Nylon 3 synthesized by ring opening polymerization with a metal-free catalyst
Hongjun
Yang
a,
Junpeng
Zhao
a,
Manqing
Yan
a,
Stergios
Pispas
b and
Guangzhao
Zhang
*ac
aHefei National Laboratory for Physical Sciences at Microscale, Department of Chemical Physics, University of Science and Technology of China, Hefei, P. R. China 230026. E-mail: gzzhang@ustc.edu.cn; Fax: +86-551-3606743; Tel: +86-551-3606763
bTheoretical and Physical Chemistry Institute, National Hellenic Research Foundation, 48 Vassileos Constantinou Ave., 11635, Athens, Greece
cFaculty of Materials Science and Engineering, South China University of Technology, Guangzhou, P. R. China 510640
First published on 19th October 2011
Abstract
We have synthesized nylon 3 via ring opening polymerization of 2-azetidinone (β-lactam) with 1-tert-butyl-4,4,4-tris(dimethylamino)-2,2-bis[tris(dimethylamino)- phosphoranylidenamino]-2Λ5,4Λ5-catenadi(phosphazene) (t-BuP4) as the catalyst in a mixture of dimethylacetamide (DMAc) and LiCl. The polymers have been characterized by nuclear magnetic resonance spectroscopy (NMR), Fourier transform infrared spectroscopy (FTIR), X-ray diffraction (XRD), laser light scattering (LLS) and viscometry. The synthesized nylon 3 is a linear and crystalline polymer with a molecular weight as high as 105 g mol−1. The intrinsic viscosity ([η]) relates to the weight average molecular weight (Mw) as [η] = 1.02 × 10−4Mw0.91. The effects of solvent, temperature and catalyst concentration on the polymerization have been examined. The molecular weight and yield increases with the amount of LiCl in the polymerization mixture, but both of them decrease with temperature at a temperature above 50 °C. As the catalyst concentration increases, the yield and the molecular weight of nylon 3 decrease. The possible mechanism for the initiation of polymerization is discussed.
Introduction
Nylon 3 or polyamide 3 with a β-amino acid skeleton, which is different from other commercial nylons in the amide linkages, usually exhibits interesting properties such as high capacity of moisture up-take, high glass transition temperature and high crystallinity.1–5Nylon 3 fibers are very close to silk in properties. Cationic nylon 3 was reported to have activities similar to those of natural host-defense peptides,6–11 so it is a promising material for tissue engineering and drug delivery. Furthermore, nylon 3 has been used as formaldehyde scavenger and polyoxymethylene stabilizer.4Nylon 3 was first synthesized by hydrogen transfer polymerization of acrylamide.12–15 Such nylon 3 is branched, and the presence of an olefinic double bond at its chain end usually decreases its thermal stability.16 The large amount of metal-based initiator used in this procedure is hard to remove after polymerization, which can drastically decreases their thermal stability and other physical properties. In addition, since most of the initiators do not dissolve in a common solvent, the polymerization is heterogeneous and difficult to control.
A series of mono-, bi-, tri- and tetra-substituted poly(β-lactams) have been successfully synthesized by ring opening polymerization with metal catalysts.17–21 However, nylon 3 can not be synthesized viapolymerization of unsubstituted β-lactam with such a metal catalyst. Recently, nylon 3 was synthesized by ring opening polymerization of unsubstituted β-lactam catalyzed by enzyme.22 Unfortunately, its molecular weight was very low. In this work, we have synthesized high molecular weight unsubstituted nylon 3 by the use of a metal-free catalyst, that is, (1-tert-butyl-4,4,4-tris(dimethylamino)-2,2-bis[tris(dimethylamino)-phosphoranylidenamino]-2Λ5,4Λ5-catenadi(phosphazene) (t-BuP4), which was revealed to be a very strong base.23,24 Recently, it was reported that t-BuP4 demonstrates efficiency in catalyzing ring opening polymerizations of oxyalkylenes,25–27ε-lactams,28 cyclopropane-1,1-dicarboxylates,29β-lactones30 and cyclic esters.31t-BuP4 was also used as a complexing agent for butyl lithium to synthesize block polymer.32 Particularly, it is soluble in some common solvents and can work in mild conditions.
Experimental section
Materials
The synthesis of β-lactam (2-azetidinone) is described in detail elsewhere.12t-BuP4 and anhydrous lithium chloride (LiCl) from Aldrich were used as received. Dimethylacetamide (DMAc) from Sinopharm was refluxed over calcium hydride and distilled under reduced pressure prior to use. Formic acid or HCOOH (98%) and other reagents from Sinopharm were used as received.Polymerization
LiCl (0.5 g) was dried in a vacuum at 40 °C for 3 days in a 50 mL round bottom flask. Subsequently, dried DMAc (30 mL) was introduced, and the solution was stirred for 30 min. Then, 20 mL of DMAc was distilled out so that traces of water in LiCl were removed together with DMAc. The remaining solution together with β-lactam was introduced into a Pyrex vial under dry nitrogen. After degassing by three freeze-pump-thaw cycles, t-BuP4 was added under dry nitrogen, and the polymerization was conducted at 25 °C for a certain time. Afterwards, the solution was poured into a large amount of acetone/water (5/1 v/v) mixture. The resulting polymer was collected and washed with the acetone/water mixture, and then dried in vacuum. Nylon 3 samples presented in this work are designated as Ny1 to Ny6 (Table 1).| Sample | r a | T b (°C) | t c (min) | Yield (%) | M w × 10−4 (g mol−1) d | 〈Rg〉 (nm) | 〈Rh〉 (nm) | 〈Rg〉/〈Rh〉 |
|---|---|---|---|---|---|---|---|---|
| a Molar ratio of t-BuP4 toβ-lactam. b Reaction temperature. c Reaction time. d Determined by SLS. | ||||||||
| Ny1 | 1/200 | 25 | 180 | 96 | 1.12 | 8 | 4 | 2.0 |
| Ny2 | 1/333 | 25 | 180 | 86 | 1.22 | 9 | 4 | 2.0 |
| Ny3 | 1/500 | 25 | 180 | 80 | 1.27 | 10 | 5 | 2.0 |
| Ny4 | 1/2000 | 25 | 180 | 74 | 10.5 | 38 | 17 | 2.2 |
| Ny5 | 1/2000 | 50 | 180 | 90 | 12.8 | 40 | 18 | 2.2 |
| Ny6 | 1/2000 | 80 | 180 | 54 | 7.66 | 29 | 13 | 2.2 |
Characterizations
Proton nuclear magnetic resonance (1H NMR) spectra were recorded on a Bruker AV400 NMR spectrometer using deuterated chloroform (CDCl3) and deuterated trifluoroacetic acid (TFA-d3) mixture (CDCl3/TFA-d3, 5/1, v/v) as the solvent. Carbon 13 nuclear magnetic resonance (13C NMR) spectra were recorded on the same spectrometer using trifluoroacetic acid (TFA-d3) as the solvent. FTIR spectra were recorded on a Bruker VECTOR-22 IR spectrometer. The spectra were collected at 64 scans with a spectral resolution of 4 cm−1 by the KBr technique. XRD patterns were recorded on a TTRAX3 diffractometer. CuKa radiation source was operated at 40 kV and a current of 200 mA. Patterns were recorded by monitoring the diffractions in the range 5°–65°. The scan speed was 1° min−1. XRD data were analyzed by use of Origin 8.0 Peak Fitting Module. Each peak was modeled by using a Gaussian peak shape to calculate the crystallinity.33 Intrinsic viscosity was measured using Ubbelohde viscometer in formic acid at 25 °C. LLS measurements were conducted on an ALV/DLS/SLS-5022F spectrometer with a multi-τ digital time correlation (ALV5000) and a cylindrical 22 mW UNIPHASE He-Ne laser (λ0 = 632 nm) as the light source. The weight-average molar mass (Mw), root-mean-square radius of gyration (〈Rg〉) and the second virial coefficient (A2) were obtained from Zimm plots by static light scattering (SLS) with the scattering angle (θ) varying from 20° to 150°. The hydrodynamic radius distribution f(Rh) and average hydrodynamic radius (〈Rh〉) were measured by dynamic light scattering (DLS) at θ = 20°. The specific refractive index increment (dn/dC) of nylon 3 in HCOOH measured on a Jianke differential refractometer at 632 nm is 0.167 mL g−1 at 25 °C. Themogravimetric analysis (TGA) was performed on a TA SDT Q600 instrument under nitrogen atmosphere at a heating rate of 10 °C min−1 in the range 0 to 800 °C. Differential scanning calorimetry (DSC) was conducted in dry nitrogen atmosphere using a DSC Q2000. After the sample was heated from 25 °C to 200 °C with a rate of 10 K min−1, it was cooled to room temperature with a rate of 200 K min−1, so that the effect of thermal history was eliminated. The sample was re-heated from 25 °C to 200 °C with a rate of 10 K min−1, and the glass transition temperature (Tg) was taken as that centered at the transition.Results and discussion
The ring opening polymerization of β-lactam was first investigated in DMAc containing 10 wt% of LiCl at room temperature. The molar ratio (r) of monomer to t-BuP4 was 2000/1. Fig. 1 shows typical 1H NMR spectra of the β-lactam and nylon 3 (sample Ny4). The signals due to the protons in –NH–CH2– and –COCH2– can be observed at 3.6 ppm (c) and 2.8 ppm (d), respectively. The signal due to the secondary amide group is at 8.1 ppm.1 All the chemical shifts in the polymer agree with those in nylon 3 synthesized by hydrogen transfer polymerization, indicating that t-BuP4 can catalyze the ring opening polymerization of β-lactam. On the other hand, the bands at 3070 cm−1 and 1530 cm−1 assigned to amide and the band at 1640 cm−1 to carbonyl are observed in FTIR spectra (not shown),18 further indicating the polymerization of β-lactam. Note that the spectrum in Fig. 1 does not show the signals of primary amide (δ = 7.40 and δ = 6.80) due to branching, implying that nylon 3 synthesized here has a linear structure.1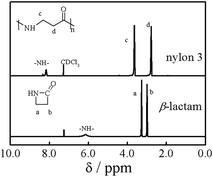 | ||
| Fig. 1 1H NMR spectra of β-lactam and nylon 3 (sample Ny4) in CDCl3/TFA-d3 (5/1, v/v). | ||
Fig. 2 shows the 13C NMR spectrum of nylon 3 (sample Ny4). Besides the signals about the solvent, we can observe three peaks corresponding to the carbons of linear nylon 3. No any peaks due to branching appear. The facts further indicate that the nylon 3 is linear.
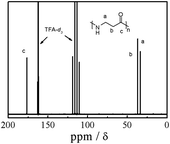 | ||
| Fig. 2 13C NMR spectrum of nylon 3 (sample Ny4) in CDCl3. | ||
Effects of solvent, temperature and catalyst concentration on the polymerization have been further examined. DMAc containing 5–10 wt % of LiCl is the best solvent for synthesis of nylon 3. The addition of LiCl to DMAc can improve the polymer solubility so that the molecular weight and yield increases with the amount of LiCl above its solubility. In DMAc without LiCl or in dimethyl sulfoxide (DMSO), precipitation occurs in the initiation stage, and only low molecular weight polymers can be obtained at a limited yield. We also examined the melt polymerization of β-lactam in bulk at 110 °C. Likewise, either the yield or molecular weight of the polymer obtained was low.
We have studied the temperature effect in the range of 25 to 80 °C. At a temperature below 50 °C, white gel is formed during the polymerization, leading to a heterogeneous polymerization system. At a temperature above 50 °C, the system is homogeneous during polymerization. On the other hand, as shown in Table 1, either the molecular weight or yield dramatically decreases with temperature. It is known that t-BuP4 forms a bulky cation during the polymerization, which can suppress the formation of cyclic oligomers.34 However, the bulky cation is probably not stable at a higher temperature so that the reaction equilibrium shifts to cyclic oligomers, leading the molecular weight and yield to decrease.
Catalyst concentration has a considerable effect on the polymerization. Table 1 also shows the dependence of the molecular weight and yield on the molar ratio (r) of t-BuP4 toβ-lactam. As r increases, the concentration of active species increases, so that the polymerization rate increases. However, the molecular weight decreases. This is understandable because the polymerized monomers per active species decrease with the number of active species. A high molecular weight (>105) is achieved at the catalyst/monomer ratio of 1/2000 (mol/mol).
Fig. 3 shows the time dependence of monomer conversion and ln([M]0/[M]), where [M]0 and [M] are the initial monomer concentration and the concentration at a certain time. The polymerization was carried out in DMAc containing 10 wt% of LiCl at 25 °C. The concentration of β-lactam is 1.4 mol L−1, and the molar ratio (r) of catalyst to monomer is 1/2000. The system turns to light yellow as soon as t-BuP4 is added, an indication of formation of lactamate anion or acyllactam anion, implying that the polymerization follows an anionic polymerization mechanism. As expected, the conversion increases with reaction time after t-BuP4 is added. Half of the monomer molecules are converted into polymer chains within 10 min, indicating the high activity of the catalyst. The linear time dependence of ln([M]0/[M]) in the first 10 min demonstrates that the polymerization is of first-order. Namely, the concentration of active species is fairly constant. Conversions close to 50% are achieved at the end of this period.
![Time dependence of conversion and ln([M]0/[M]) at 25 °C.](/image/article/2011/PY/c1py00334h/c1py00334h-f3.gif) | ||
| Fig. 3 Time dependence of conversion and ln([M]0/[M]) at 25 °C. | ||
It should be noted that the polymerization of β-lactam catalyzed by t-BuP4 does not have an induction period, unlike those with metal catalyst. We characterized the product by FTIR ten seconds after t-BuP4 was added, where the polymerization was terminated by addition of excess of menthol. The occurrence of a band at 1540 cm−1 assigned to amide II in FITR spectra (not shown) indicates that the amide group has already changed from cis to trans configuration due to ring opening in a such short time.18 The rapid conversion demonstrates that t-BuP4 is an efficient promoter for the β-lactam polymerization. Actually, the polymerization catalyzed by t-BuP4 even does not need any other activator. This is quite different from the ring-opening polymerization of β-lactam with metal catalyst, where N-benzoyl derivative, which is much more electrophilic than a β-lactam, is usually used as an activator.
Based on the mechanism proposed before,35 we suggest that the initiation of ring opening polymerization of β-lactam catalyzed by t-BuP4 involves nucleophilic attack of a lactamate anion on the carbonyl group of an imide at the polymer chain end (Scheme 1). Specifically, the initiation involves the following steps. (1) At the beginning, lactam reacts with t-BuP4 (B) to yield a lactamate anion. (2) The lactamate anion reacts with monomer to form an aminic anion with ring opening. (3) The unstable aminic anion quickly abstracts a proton from β-lactam to yield dimeric lactam and regenerate lactamate anion, which acts as the growth center of polymerization. Note that unlike the case of anionic ring opening polymerization with metal catalysts, the lactamate anion transforms into an aminic anion immediately, so that no apparent induction period is observed.
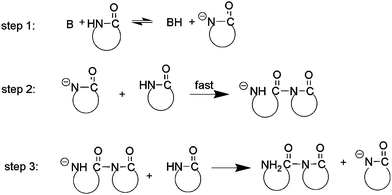 | ||
| Scheme 1 Initiation process of the polymerization. | ||
Fig. 4 shows a typical XRD pattern of nylon 3 prepared in this study (sample Ny4). Two peaks concerning the crystalline region can be observed, that is, α form at 2θ = 19.9° and 2θ = 23.6°, corresponding to the crystalline planes (200) and (002) + (202), respectively. Such forms were also observed in nylon 3 synthesized by hydrogen transfer polymerization.2 The crystallinity measured by XRD here is ∼40%. Note that the crystallinity of nylon has dependence on process or condition.
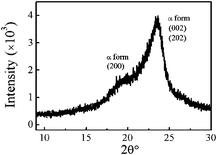 | ||
| Fig. 4 XRD pattern of nylon 3 (sample Ny4). | ||
Fig. 5 shows typical TGA and DSC curves of nylon 3. The thermal degradation starts at about 350 °C and the 50% of weight loss is at about 380 °C. The temperatures are higher than those reported before.33 This is because nylon 3 synthesized in this work has a higher molecular weight. Note that we can observe 8% of weight loss at about 150 °C. This is due to the release of water molecules which bind with the amide groups in nylon 3 viahydrogen bonding. The DSC curve shows that nylon 3 has a glass transition temperature (Tg) around 126 °C. Note that DSC measurement was conducted in the range from 25 °C to 200 °C. Further increasing the temperature leads nylon 3 to degrade, so that we cannot observe a melting peak before its decomposition.
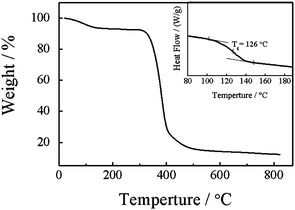
Fig. 6 shows a typical Zimm plot for nylon 3 polymers (sample Ny3 in Table 1). The molecular weight and the average radius of gyration 〈Rg〉 measured are 1.05 × 105g mol−1 and 38 nm, respectively. The positive A2 value (∼7.6 × 10−4 mol mL g−2) indicates that HCOOH is a good solvent for nylon 3. Actually, depending on the synthesis conditions, Mw ranges from 1.12 × 104 to 1.28 × 105 g mol−1, and 〈Rg〉 is in the range 8 to 40 nm (Table 1).
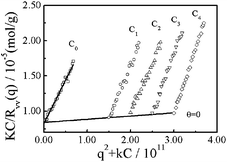 | ||
| Fig. 6 Zimm plot for nylon 3 polymer (sample Ny4) in formic acid at 25 °C, where polymer concentration ranges from 3.0 × 10−3 to 6.0 × 10−3 g mL−1. | ||
Fig. 7 shows a typical hydrodynamic radius distribution for sample Ny3. The average hydrodynamic radius 〈Rh〉 is 17 nm. In this work, depending on the synthesis conditions, the average hydrodynamic radius ranges from 4 to 18 nm (Table 1). It is known that for uniform non-draining sphere, hyperbranched cluster, and random coil, 〈Rg〉/〈Rh〉 values are ∼0.774, 1.0–1.2, and 1.5–1.8, respectively.36–39 Here, 〈Rg〉/〈Rh〉 is about 2.0–2.4 for nylon 3 (Table 1), indicating that the polymer has a rigid-like structure.
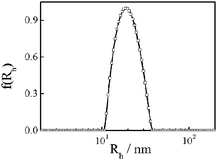 | ||
| Fig. 7 Hydrodynamic radius distribution (f(Rh)) of sample Ny4 in formic acid. | ||
We also measured the viscosity of the nylon 3 samples. Fig. 8 shows the relation between intrinsic viscosity [η] and weight average molecular weight (Mw) measured by SLS. Clearly, Mw is scaled to [η]. Namely, it follows the Mark-Houwink equation, [η] = KMα, where K and α are constants, which depend on the nature of the polymer and solvent, as well as temperature. It is well known that α = 0.5 for a polymer in a theta solvent. In a good solvent, 0.5 < α < 0.8 for a flexible polymer, α > 0.8 for a semiflexible polymer, and 1.8 < α < 2.0 for a rigid polymer.40 From the slope and intercept of the line in Fig. 5, we have α = 0.91 and K = 1.02 × 10−4 mL g−1, respectively. Like other polyamides,41nylon 3 has a relatively higher α value, further indicating that the chains have some rigid character, which is consistent with the conclusion from 〈Rg〉/〈Rh〉 values. The intramolecular and intermolecular hydrogen bonding in nylon 3 should be responsible for the chain rigidity.
![Specific viscosity [η] vs. weight molecular weight (Mw) relation.](/image/article/2011/PY/c1py00334h/c1py00334h-f8.gif) | ||
| Fig. 8 Specific viscosity [η] vs. weight molecular weight (Mw) relation. | ||
Conclusions
In conclusion, we have synthesized nylon 3 viaring-opening polymerization with t-BuP4 in a mixture of dimethylacetamide (DMAc) and LiCl. The polymer is linear and crystalline. The absolute weight average molecular weight can be as high as 105 g mol−1. The intrinsic viscosity ([η]) is related to the weight average molecular weight (Mw) by [η] = 1.02 × 10−4Mw0.91. t-BuP4 is an effective catalyst for the anionic polymerization of β-lactam, and the polymerization can be conducted in mild conditions. LiCl can improve the polymer solubility so that it can increase its molecular weight and yield. An increase in temperature can increase the polymerization rate, but decreases the molecular weight and yield. As t-BuP4 concentration increases, the yield and molecular weight of nylon 3 decrease. The initiation of the polymerization follows anionic ring opening mechanism.Acknowledgements
The financial support of the National Distinguished Young Investigator Fund (20725414) and Ministry of Science and Technology of China (2012CB933800) is acknowledged.References
- U. Morgenstern and W. Berger, Makromol. Chem., 1992, 193, 2561–2569 CrossRef CAS.
- J. Masamoto, Memoirs of the Fukui Institute of Technology, 2003, 33, 291–296 Search PubMed.
- J. Masamoto, in Polymeric Material Encyclopedia, ed. J. C. Salamone, CRC Press, Inc., 1996, vol. 6, p. 4672 Search PubMed.
- J. Masamoto, in Advanced Polymer Progressing Operations, ed. N. P. Cheremisinoff, William Andrew Publishing/Noyes, New Jersey, 1998, p. 109 Search PubMed.
- R. Graf, G. Lohaus, K. Boerner, E. Schmidt and H. Bestian, Angew. Chem., Int. Ed. Engl., 1962, 1, 481–488 CrossRef.
- J. H. Zhang, S. H. Gellman and S. S. Stahl, Macromolecules, 2010, 43, 5618–5626 CrossRef CAS.
- M. T. Dohm, B. P. Mowery, A. M. Czyzewski, S. S. Stahl, S. H. Gellman and A. E. Barron, J. Am. Chem. Soc., 2010, 132, 7957–7967 CrossRef CAS.
- J. H. Zhang, D. A. Kissounko, S. E. Lee, S. H. Gellman and S. S. Stahl, J. Am. Chem. Soc., 2009, 131, 1589–1597 CrossRef CAS.
- B. P. Mowery, A. H. Lindner, B. Weisblum, S. S. Stahl and S. H. Gellman, J. Am. Chem. Soc., 2009, 131, 9735–9745 CrossRef CAS.
- M. R. Lee, S. S. Stahl, S. H. Gellman and K. S. Masters, J. Am. Chem. Soc., 2009, 131, 16779–16789 CrossRef CAS.
- J. J. Cheng and T. J. Deming, J. Am. Chem. Soc., 2001, 123, 9457–9458 CrossRef CAS.
- K. Hashimoto, K. Hotta, M. Okada and S. Nagata, J. Polym. Sci., Part A: Polym. Chem., 1995, 33, 1995–1999 CrossRef CAS.
- J. Masamoto, K. Sasaguri, C. Ohizumi, K. Yamaguchi and H. Kobayashi, J. Appl. Polym. Sci., 1970, 14, 667–680 CrossRef CAS.
- J. P. Kennedy and T. Otsu, J. Macromol. Sci., Rev. Macromol. Chem., 1972, C(6), 237–283 Search PubMed.
- T. Iwamura, I. Tomita, M. Suzuki and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 465–472 CrossRef CAS.
- J. D. Glickson and J. Applequist, Macromolecules, 1969, 2, 628–634 CrossRef CAS.
- K. Hashimoto, Prog. Polym. Sci., 2000, 25, 1411–1462 CrossRef CAS.
- H. Bestian, Angew. Chem., Int. Ed. Engl., 1968, 7, 278–285 CrossRef CAS.
- K. Hashimoto, T. Oi, J. Yasuda, K. Hotta and M. Okada, J. Polym. Sci., Part A: Polym. Chem., 1997, 35, 1831–1838 CrossRef CAS.
- K. Hashimoto, J. Yasuda and M. Kobayashi, J. Polym. Sci., Part A: Polym. Chem., 1999, 3, 909–915 CrossRef.
- M. Fournier and R. E. Prud'homme, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 761–769 CrossRef CAS.
- L. Schwab, R. Kroon, A. J. Schouten and K. Loos, Makromol. Chem., Rapid Commun., 2008, 2, 794–797 CrossRef.
- R. Schwesinger, C. Hasenfratz, H. Schlemper, L. Walz, E. M. Peters, K. Peters and H. G. Schnering, Angew. Chem., Int. Ed. Engl., 1993, 32, 1361–1366 CrossRef.
- J. Tang, J. Dopke and J. G. Verkade, J. Am. Chem. Soc., 1993, 115, 5015–5020 CrossRef CAS.
- J. Zhao, G. Mountrichas, G. Zhang and S. Pispas, Macromolecules, 2009, 42, 8661–8668 CrossRef CAS.
- J. Zhao, G. Mountrichas, G. Zhang and S. Pispas, Macromolecules, 2010, 43, 1771–1777 CrossRef CAS.
- H. Schmalz, M. G. Lanzendorfer, V. Abetz and A. H. E. Müller, Macromol. Chem. Phys., 2003, 204, 1056–1071 CrossRef CAS.
- W. Memeger, G. C. Campbell and F. Davidson, Macromolecules, 1996, 29, 6475–6480 CrossRef CAS.
- N. Illy, S. Boileau, W. Buchmann, J. Penelle and V. Barbier, Macromolecules, 2010, 43, 8782–8789 CrossRef CAS.
- J. D. Winter, O. Coulembier, P. Gerbaux and P. Dubois, Macromolecules, 2010, 43, 10291–10296 CrossRef.
- L. Zhang, F. Nederberg, R. C. Pratt, R. M. Waymouth, J. L. Hedrick and C. G. Wade, Macromolecules, 2007, 40, 4154–4158 CrossRef CAS.
- A. A. Toy, S. Reinicke, A. H. E. Müller and H. Schmalz, Macromolecules, 2007, 40, 5241–5244 CrossRef CAS.
- E. Wolne and B. Stoll, Colloid Polym. Sci., 1980, 258, 300–313 Search PubMed.
- L. Zhang, F. Nederberg, M. J. Messman, R. C. Pratt, J. L. Hedrick, G. Charles and C. G. Wade, J. Am. Chem. Soc., 2007, 129, 12610–12611 CrossRef CAS.
- J. Penelle, in Handbook of Ring-Opening Polymerization, ed. P. Dubois, P. Degée, O. Coulembier and J. M. Raquez, Wiley-VCH, Weinheim, Germany, 2009, p. 186 Search PubMed.
- W. Burchard, in Light Scattering Principles and Development, ed. W. Brown, Clarendon Press, Oxford, 1996, p. 439 Search PubMed.
- W. Burchard, M. Schmidt and W. H. Stockmayer, Macromolecules, 1980, 13, 1265–1272 CrossRef CAS.
- J. P. Douglas, J. Roovers and K. F. Freed, Macromolecules, 1990, 23, 418–422 CrossRef.
- B. Chu, Z. Wang and J. Yu, Macromolecules, 1991, 24, 6832–6838 CrossRef CAS.
- E. Charles, J. Carraher, in Polymer Chemistry, Marcel Dekker Inc., New York, 6th edn 2003, p. 1 Search PubMed.
- J. E. Mark, in Polymer Data Handbook, Oxford University Press, New York, 1999, p. 181 Search PubMed.
| This journal is © The Royal Society of Chemistry 2011 |