emission band in the visible region (520 nm), attributed to the BODIPY core (Fig. 4). These results suggest an intramolecular excitation energy transfer process (intra-EET) from the donor pyrene to the acceptor BODIPY. The excitation energy transfer from pyrene to BODIPY is not total since a remnant emission from the pyrene fluorophore is observed in the emission spectrum. The mechanism of the intra-EET process can be assigned to through-bond or through-space interactions. Although the spectral overlap between the emission of the donor pyrene and the S0-S1 absorption of the BODIPY is small, the possibility of Förster type EET through the S0-S2 and higher energies absorption bands of the BODIPY (see Fig. S2, ESI†) has been reported.16 Furthermore, the mutual orientation of the transition dipole moments of the chromophores in compound 6 is also adequate. To properly asses the operating mechanism in the intra-EET process we have measured the fluorescence spectra at low temperatures (down to 77 K, Fig. S3, ESI†). At low temperatures the electron transfer (involved in the through-bond EET) should be stopped while the EETvia dipole–dipole coupling should still take place. The intra-EET of compound 6 disappeared lowering the sample temperature, confirming that the intra-EET process in compound 6 takes place through-bond.17
As a consequence, the visible BODIPY fluorescence emission of this bichromophoric system can be monitored far away from the excitation of the pyrene at the UV, reducing the detection of the excitation light scattering and improving the use of this dye as a fluorescent probe, mainly in biochemical systems where the size of biological entities (proteins, membranes, cells, etc) can induce high scattering of the excitation light.
While the absorption spectrum of compound 6 was nearly solvent independent, the fluorescence properties of this derivative presented a marked dependency on the nature of the solvent. The theoretically predicted geometries in apolar (cyclohexane) and polar (methanol) media are very close, with similar twist angles for the pyrene in both the ground and excited states. Fig. 5 shows the fluorescence spectrum of compound 6 in several polar and polar/protic solvents after direct excitation at the BODIPY core in the visible region (480 nm). Besides the normal emission of the BODIPY core at 510 nm, which was strongly quenched, a new emission band appeared at longer wavelengths. The intensity and position of the new band depended on the solvent. In ethyl acetate this emission band was intense and placed at around 650 nm with a fluorescence lifetime of 3.5 ns. The new red band became less intense in more polar and protic solvent (in F3-ethanol practically disappears) and progressively shifted to lower energies (around 700 nm in acetone or methanol). In these solvents, the decay curves are analyzed as biexponentials with a short lifetime of 50 ps and a moderate lifetime of 1 ns with similar statistical weights. As occurs in compound 5, the HOMO is placed in the pyrene, whereas the LUMO in the BODIPY (Fig. S1, ESI†).
 | ||
| Fig. 5 Fluorescence spectra, exciting at 480 nm (A) and 340 nm (B), of compound 6 in ethyl acetate (a), acetone (b), ethanol (c), methanol (d), and F3-ethanol (e). | ||
These results clearly show that upon excitation there is an important interaction between both chromophoric systems in polar media giving rise to a new emission band. Taking into account the solvatochromic shift of this band (from 650 in ethyl acetate to 700 nm in methanol), the new emitting species would be characterized by an important dipole moment, and its excited state is stabilized in polar media. Excimer emission of the pyrene is discarded since the measurements are carried out in diluted solutions. At first impression, the formation of an exciplex is rather improbable in view of the excited state optimized geometry, since pyrene and BODIPY are located in planes twisted 72°, therefore, avoiding any cofacial arrangements between them. However, it has been previously suggested that exciplex-like emission occurs even in rigid donor–acceptor systems.18 In these cases, the intramolecular charge transfer state can convert into an exciplex-like state upon a Coulombic interaction (harpooning effect),19 causing a weak and red-shifted emission. Probably this approach occurs in compound 6, where the red emission detected in polar media could be attributed to an exciplex-like emission with a marked charge transfer character. Surprisingly, this emission was slightly favored at low temperatures, matching results previously reported.18c
In fact, in compound 6, the BODIPY core may serve as an electron acceptor (A) while the pyrene ring is somewhat electron donor (D). Molecules with such as D–A structures often show interesting optical properties, e.g., solvatochromism.20 In nonpolar solvents, the LE state of the BODIPY luminogen emits intense green light. Increasing the solvent polarity brings the luminogens from the LE states to the intramolecular charge transfer (ICT) state, and probably afterwards to the exciplex, causing a large bathochromic shift in the emission color and also a dramatic decrease in the emission efficiency.20,21
To examine whether the ICT process is involved in 6 in polar solvents, we systematically changed the polarity of the media by admixing polar THF and nonpolar hexane in different ratios (Fig. 6). When the hexane fraction (fh) in the THF/hexane mixture increased from 0 to 90%, the emission color of 6 changed gradually from magenta to orange, then yellow, and finally to green.
 | ||
| Fig. 6 Photographs (under UV illumination) and emission spectra of 6 (under visible light excitation) in THF/hexane mixtures with different fractions of hexane (fh, vol %). Solution concentration: 10 μM. | ||
The visible observation was verified by the spectroscopic analysis. The absorption spectrum of 6 in THF solution was similar to that recorded in c-hexane (Fig. 4). However, the emission of 6 was very sensitive to solvent polarity change (Fig. 6). In pure THF, the emission spectrum of 6 (λexc = 480 nm) was dominated by its ICT emission at 626 nm. Its LE emission appeared as a small shoulder at 531 nm. When the fh value in the solvent mixture was progressively increased from 0 to 70%, the ICT peak moved continuously to 603 nm, while the LE peak remained the same. Both the ICT and LE peaks were intensified upon hexane addition. In the solvent mixture with 80 vol % hexane, the LE emission was dominated in the spectrum, while the ICT emission diminished to a weak shoulder. The ICT peak disappeared when the hexane content reached 90 vol %, and the spectrum only exhibited an LE emission at 531 nm (Fig. 6). Indeed, in pure hexane, the emission at 532 nm was characterized by a fluorescence quantum yield that was 4.6-fold higher than that in pure THF (from 0.028 in THF to 0.13 in hexane).
The fluorescence behavior under UV excitation (340 nm) was also strongly dependent on the solvent. In polar media, the dominant band was the emission from the pyrene. The emission from the LE state of the BODIPY core, previously detected in apolar solvents, is no longer obtained in polar media and just a weak signal from the ICT state (except in ethyl acetate where it was more intense) was detected (Fig. 5). In a previous work about a similar system but with the pyrene placed perpendicular to the BODIPY due to steric reasons, no emission from the pyrene was detected due to efficient intra-EET in all the solvents.15 However, in compound 6, where the pyrene had some freedom to oscillate, the intra-EET process most likely populates the LE state of the BODIPY but the further stabilization of the ICT state in the polar media quenches the LE state.
Summarizing, compound 6 is a very versatile dye: in apolar media the visual emission of the BODIPY core can be monitored far away from the excitation of the pyrene chromophore in the UV by means of a through-bond intra-EET process, whereas in polar media the green–red emission from a ICT state can be modulated depending on the nature of the solvent. So, this compound can be used as fluorescence probe to monitor environmental polarity. Besides, the blue emission from the pyrene can be achieved in polar/protic solvents under UV excitation and blue and red emission can coexist in media with moderate polarity (i.e. ethyl acetate).
Conclusions
The emission from BODIPY dyes can be modulated by the incorporation of appropriate substituents at the meso 8-position. The attachment of different aromatic groups (triphenylamine, thiophenep-substituted phenyl, and pyrene) can induce new processes which could be sensitive to solvent properties. In this way new fluorescence probes based on BODIPY fluorophore can be developed.The bichromophoric pyrene-BODIPY system shows an intramolecular energy transfer process in apolar media with an interesting green emission after UV excitation. In polar media, the formation of an ICT- state, which probably evolves to an exciplex state due to the harpooning effect, shifts the emission band to the red. Blue emission from the pyrene chromophore can be achieved after UV irradiation when the energy transfer process is blocked and the emission from the ICT exciplex is quenched, for instance in polar/protic media. Consequently, blue/green/red emissions can be obtained from this derivative depending on excitation conditions and on the solvent.
Acknowledgements
This work was supported by Ministerio de Ciencia e Innovación, projects MAT2007-65778-C02-02 and MAT2010-20646-C04-03. Part of this work was supported by Grant GTO-2007-C02-69094 (CONCyTEG, Mexico). V. Martinez thanks the Gobierno Vasco (IT339-10) for postdoctoral contract.References
- (a) E. Yariv and R. Reisfeld, Opt. Mater., 1999, 13, 49–54 CrossRef CAS; (b) M. Ahmad, T. A. King, D-K. Ko, B. H. Cha and J. Lee, J. Phys. D: Appl. Phys., 2002, 35, 1473–1476 Search PubMed; (c) A. Costela, I. García-Moreno and R. Sastre, Phys. Chem. Chem. Phys., 2003, 5, 4745–4763 RSC; (d) Y. Yang, M. Wang, G. Qian, Z. Wang and X. Fan, Opt. Mater., 2004, 24, 621–628 Search PubMed; (e) A. Costela, I. García-Moreno, M. Pintado-Sierra, F. Amat-Guerri, R. Sastre, M. Liras, F. López Arbeloa, J. Bañuelos and I. López Arbeloa, J. Phys. Chem. A, 2009, 113, 8118–8124 CrossRef CAS.
- (a) R. Y. Lai and A. J. Bard, J. Phys. Chem. B, 2003, 107, 5036–5042 CrossRef CAS; (b) F. López Arbeloa, J. Bañuelos, V. Martínez, T. Arbeloa and I. López Arbeloa, Int. Rev. Phys. Chem., 2005, 24, 339–374 CrossRef CAS; (c) A. D. Quartarolo, N. Russo and E. Sicilia, Chem.–Eur. J., 2006, 12, 6797–6803 CrossRef; (d) A. Cui, X. Peng, J. Fan, X. Chen, Y. Wu and B. Chuo, J. Photochem. Photobiol., A, 2007, 186, 85–92 CrossRef CAS; (e) W. Qin, T. Rohand, W. Dehaen, J. N. Clifford, K. Driesen, D. Beljonne, B. Van Averbeke, M. Van der Auweraer and N. Boens, J. Phys. Chem. A, 2007, 111, 8588–8597 CrossRef CAS; (f) M. A. H. Alamiry, A. C. Benniston, G. Copley, K. J. Elliot, A. Harriman, B. Stewart and Y-G. Zhi, Chem. Mater., 2008, 20, 4024–4032 CrossRef CAS; (g) F. López Arbeloa, J. Bañuelos, V. Martínez, T. Arbeloa and I. López Arbeloa, Trends in Physical Chemistry, 2008, 13, 101–122 Search PubMed.
- (a) A. A. Gorman, I. Hamblett, T. A. King and M. D. Rahn, J. Photochem. Photobiol., A, 2000, 130, 127–132 CrossRef CAS; (b) T. G. Pavlopoulos, Prog. Quantum Electron., 2002, 26, 193–224 CrossRef CAS.
- (a) A. Loudet and K. Burgess, Chem. Rev., 2007, 107, 4891–4932 CrossRef CAS; (b) T. E. Wood and A. Thompson, Chem. Rev., 2007, 107, 1831–1861 CrossRef CAS; (c) G. Ulrich, R. Ziessel and A. Harriman, Angew. Chem. Int. Ed., 2008, 47, 1185–1201 Search PubMed; (d) A. C. Benniston and G. Copley, Phys. Chem. Chem. Phys., 2009, 11, 4124–4131 RSC; (e) M. Benstead, G. H. Mehl and R. W. Boyle, Tetrahedron, 2001, 67, 3573–3601 Search PubMed.
- (a) K. Umezawa, A. Matsui, Y. Nakamura, D. Citterio and K. Suzuki, Chem.–Eur. J., 2009, 15, 1096–1106 CAS; (b) M. J. Ortiz, I. García-Moreno, A. R. Agarrabeitia, G. Durán-Sampedro, A. Costela, R. Sastre, F. López Arbeloa and I. López Arbeloa, Phys. Chem. Chem. Phys., 2010, 12, 7804–7811 RSC; (c) V. R. Donuru, S. Zhu, S. Green and H. Liu, Polymer, 2010, 51, 5359–5368 CAS; (d) V. Leen, W. Qin, W. Yang, J. Cui, C. Xu, X. Tang, W. Liu, K. Robeyns, L. Van Meervelt, D. Beljonne, R. Lazzaroni, C. Tonnele, N. Boensa and W. Dehaen, Chem.–Asian J., 2010, 5, 2016–2026 Search PubMed.
- (a) J. H. Boyer, A. Haag, M. L. Soong, K. Thangaraj and J. H. Boyer, Appl. Opt., 1991, 30, 3788–3789 CrossRef CAS; (b) J. H. Boyer, A. M. Haag, G. Sathyamoorthi, M. L. Soong, K. Thangaraj and T. G. Pavlopoulos, Heteroat. Chem., 1993, 4, 39–49 CrossRef CAS.
- (a) R. W. Wagner, J. S. Lindsey, J. Seth, V. Palaniappan and D. F. Bocian, J. Am. Chem. Soc., 1996, 118, 3996–3997 CrossRef CAS; (b) R. K. Lammi, A. Ambroise, T. Balasubramanian, R. W. Wagner, D. F. Bocian, D. Holten and J. S. Lindsey, J. Am. Chem. Soc., 2000, 122, 7579–7591 CrossRef CAS; (c) F. D’Souza, P. M. Smith, M. E. Zandler, A. L. McCarty, M. Itou, Y. Araki and O. Ito, J. Am. Chem. Soc., 2004, 126, 7898–7907 CrossRef CAS; (d) A. Harriman, L. J. Mallon, S. Goeb and R. Ziessel, Phys. Chem. Chem. Phys., 2007, 9, 5199–5201 RSC; (e) T. Rousseau, A. Cravino, E. Ripaud, P. Leriche, S. Rihn, A. De Nicola, R. Ziessel and J. Roncali, Chem. Commun., 2010, 46, 5082–5084 RSC; (f) S. Kolemen, Y. Cakmak, S. Erten-Ela, Y. Altay, J. Brendel, M. Thelakkat and E. U. Akkaya, Org. Lett., 2010, 12, 3812–3815 CrossRef CAS; (g) O. A. Bozdemir, Y. Cakman, F. Sozmen, T. Ozdewmir, A. Siemiarczuk and E. U. Akkaya, Chem.–Eur. J., 2010, 16, 6346–6351 CrossRef CAS.
- (a) C. Goze, G. Ulrich, L. Charbonnière, M. Cesario, T. Prangé and R. Ziessel, Chem.–Eur. J., 2003, 9, 3748–3755 CrossRef CAS; (b) J. L. Bricks, A. Kovalchuk, C. Trieflinger, M. Nofz, M. Büschel, A. I. Tolmachev, J. Daub and K. Rurack, J. Am. Chem. Soc., 2005, 127, 13522–13529 CrossRef CAS; (c) T. Kálai and K. Hideg, Tetrahedron, 2006, 62, 10352–10360 CrossRef CAS; (d) H. Sunahara, Y. Urano, H. Kojima and T. Nagano, J. Am. Chem. Soc., 2007, 129, 5597–5604 CrossRef CAS; (e) H. Lu, S. Zhang, H. Liu, Y. Wang, Z. Shen, C. Liu and X. You, J. Phys. Chem. A, 2009, 113, 14081–14086 CrossRef CAS; (f) X. Qian, Y. Xiao, Y. Xu, X. Guo, J. Qian and W. Zhu, Chem. Commun., 2010, 46, 6418–6436 RSC; (g) O. A. Bozdemir, R. Guliyev, O. Buyukcakir, S. Onur, S. Kolemen, G. Gulseren, T. Nalbantoglu, H. Boyaci and E. U. Akkaya, J. Am. Chem. Soc., 2010, 132, 8029–8036 CrossRef CAS.
- (a) R. P. Haugland, Handbook of Fluorescent Proves and Research Chemicals, Molecular Probes, Eugene, OR, 1999 Search PubMed; (b) M. L. Metzker, J. Lu and R. A. Gibbs, Science, 1996, 271, 1420–1422 CAS; (c) S. A. Farber, M. Pack, S-Y. Ho, I. D. Johnson, D. S. Wagner, R. Dosch, M. C. Mullins, H. S. Hendrickson, E. K. Hendrickson and M. E. Halpern, Science, 2001, 292, 1385–1388 CrossRef CAS; (d) J-S. Lee, N-Y. Kang, Y. K. Kim, A. Samanta, S. Feng, H. K. Kim, M. Vendrell, J. H. Park and Y-T. Chang, J. Am. Chem. Soc., 2009, 131, 10077–10082 CrossRef CAS.
- (a) E. Peña-Cabrera, A. Aguilar-Aguilar, M. González-Domínguez, E. Lager, R. Zamudio-Vázquez, J. Godoy-Vargas and F. Villanueva-García, Org. Lett., 2007, 9, 3985–3988 CrossRef CAS; (b) E. Lager, J. Liu, A. Aguilar-Aguilar, B. Z. Tang and E. Pena-Cabrera, J. Org. Chem., 2009, 74, 2053–2058 CrossRef CAS.
- (a) I. J. Arroyo, H. Rongrong, G. Merino, B. Z. Tang and E. Peña-Cabrera, J. Org. Chem., 2009, 74, 5719–5722 CrossRef CAS; (b) C. F. A. Gómez-Durán, I. García-Moreno, A. Costela, V. Martín, R. Sastre, R. Sastre, J. Bañuelos, F. López Arbeloa, I. López Arbeloa and E. Peña-Cabrera, Chem. Commun., 2010, 46, 5103–5105 RSC.
- (a) F. Li, S. I. Yang, Y. Ciringh, J. Seth, C. H. Martin, D. L. Singh, D. Kim, R. R. Birge, D. F. Bocian, D. Holten and J. S. Lindsey, J. Am. Chem. Soc., 1998, 120, 10001–10017 CrossRef CAS; (b) A. Burghart, H. Kim, M. B. Welsch, L. H. Thoresen, J. Reibenspies and K. Burgess, J. Org. Chem., 1999, 64, 7813–7819 CrossRef CAS; (c) J. Chen, A. Burghart, A. Derecskei-Kovacs and K. Burgess, J. Org. Chem., 2000, 65, 2900–2906 CrossRef CAS; (d) H. L. Kee, C. Kirmaier, L. Yu, P. Thamyongkit, W. J. Youngblood, M. E. Calder, L. Ramos, B. C. Noll, D. F. Bocian, W. R. Scheidt, R. R. Birge, J. S. Lindsey and D. Holten, J. Phys. Chem. B, 2005, 109, 20433–20443 CrossRef CAS.
- (a) J. Bañuelos, F. López Arbeloa, V. Martínez, T. Arbeloa, F. Amat-Guerri, M. Liras and I. López Arbeloa, Chem. Phys. Lett., 2004, 385, 29–35 CrossRef CAS; (b) S. Badré, V. Monnier, R. Méallet-Renault, C. Dumas-Verdes, E. Y. Schmidt, A. I. Mikhaleva, G. Laurent, G. Levi, A. Ibañez, B. A. Trofimov and R. B. Pansu, J. Photochem. Photobiol., A, 2006, 183, 238–246 CrossRef CAS; (c) Q. Zheng, G. Xu and P. N. Prasad, Chem.–Eur. J., 2008, 14, 5812–5819 CrossRef CAS.
- P. Leriche, P. Frére, A. Cravino, O. Alévêque and J. Roncali, J. Org. Chem., 2007, 72, 8332–8336 CrossRef CAS.
- R. Ziessel, C. Goze, G. Ulrich, M. Césario, P. Retailleau, A. Harriman and J. P. Rostron, Chem.–Eur. J., 2005, 11, 7366–7378 CrossRef CAS.
- (a) A. Harriman, G. Izzet and R. Ziessel, J. Am. Chem. Soc., 2006, 128, 10231–10239 CrossRef CAS; (b) A. Harriman, L. J. Mallon, S. Goeb, G. Ulrich and R. Ziessel, Chem.–Eur. J., 2009, 15, 4553–4564 CrossRef CAS.
- (a) C-W. Wan, A. Burghart, J. Chen, F. Bergström, L. B-A. Johansson, M. F. Wolford, T. G. Kim, M. R. Topp, R. M. Hochtrasser and K. Burgess, Chem.–Eur. J., 2003, 9, 4430–4441 CrossRef CAS; (b) T. G. Kim, J. C. Castro, A. Loudet, J.G-S. Jiao, R. M. Hochtrasser, K. Burgess and M. R. Topp, J. Phys. Chem. A, 2006, 110, 20–27 CrossRef CAS; (c) M. A. H. Alamiry, A. Harriman, L. J. Mallon, G. Ulrich and R. Ziessel, Eur. J. Org. Chem., 2008, 2774–2782 Search PubMed; (d) R. Ziessel and A. Harriman, Chem. Commun., 2011, 47, 611–631 RSC.
- (a) T. Scherer, I. H. M. Van Stokkum, A. M. Brouwer and J. W. Verhoeven, J. Phys. Chem., 1994, 98, 10539–10549 CrossRef; (b) B. Wegewijs, J. W. Verhoeven and S. E. Braslavsky, J. Phys. Chem. A, 1996, 100, 8890–8894 Search PubMed; (c) S. Mula, K. Elliot, A. Harriman and R. Ziessel, J. Phys. Chem. A, 2010, 114, 10515–10522 Search PubMed.
- (a) J. W. Verhoeven, T. Scherer and R. J. Willemse, Pure Appl. Chem., 1993, 65, 1717–1722 CrossRef CAS; (b) J. W. Verhoeven, Pure Appl. Chem., 1990, 62, 1585–1596 CrossRef CAS; (c) A. C. Benniston, A. Harrimann, V. L. Whittle, M. Zelzer and R. W. Harrington, Photochem. Photobiol. Sci., 2010, 9, 1009–1017 RSC.
- R. Hu, E. Lager, A. Aguilar-Aguilar, J. Liu, J. W. Y. Lam, H. H. Y. Sung, I. D. Williams, Y. Zhong, K. S. Wong, E. Peña-Cabrera and B. Z. Tang, J. Phys. Chem. C, 2009, 113, 15845 CrossRef CAS.
- (a) F. López Arbeloa, T. López Arbeloa and I. López Arbeloa, Trends Photochem. Photobiol., 1994, 3, 145–155 Search PubMed; (b) Z. R. Grabowski, K. Rotkiewicz and W. Rettig, Chem. Rev., 2003, 103, 3899–4032 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Complementary photophysical data and theoretical results. See DOI: 10.1039/c1ra00020a |
| This journal is © The Royal Society of Chemistry 2011 |