DOI:
10.1039/C1PY00320H
(Paper)
Polym. Chem., 2011,
2, 2878-2887
Stable azlactone-functionalized nanoparticles prepared from thermoresponsive copolymers synthesized by RAFT polymerization†
Received
15th July 2011
, Accepted 14th September 2011
First published on 17th October 2011
Abstract
A new class of stable azlactone-functionalized thermoresponsive nanoparticles have been synthesized and characterized. Such particles are based on well-defined copolymers of poly(N-isopropylacrylamide) (PNIPAM), poly(N,N-dimethylacrylamide) (PDMA) and poly(2-vinyl-4,4-dimethylazlactone) (PVDM) copolymers synthesized using reversible addition–fragmentation chain transfer (RAFT) polymerization. A well-defined PNIPAM macromolecular RAFT agent and a PDMA macromolecular RAFT agent of low polydispersities (PDIs = 1.04–1.12) were used to mediate the copolymerizations of DMA/VDM and of NIPAM/VDM, respectively, resulting in copolymers of Mn ranging from 19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 g mol−1 to 64600 g mol−1 and PDIs ranging from 1.04 to 1.20. Depending on the number-average polymerization degrees of NIPAM, DMA and VDM, lower critical solution temperatures (LCSTs) ranging from 36 °C to 44 °C were measured. Two block copolymers PNIPAM46-b-P(VDM6-co-DMA65) and PDMA23-b-P(VDM10-co-NIPAM46) have a LCST that occurs at the physiological temperature. Above the LCST, the resulting nanoparticles were covalently stabilized by reacting a diamine with azlactone rings. The size exclusion chromatography (SEC) analyses revealed that all unimers are incorporated into the core-crosslinked structures and into the shell-crosslinked structures. This new strategy of crosslinking nanoparticles based on thermoresponsive copolymers is of particular interest as it is highly efficient. The azlactone groups remaining in core-crosslinked nanoparticles are suitable to react with dansylhydrazine as shown by SEC analysis using UV detection.
500 g mol−1 to 64600 g mol−1 and PDIs ranging from 1.04 to 1.20. Depending on the number-average polymerization degrees of NIPAM, DMA and VDM, lower critical solution temperatures (LCSTs) ranging from 36 °C to 44 °C were measured. Two block copolymers PNIPAM46-b-P(VDM6-co-DMA65) and PDMA23-b-P(VDM10-co-NIPAM46) have a LCST that occurs at the physiological temperature. Above the LCST, the resulting nanoparticles were covalently stabilized by reacting a diamine with azlactone rings. The size exclusion chromatography (SEC) analyses revealed that all unimers are incorporated into the core-crosslinked structures and into the shell-crosslinked structures. This new strategy of crosslinking nanoparticles based on thermoresponsive copolymers is of particular interest as it is highly efficient. The azlactone groups remaining in core-crosslinked nanoparticles are suitable to react with dansylhydrazine as shown by SEC analysis using UV detection.
Introduction
Self-assembled particles based on amphiphilic block copolymers have attracted attention due to their large number of potential applications, including surface modification, removal of pollutants and nanoreactors.1 However, their stability depends on various parameters such as their concentration, the temperature and the solvent. Thus, the stabilization of these structures via crosslinking individual components of the copolymer chains is of particular interest as it has the potential to increase the range of applications, particularly for drug delivery and in other biomedical fields.2 Stabilization of self-assembled particles via crosslinking has previously been reviewed.3–8 It has been achieved using a range of organic reactions with the most significant methods being the condensation reactions between carboxylic acid groups or activated ester groups and diamines, quaternization of the amine groups, UV-induced cycloaddition and the use of the Huisgen 1,3-dipolar cycloaddition (“Click”) reaction. The latter methods involve the incorporation of specific functionalities within the copolymer. This can be achieved using controlled/living radical synthetic methodologies such as nitroxide-mediated polymerisation9 (NMP), atom transfer radical polymerization10 (ATRP) and reversible addition–fragmentation chain transfer polymerization11,12 (RAFT). These methodologies have greatly facilitated the synthesis of (co)polymers with specific functionalities and control over molecular weight and macromolecular structure. Of the techniques listed, RAFT polymerization is arguably the most versatile as it is applicable to most commercially available and novel monomers by careful selection of the RAFT agent.
In this study, the RAFT polymerization has been applied to target a new class of core–shell particles based on block copolymers containing a thermoresponsive part (poly(N-isopropylacrylamide)—PNIPAM), a hydrophilic part (poly(N,N-dimethylacrylamide)—PDMA) and a polymer reactive towards amines (poly(2-vinyl-4,4-dimethylazlactone)—PVDM). PNIPAM exhibits a lower critical solution temperature (LCST), which undergoes a rapid, reversible coil-to-globule transition at around 31–32 °C in aqueous solution13 as the water molecules surrounding the amide group together with hydrogen bonds are eliminated. A number of examples of thermoresponsive core–shell particles based on PNIPAM synthesized using RAFT have been reported in the literature and are the subject of a recent review.14 However, only a few studies have been reported on the RAFT polymerization of polyacrylamide based thermoresponsive copolymers reactive towards amines.15–20 The reactive functionalities towards amines used in these reported studies are activated esters such as the N-hydroxysuccinimide ester15–19 and the pentafluorophenyl ester.20 The main drawback of using such functionalities is the formation of small molecule byproducts after reaction with amines. In our study, the azlactone ring has been chosen as reactive functionality towards amines. The advantages of such electrophile ring are numerous in comparison with the N-hydroxysuccinimide ester15–19 and the pentafluorophenyl ester.20 First, the azlactone ring displays a high reactivity towards amino-functionalized molecules at room temperature by means of a ring-opening addition reaction without the addition of catalysts or the formation of byproducts.21 This has led to the use of solid supported PVDM as a scavenger for amine impurities22–27 and has the ability to react with the free amine groups of biomolecules, leading to interest in potential bioapplications.28–33 Moreover, the azlactone functionality is resistant to hydrolysis at neutral pH: this is a considerable advantage compared to the succinimide group.32
In this contribution, we report the synthesis and characterization of a new class of thermoresponsive core–shell particles based on PNIPAM-b-P(VDM-co-DMA) block copolymers and on PDMA-b-P(VDM-co-NIPAM) block copolymers where the PVDM is the functional polymer containing the azlactone ring used to react with a diamine to form stable core–shell particles by crosslinking in aqueous solution. This “ideal” and original crosslinking strategy is non-toxic, is facile under mild conditions (i.e. ambient temperature and in aqueous solution), does not necessitate the addition of a co-reagent, does not generate small molecule byproducts and is efficient. This approach is really a major advantage over previously reported approaches.3–8 The potential reactivity of the crosslinked self-assembled azlactone-functionalized particles is studied towards dansylhydrazine. To the best of our knowledge, this is the first work concerning the incorporation of the azlactone functionality into thermosensitive copolymers that can self-assemble into core–shell nanoparticles which are efficiently stabilized by covalent crosslinking without the presence of a co-reagent and without the formation of a byproduct.
Experimental
Reagents
N-Isopropylacrylamide (NIPAM) was obtained from Aldrich (97% purity) and was recrystallised from petroleum ether (bp 40–60 °C) prior to use. Sodium hydroxide (≥98%, Sigma-Aldrich), 2-methylalanine (98%, Aldrich), 2,6-di-tert-butyl-p-cresol (≥99%, Aldrich), acryloyl chloride (97%, Aldrich), concentrated hydrochloric acid (37%, Sigma-Aldrich), triethylamine (≥99%, Sigma-Aldrich), ethyl chloroformate (≥98%, Fluka), dansylhydrazine (Fluka, 97%), 2,2′-(ethylenedioxy)-bis(ethylamine) (Aldrich, 98% purity), and 4,4′azobis(4-cyanovaleric acid) (ACPA, Aldrich ≥ 98%) were used as received. 2,2-Azobis(2-methylpropionitrile) (AIBN, 98% purity) was obtained from Aldrich and recrystallized from methanol prior to use. N,N-Dimethylacrylamide (DMA) was obtained from Aldrich (98% purity) and distilled under vacuum prior to use. Dimethylformamide (DMF), acetone, dioxane, and dichloromethane were obtained from Aldrich and distilled prior to use. Pure water used for dynamic light scattering measurements was obtained from a Millipore Direct Q system and had a conductivity of 18.2 MΩ cm at 25 °C. The RAFT agent methyl-2-(n-butyltrithiocarbonyl) propanoate (MBTTCP) was synthesized according to a literature procedure19 and purified viacolumn chromatography using a 95/5 (v/v) mixture of petroleum ether/ethyl acetate as eluent, yield = 64%.
Synthesis of 2-vinyl-4,4-dimethylazlactone (VDM)
Synthesis of N-acryloyl-2-methylalanine.
To a solution of 1.768 g (4.42 × 10−2 mol) of sodium hydroxide in 4.4 mL of water, prealably cooled to 0 °C using an ice bath, are slowly added 2 g (1.94 × 10−2 mol) of 2-methylalanine and 2.0 mg (9.07 × 10−6 mol) of 2,6-di-tert-butyl-p-cresol. When the solution is homogeneous, 2 g (2.21 × 10−2 mol) of acryloyl chloride were added dropwise under stirring and keeping the temperature at 0 °C using an ice bath. After complete addition of acryloyl chloride, the stirring is continued for 3 h. To this solution was slowly added 2.3 mL of concentrated hydrochloric acid. During the addition a white solid is formed. The reaction solution was stirred for 30 min. The solid was filtered and was recrystallized from a mixture of ethanol and water (1/1 in volume). The white solid was filtered and dried under vacuum. Yield: 60%. mp 201–202 °C. 1H NMR (DMSO d6), δ in ppm: 1.36 (s, C(CH3)2); 5.56 (dd, Htrans, JHtrans–Hcis = 2.1 Hz, JHtrans–Hgem = 10.1 Hz); 6.08 (dd, Hcis, JHcis–Htrans = 2.1 Hz, JHcis–Hgem = 17.1 Hz); 6.25 (dd, Hgem, JHgem–Htrans = 10.1 Hz, JHgem–Hcis = 17.1 Hz); 8.29 (s, NH–); 12.20 (s, COOH).
Synthesis of VDM.
0.4 g (2.54 × 10−3 mol) of N-acryloyl-2-methylalanine and 0.38 g (3.75 × 10−3 mol) of triethylamine were added to 10 mL of acetone. The resulting solution was cooled at 0 °C using an ice bath and 0.277 g (2.5 × 10−3 mol) of ethyl chloroformate was added dropwise. The solution was stirred for 3 h at 0 °C. The solution was then filtered and the white solid was washed with acetone. The filtrate was concentrated under vacuum and the filtrate residue was distilled under vacuum (bp 47 °C at 4 mmHg) to give a colorless oil. Yield: 49%. FTIR, ν in cm−1: 1823; 1670; 1651; 1070. 1H NMR (CDCl3), δ in ppm: 1.47 (s, C(CH3)2); 5.96 (dd, Htrans, JHtrans–Hcis = 2.0 Hz, JHtrans–Hgem = 9.9 Hz); 6.23 (dd, Hcis, JHcis–Htrans = 2.0 Hz, JHcis–Hgem = 17.6 Hz); 6.27 (dd, Hgem, JHgem–Htrans = 9.9 Hz, JHgem–Hcis = 17.6 Hz).13C NMR (CDCl3), δ in ppm: 24.50 (C(CH3)2); 64.61 (C(CH3)2); 123.88 (CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH); 128.81 (CH2CH); 158.92 (CN); 180.53 (CO).
CH); 128.81 (CH2CH); 158.92 (CN); 180.53 (CO).
Characterization
Size exclusion chromatography (SEC).
PDMA
homopolymers and copolymers were characterized on a SEC system operating in DMF eluent at 60 °C fitted with a guard column (PL Gel 5 μm) and two Polymer Laboratories PL Mixed D columns, a Waters 410 differential refractometer (DRI) and a Waters 481 UV detector operating at 309 nm. The instrument was operated at a flow rate of 1.0 mL min−1 and was calibrated with narrow linear polystyrene standards ranging in molecular weight from 460000 g mol−1 to 580 g mol−1. Molecular weights and polydispersity indices (PDIs) were calculated using Waters EMPOWER software.
PNIPAM homopolymers were characterized on a SEC system operating in DMF eluent at 60 °C fitted with a guard column (PL Gel 5 μm) and two Polymer Laboratories PolarGel columns, a Waters 410 differential refractometer (DRI) and a Waters 481 UV detector operating at 309 nm. The instrument was operated at a flow rate of 1.0 mL min−1 and was calibrated with narrow linear polystyrene standards ranging in molecular weight from 460000 g mol−1 to 580 g mol−1. Molecular weight and polydispersity indices (PDIs) were calculated using Waters EMPOWER software.
Dynamic light scattering (DLS) analysis.
Instrumentation.
Dynamic light scattering measurements were made in order to determine the hydrodynamic diameter (Dh) and particle size distribution of copolymers and of resulting core–shell particles over temperatures ranging from 20 °C to 65 °C. A Malvern Instruments Nanosizer fitted with a 4 mW He–Ne laser operating at 633 nm with dual angle detection (13°, 173°) was used for all measurements and the data were processed using the CONTIN method of analysis. Temperature trend measurements were recorded starting from 20 °C and at increasing temperature increments of 2 °C until 65 °C with an equilibration time of two minutes. Individual measurements were made at 25 °C, 50 °C and 60 °C after an equilibration time of two minutes.
A typical sample preparation for dynamic light scattering.
A quantity of copolymer (1.9 × 10−3 g) was measured into a clean glass vial and Millipore deionised water (9 mL) was added. The copolymer was left to dissolve for 2 hours stirring at room temperature, after which the solution was filtered using a glass syringe and a series of syringe filters of decreasing pore size (AT Chromato, 0.45 μm, Whatman Inorganic 0.2 μm and 0.1 μm respectively) into a fresh and clean glass cuvette for analysis.
UV-Vis spectroscopy characterization.
UV-vis measurements were performed on a Varian Cary 100 Scan spectrometer fitted with a dual cell Peltier accessory for heating. Samples of copolymers in deionized water of concentration 1 g L−1 were prepared and left to dissolve over a period of two hours at room temperature. The sample was introduced into a quartz cuvette and placed in the Peltier accessory. The temperature was adjusted manually and the sample allowed to equilibrate for five minutes before two measurements of the transmittance at 500 nm were made.
Infra-red spectroscopy characterization.
Infra-red spectra of copolymers were recorded using a ThermoElectron Corp. spectrometer operating with an attenuated total reflection (ATR solid) gate. Spectra were analyzed with OMNIC software.
A typical procedure for the synthesis of PNIPAM macroCTA mediated by MTTCP.
A magnetic stirrer was charged to a Schlenk tube along with NIPAM (11.2207 g, 9.93 × 10−2 mole), MBTTCP (0.3404 g, 1.35 × 10−3 mole), AIBN (0.0224 g, 1.36 × 10−4 mole) and DMF solvent (22 mL) in the ratio of [NIPAM]0/[MBTTCP]0/[AIBN]0 = 74/1/0.1. The polymerization mixture was deoxygenated by freeze–pump–thawing. When no more bubbles were observed in the reaction mixture, the reaction vessel was switched to a nitrogen atmosphere and placed in a thermostatted oil bath at 70 °C to initiate the polymerization. The immersion in the oil bath defines t = 0 and a sample was removed using a deoxygenated syringe as a reference point. Small samples were periodically withdrawn from the reaction mixture using a deoxygenated syringe and subjected to 1H-NMR spectroscopy to determine monomer conversion and SEC analysis to determine the molecular weight and the polydispersity index. The reaction was stopped after 140 minutes and monomer conversion was determined to be 62%. DMF was removed using a rotary evaporator and the polymer purified by a series of precipitations in cold petroleum ether, filtered and dried in a vacuum oven at 25 °C. Mn (SEC) = 5600 g mol−1, PDI (SEC) = 1.10. DPn (1H-NMR) = 46. 1H NMR (CDCl3): δ (ppm): 6.24 (1H, –NH–CH(CH3)2), 4.00 (1H, –NH–CH(CH3)2), 3.66 (3H, CH3–OCO), 3.35 (2H, –S–C(S)S–CH2–C3H7), 1.14 (6H, –NH–CH(CH3)2 and 4H, –S–C(S)S–CH2–(CH2)2–CH3), 0.95 (3H, –S–C(S)S–(CH2)3–CH3).
A typical procedure for the synthesis of PDMA macroCTA mediated by MTTCP.
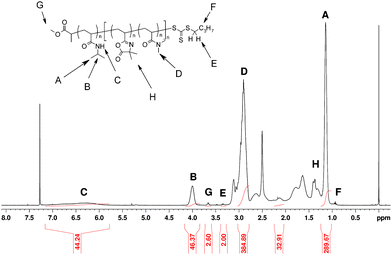
A magnetic stirrer was charged to a Schlenk tube along with DMA (4.02 g, 4.1 × 10−2 mol), MBTTCP (0.402 g, 1.6 × 10−3 mol), AIBN (2.52 × 10−2 g, 1.54 × 10−4 mol), DMF (0.1 mL) used as internal standard and dioxane solvent (8 mL) in the ratio of [DMA]0/[MBTTCP]0/[AIBN]0 = 26/1/0.1. The polymerization mixture was deoxygenated by freeze–pump–thawing. When no more bubbles were observed in the reaction mixture, the reaction vessel was switched to a nitrogen atmosphere and placed in a thermostatted oil bath at 70 °C to initiate the polymerization. The immersion in the oil bath defines t = 0 and a sample was removed using a deoxygenated syringe as a reference point. The polymerization was stopped after 90 minutes and the reaction mixture was subjected to 1H-NMR spectroscopy to determine monomer conversion and SEC analysis to determine the molecular weight and the polydispersity index. The monomer conversion was determined to be 85%. Dioxane and DMF were removed using a rotary evaporator and the polymer was purified by a series of precipitations in cold petroleum ether, filtered and dried in a vacuum oven at 25 °C. Mn (SEC) = 5300 g mol−1, PDI (SEC) = 1.05. DPn (1H-NMR) = 22. The intensity of the signal at 3.37 ppm (corresponding to two protons PDMA–S–C(S)S–CH2–C3H7) at the chain end was compared to the intensity of the signal at 2.93 ppm (corresponding to the methyl protons on the PDMA, –CO–N(H)– (CH3)2) in order to determine the number of DMA units in the chain.
A standard procedure for the copolymerization of DMA and VDM mediated by PNIPAM macroCTA agent.
A magnetic stir bar was charged to a Schlenk tube along with the PNIPAM macroRAFT agent (5.027 × 10−1 g, 8.9 × 10−5 mol), DMA (1.1725 g, 1.18 × 10−2 mol), VDM (9.28 × 10−2 g, 6.67 × 10−4 mol), AIBN (4.2 × 10−3 g, 2.56 × 10−5 mol) and dioxane (3 mL). The mixture was deoxygenated by a series of freeze–pump–thaw cycles. When no further oxygen was produced, the reactor vessel was switched to an inert argon atmosphere and the Schlenk tube placed in a thermostatted oil bath at 70 °C. The immersion of the Schlenk tube within the oil bath defines t = 0. The polymerization was stopped after 50 minutes by removing the Schlenk tube from the oil bath and opening the mixture to air. The synthesized copolymer was dissolved in acetone and precipitated into cold diethyl ether, filtered and dried in a vacuum oven at 25 °C. The copolymer was characterized by 1H NMR spectroscopy, IR spectroscopy and SEC analysis. Mn (SEC) = 33500 g mol−1, PDI (SEC) = 1.17. FTIR: ν(CO) (azlactone) 1815 cm−1, ν(CN) (azlactone) 1628 cm−1, ν(CO) (DMA) 1616 cm−1. 1H NMR (CDCl3): δ (ppm): 6.24 (1H, –NH–CH(CH3)2), 4.00 (1H, –NH–CH(CH3)2), 3.66 (3H, CH3–OCO), 3.35 (2H, –S–C(S)S–CH2–C3H7), 2.91 (6H, –N(CH3)2), 1.40 (6H, –OCO–C(CH3)2–N), 1.14 (6H, –NH–CH(CH3)2 and 4H, –S–C(S)S–CH2–(CH2)2–CH3), 0.95 (3H, –S–C(S)S–(CH2)3–CH3). In order to determine the number of DMA units in block copolymers, the total intensity of the signals of the methyl protons at 2.91 ppm (corresponding to the methyl protons of PDMA, –N(CH3)2) and at 1.40 ppm (corresponding to the methyl protons of PVDM, –OCO–C(CH3)2–N) was compared with the intensity of the signal at 4.00 ppm (corresponding to the methine proton of PNIPAM, –NH–CH(CH3)2).
A standard procedure for the copolymerization of VDM and NIPAM mediated by PDMA macroCTA agent.
A magnetic stir bar was charged to a Schlenk tube along with the PDMA macroRAFT agent (3 × 10−1 g, 1.19 × 10−4 mol), VDM (0.163 g, 1.17 × 10−3 mol), NIPAM (1.07 g, 9.47 × 10−3 mol), ACPA (6.0 × 10−3 g, 2.14 × 10−5 mol), DMF used as internal standard (0.1 mL) and dioxane (3.5 mL). The mixture was deoxygenated by a series of freeze–pump–thaw cycles. When no further oxygen was produced, the reactor vessel was switched to an inert argon atmosphere and the Schlenk tube placed in a thermostatted oil bath at 70 °C. The immersion of the Schlenk tube within the oil bath defines t = 0. The polymerization was stopped after 100 minutes by removing the Schlenk tube from the oil bath and opening the mixture to air. The VDM conversion and the NIPAM conversion were determined to be 100% and 58% respectively. Dioxane and DMF were removed using a rotary evaporator and the synthesized copolymer was dissolved in acetone and precipitated into cold diethyl ether, filtered and dried in a vacuum oven at 25 °C. The copolymer was characterized by 1H NMR spectroscopy, IR spectroscopy and SEC analysis. Mn (SEC) = 19500 g mol−1, PDI (SEC) = 1.04. FTIR: ν(CO) (azlactone) 1815 cm−1, ν(CN) (azlactone) 1628 cm−1, ν(CO) (DMA) 1616 cm−1. 1H NMR (CDCl3): δ (ppm): 6.24 (1H, –NH–CH(CH3)2), 4.00 (1H, –NH–CH(CH3)2), 3.66 (3H, CH3–OCO), 3.35 (2H, –S–C(S)S–CH2–C3H7), 2.93 (6H, –N(CH3)2), 1.40 (6H, –OCO–C(CH3)2–N), 1.14 (6H, –NH–CH(CH3)2 and 4H, –S–C(S)S–CH2–(CH2)2–CH3), 0.95 (3H, –S–C(S)S–(CH2)3–CH3). In order to determine the number of NIPAM units in block copolymers, the intensity of the signal at 4.00 ppm (corresponding to the methine proton of PNIPAM, –NH–CH(CH3)2), and the intensity of the signal at 1.40 ppm (corresponding to the methyl protons of PVDM, –OCO–C(CH3)2–N) were compared with the intensity of the signal at 2.93 ppm (corresponding to the methyl protons of PDMA, –N(CH3)2).
A standard procedure for the preparation of crosslinked core–shell nanoparticles and their reactivity towards dansylhydrazine.
A measured quantity of copolymer PDMA23-b-P(VDM10-co-NIPAM46) (23 mg) was placed in a round bottom flask and dissolved in deionized water (20 mL) by magnetically stirring at ambient temperature over a period of two hours. When this time had elapsed the flask containing the solution of copolymer was placed in a thermostatted oil bath preheated to 40 °C in order to form the self-assembled structures. When the reaction mixture had reached this temperature, 2,2′ethylenedioxy-bis(ethylamine) (2 mL) was added and the mixture was stirred for a further 5 h at 40 °C. After this time had elapsed the flask was removed from the oil bath, cooled at room temperature and water removed via lyophilisation. The crosslinked core–shell particles were characterized by SEC and DLS.
The crosslinked core–shell particles obtained from copolymer PDMA23-b-P(VDM10-co-NIPAM46) (20 mg) were placed in a round bottom flask in DMF (2 mL). Then, 2 mg of dansylhydrazine were added and the mixture was stirred for 3 days at 40 °C. The resulting solution was analyzed by SEC.
Results and discussion
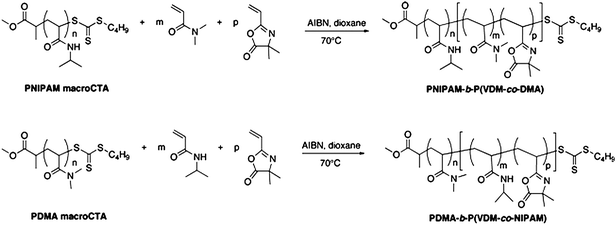
In order to synthesize PNIPAM-b-P(VDM-co-DMA) and PDMA-b-P(VDM-co-NIPAM) block copolymers, first blocks (PNIPAM and PDMA) have been previously synthesized using MTTCP as a RAFT agent. In the second step, the resulting PNIPAM and the resulting PDMA have been used as macromolecular chain transfer agents (macroCTAs) to mediate the copolymerization of VDM/DMA and of VDM/NIPAM respectively. The amount of VDM in the second block was kept low in order to maintain the P(VDM-co-DMA) block hydrophilic and to keep the LCST of the P(VDM-co-NIPAM) block near 32 °C.
Synthesis of PNIPAM macroCTA and of a PDMA macroCTA
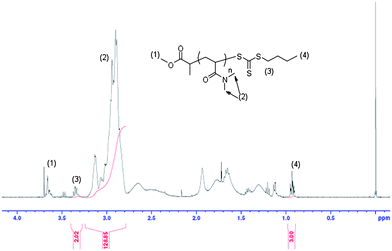
A recent review14 shows that the RAFT polymerization of NIPAM in both aqueous and organic media has been the subject of various studies. We have chosen the method reported by our group, and Monteiro's group to prepare well-defined PNIPAM using methyl-2-(n-butyltrithiocarbonyl) propanoate (MBTTCP) as RAFT agent (Scheme 1).19,34 PNIPAMs with controlled number-average molecular weights and narrow polydispersities were obtained (Fig. S3–S4 in the ESI†). The presence of the trithio-moiety at the PNIPAM chain end was confirmed by the presence of a peak on the UV detector at 309 nm (Fig. 1), corresponding to the CS bond, thus indicating that this product could be used as a macroCTA to mediate further polymerizations. Comparatively little work has been performed describing the detailed RAFT polymerization of DMA.35,36 In this study, the PDMA macroCTA was synthesized by RAFT polymerization of DMA using MTTCP (Scheme 1). After 90 min, the DMA conversion was calculated to be 85% as determined by 1H NMR spectroscopy by comparing the integrals of vinylic protons from DMA at 5.55 ppm with the integral of CHO from DMF at 8 ppm. The DMA degree of polymerization (DPn) was also determined by 1H NMR spectroscopy of the precipitated polymer by assuming that all chains are functionalized by a trithiocarbonate. The intensity of the signal at 3.35 ppm (corresponding to two protons PDMA–S–C(S)S–CH2–C3H7) at the chain end was compared to the intensity of the broad signal at 2.91 ppm (corresponding to the six protons on the PDMA, –CO–N(CH3)2) to calculate 22 DMA units in the chain (Fig. 2). Moreover, the SEC chromatograms using RI and UV detections showed unimodal traces (Fig. 3). As with the PNIPAM macroCTA, the presence of the trithio-moiety at the PDMA chain end was confirmed by the presence of a peak on the UV detector at 309 nm, corresponding to the CS bond. Both trithiocarbonyl-functionalized PNIPAM and PDMA have then been employed as macroCTAs.
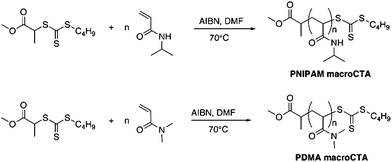 |
| | Scheme 1
RAFT polymerizations of NIPAM and DMA using AIBN as initiator and MBTTCP as RAFT agent in solution at 70 °C. | |
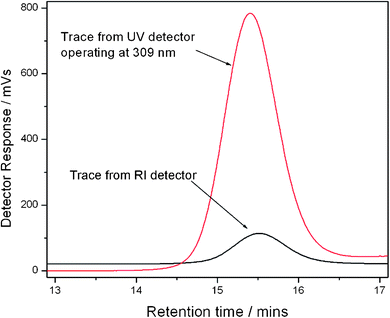 |
| | Fig. 1 Raw SEC chromatograms of the precipitated PNIPAM macromolecular chain transfer agent from the refractometer index (RI) detector and UV detector operating at 309 nm. | |
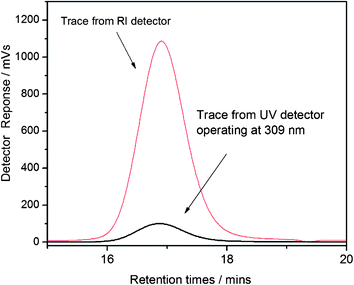 |
| | Fig. 3 Raw SEC chromatograms of the precipitated PDMA macromolecular chain transfer agent from the refractometer index (RI) detector and UV detector operating at 309 nm. | |
Synthesis of PNIPAM-b-P(VDM-co-DMA) copolymers and of PDMA-b-P(VDM-b-NIPAM) copolymers
In order to target thermoresponsive copolymers with azlactone functionality, we studied the RAFT copolymerizations of VDM with DMA and of VDM with NIPAM using PNIPAM-CTA and PDMA-CTA respectively. Lokitz et al.37 reported the RAFT polymerization of VDM and the dilute solution properties of the corresponding homopolymers prepared using either 2-cyanoisopropyl dithiobenzoate (CPDB) or 2-dodecylsulfanyl-thiocarbonylsulfanyl-2-methylpropionic acid (DMP) as RAFT chain transfer agents (CTAs). We previously described the first detailed investigation of the blocking efficiency of VDM with monomers such as styrene, methyl acrylate, and methyl methacrylate using cumyl dithiobenzoate (CDB).38 To date, only a preliminary study has been reported on the RAFT polymerization of VDM to synthesize thermosensitive and pH-sensitive copolymers.39
The previously synthesized PNIPAMs and PDMAs were used as macroCTAs to mediate the RAFT statistical copolymerization of VDM/DMA and of VDM/NIPAM in freshly distilled 1,4-dioxane at 70 °C as shown in Scheme 2. The results are shown in Table 1. The molar compositions of PNIPAM-b-P(VDM-co-DMA) and PDMA-b-P(VDM-co-NIPAM) block copolymers were determined by 1H NMR spectroscopy. For instance, the molar composition of PNIPAM46-b-P(VDM6-co-DMA65) was determined by comparing integrations of the signal corresponding to CH of PNIPAM (B, Fig. 4), of the signal corresponding to CH3 of PDMA (D, Fig. 4), and of the signal corresponding to CH3 of PVDM (H, Fig. 4). In this case, the number average degrees of polymerization were equal to 46, 6 and 65 for NIPAM, VDM and DMA respectively.
|
(Co)polymer structure |
M
n/g mol−1a |
PDIa |
DPn (NIPAM)b |
DPn (VDM)b |
DPn (DMA)b |
|
Determined from SEC in DMF calibrated with linear PSty standards using PolarGel columns (see Experimental).
Determined from 1H NMR (see Experimental).
|
| PNIPAM46 |
5640 |
1.12 |
46 |
0 |
0 |
| PNIPAM46-b-P(VDM6-co-DMA65) |
33400 |
1.10 |
46 |
6 |
65 |
| PNIPAM46-b-P(VDM4-co-DMA94) |
59200 |
1.17 |
48 |
4 |
94 |
| PNIPAM42-b-P(VDM18-co-DMA281) |
64600 |
1.20 |
42 |
18 |
281 |
|
PDMA23 |
5300 |
1.05 |
0 |
0 |
23 |
|
PDMA23-b-P(VDM10-co-NIPAM46)
|
19500 |
1.04 |
46 |
10 |
23 |
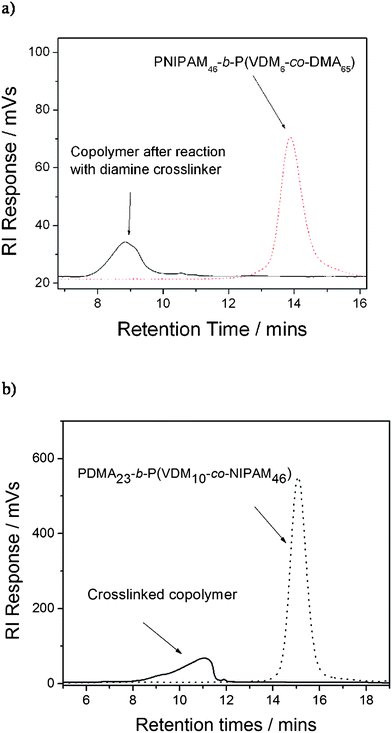
Chain extensions of PNIPAM and PDMA macroCTAs lead to well-defined copolymers as shown by the symmetrical SEC chromatograms of PNIPAM46-b-P(VDM6-co-DMA65) and of PDMA23-b-P(VDM10-co-NIPAM46) (Fig. 5). Block copolymer SEC traces shift to earlier retention times as the molecular weight of copolymers increases with respect to the PNIPAM and the PDMA macroCTAs. Moreover, the polydispersities of the copolymers remain low (less than 1.20) (Table 1). These results show that the copolymerizations are well-controlled and the block copolymers are well-defined.
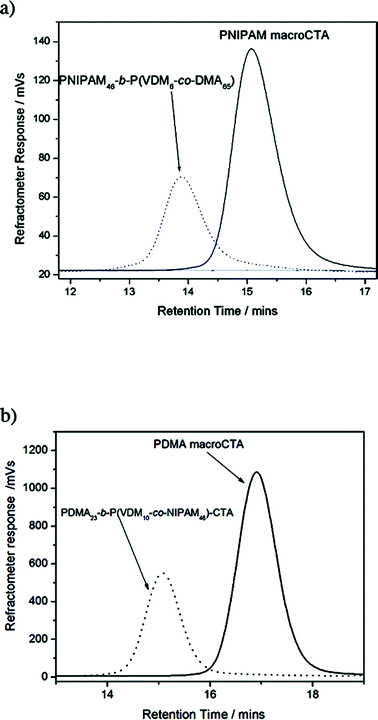 |
| | Fig. 5 Raw SEC chromatograms of (a) PNIPAM macroCTA and of the PNIPAM46-b-P(VDM6-co-DMA65) block copolymer and of (b) PDMA macroCTA and of the PDMA23-b-P(VDM10-co-NIPAM46) block copolymer. | |
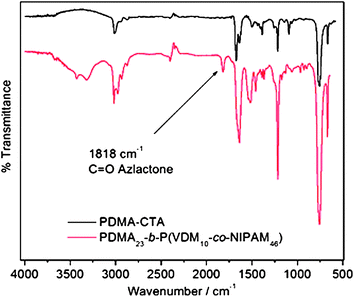
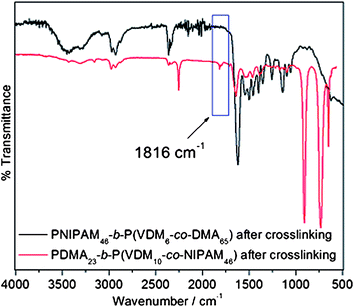
The FT-IR spectrum of the precipitated copolymer PDMA23-b-P(VDM10-co-NIPAM46) (Fig. 6) reveals the presence of bands at ν(CO) = 1815 cm−1 and ν(CN) = 1628 cm−1 which are characteristics of the azlactone ring. The same results have been obtained for PNIPAM46-b-P(VDM6-co-DMA65) block copolymers. Therefore, the azlactone group remains intact after the copolymerization and is available for further reactions with amines.
Thermoresponsive behaviour of the copolymers
The thermosensitive behaviour of the copolymers in aqueous solution was studied. The lower critical solution temperature (LCST) was determined from cloud point measurements using UV-vis spectroscopy by measuring the transmittance through a solution of the copolymer at 500 nm at temperatures ranging from 20 °C to 60 °C (Fig. S5 in the ESI†). As the temperature increases the copolymers self-assemble into core–shell particles of significantly greater particle size, resulting in a cloudy solution. LCST values for the block copolymers are shown in Table 2.
Table 2
LCST values of PNIPAM-b-P(VDM-co-DMA) and PDMA-b-P(VDM-co-NIPAM) block copolymers
|
Copolymer structure |
M
n
a/g mol−1 |
PDIa |
LCST
b/°C |
|
Determined from SEC in DMF calibrated with linear polystyrene standards using PLGel Mixed-D columns.
Determined by UV-Vis spectroscopy.
|
| PNIPAM46-b-P(VDM6-co-DMA65) |
33400 |
1.10 |
37 |
| PNIPAM46-b-P(VDM4-co-DMA94) |
59200 |
1.17 |
41 |
| PNIPAM42-b-P(VDM18-co-DMA281) |
64600 |
1.20 |
55 |
|
PDMA23-b-P(VDM10-co-NIPAM46)
|
19500 |
1.04 |
36 |
For PNIPAM-b-P(VDM-co-DMA) block copolymers, the molar composition was found to affect the temperature at which nanoparticles are formed. Increased DPn of DMA with a constant DPn of NIPAM and a constant DPn of VDM increased the LCST of the copolymer (Table 2). This trend is in agreement with previous literature13 which reports that the addition of hydrophilic constituents to a copolymer raises the LCST due to the ability of PDMA to retain water/remain hydrated and limits the formation of hydrogen bonds between the amine groups of PNIPAM, thus preventing the coil-to-globule transition from occurring. Thus, the LCST can be “tuned”13 and two block copolymers PNIPAM46-b-P(VDM6-co-DMA65) and PDMA23-b-P(VDM10-co-NIPAM46) have a LCST that occurs at physiological temperatures allowing their use for bioapplications.
Characterization and reactivity of azlactone-functionalized crosslinked nanoparticles
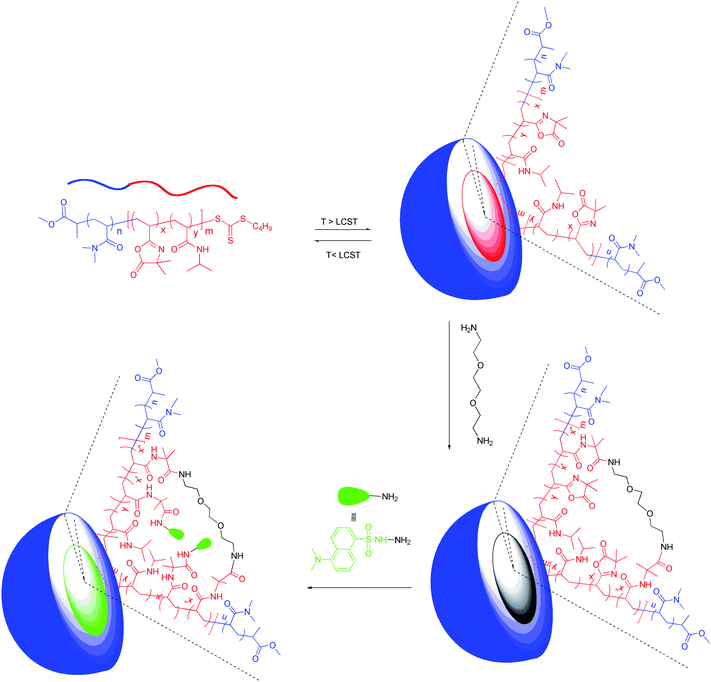
Either shell-crosslinked or core-crosslinked nanoparticles were prepared from PNIPAM46-b-P(VDM6-co-DMA65) and PDMA23-b-P(VDM10-co-NIPAM46) copolymers, respectively, using a diamine which reacts with the azlactone rings. Block copolymers were dissolved in deionized water and the temperature of solutions raised to above LCSTs to 40 °C to form nanoparticles. Finally 2,2′ethylenedioxy-bis(ethylamine) was added to react with the azlactone groups and crosslink the copolymer chains, forming covalently stabilized nanoparticles (Scheme 3). Analysis of precipitated crosslinked nanoparticles issued from PNIPAM46-b-P(VDM6-co-DMA65) and from PDMA23-b-P(VDM10-co-NIPAM46) by SEC shows a significant shift in retention time to high molecular weight and the absence of a peak corresponding to the initial copolymer indicating that an efficient crosslinking of the copolymers into core–shell particles has occurred (Fig. 7). This strategy to crosslink core–shell nanoparticles is of particular interest in comparison with previous reported ones3–8 as it is efficient, does not generate small molecule byproducts and does not need addition of catalyst or co-reagents.
The comparison of FTIR spectra of the isolated crosslinked nanoparticles issued from PNIPAM46-b-P(VDM6-co-DMA65) and PDMA23-b-P(VDM10-co-NIPAM46) block copolymers is shown in Fig. 8. It appears that when the azlactone rings are located in the shell of nanoparticles obtained from PNIPAM46-b-P(VDM6-co-DMA65), the disappearance of the band at 1815 cm−1 corresponding to the CO ester of the azlactone ring is observed. In contrast, when azlactone rings are located in the core of nanoparticles issued from PDMA23-b-P(VDM10-co-NIPAM46), this band is still present indicating that azlactone rings have been preserved. In this case, the hydrophobic core, in which azlactone groups are embedded, is therefore efficiently repelling any water penetration. This result is important as such original nanoparticles could be employed for further reaction with the amine groups of biomolecules. As the nanoparticles prepared from PDMA23-b-P(VDM10-co-NIPAM46) are of particular interest, further dynamic light scattering measurements were performed to characterize their size before and after crosslinking. At 25 °C, PDMA23-b-P(VDM10-co-NIPAM46) dissolved in water exhibits an average hydrodynamic diameter of 4 nm characteristic of unimers (Fig. 9). When the LCST is reached, this copolymer shows self-assembly behaviour to form nanoparticles with an average hydrodynamic diameter of 43 nm. Above the LCST, the resulting nanoparticles have been covalently stabilized using a diamine and the DLS analysis of the resulting core-crosslinked particles at 25 °C reveals an average hydrodynamic diameter of 29 nm (Fig. 9). This result shows that the nanoparticle structure is conserved after reducing the solution temperature below the LCST.
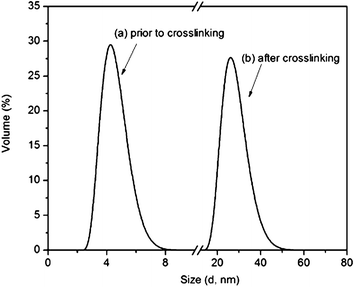 |
| | Fig. 9 Particle size distributions from dynamic light scattering of PDMA23-b-P(VDM10-co-NIPAM46) copolymers in water at 25 °C (a) prior to crosslinking (b) after core-crosslinking of the self-assembled structures with diamine. | |
A preliminary study on the reactivity of electrophile azlactone-functionalized stable nanoparticles has been performed using dansylhydrazine (Scheme 3). This molecule has been selected as it is a nucleophile that is easily detectable by UV-vis spectroscopy. The reaction between the core-crosslinked nanoparticles and the dansylhydrazine was performed in DMF at room temperature for 3 days. After the reaction, nanoparticles were analyzed by SEC using UV detection. By comparing the UV spectra extracted from SEC traces at the maximum elution volume of the core-crosslinked nanoparticles prepared from PDMA23-b-P(VDM10-co-NIPAM46) before and after reaction with dansylhydrazine, we observed a shift of the maximum wavelengths from 308.9 nm to 319.6 nm. The maximum wavelength at 308.9 nm is characteristic of trithiocarbonyl moieties at the copolymer chain end and the shift to 319.6 nm indicates that the dansylhydrazine has reacted with the core-crosslinked nanoparticlesvia the azlactone groups.
Conclusions
A series of well-defined copolymers of PNIPAM-b-P(VDM-co-DMA) and PDMA-b-P(VDM-co-NIPAM) have been synthesized using RAFT polymerization. The copolymers are thermoresponsive and the molar composition within the copolymer affects the LCST, above which the structures self-assemble to form nanoparticles. The LCST was tuned to occur at physiological temperatures with the PNIPAM46-b-P(VDM6-co-DMA65) copolymer and the PDMA23-b-P(VDM10-co-NIPAM46) copolymer. The azlactone was used to form stable core–shell particles by crosslinking either the shell or the core using a diamine in aqueous solution at 40 °C. Analysis by SEC suggested that crosslinking was efficient, with all unimers being incorporated into core–shell particles. FT-IR analyses of these stable core–shell particles revealed that some azlactone rings are still present when nanoparticles are formed from PDMA23-b-P(VDM10-co-NIPAM46). DLS analysis of the resulting core-crosslinked particles at 25 °C reveals an average hydrodynamic diameter of 29 nm. This result shows that the nanoparticle structure is conserved after reducing the solution temperature below the LCST. Moreover, the azlactone functionality is available to anchor dansylhydrazine. The interest of such azlactone-functionalized core-crosslinked particles is that they are stable at different concentrations, temperatures, and pH, and that they can further react with biomolecules such as drugs.
Acknowledgements
Authors thank Région Pays de La Loire for financial support to Martin Levere and Ministère de l'Enseignement Supérieur et de la Recherche for financial support to Hien The Ho.
Notes and references
- G. Riess, Prog. Polym. Sci., 2003, 28, 1107–1170 CrossRef CAS.
- M. H. Stenzel, Chem. Commun., 2008, 3486–3503 RSC.
- J. Rodriguez-Hérnandez, F. Chicot, Y. Gnanou and S. Lecommandoux, Prog. Polym. Sci., 2005, 30, 691–724 CrossRef.
- R. K. O'Reilly, M. J. Joralemon, K. L. Wooley and C. J. Hawker, Chem. Mater., 2005, 17, 5976–5988 CrossRef CAS.
- R. K. O'Reilly, C. J. Hawker and K. L. Wooley, Chem. Soc. Rev., 2006, 35, 1068–1083 RSC.
- E. S. Read and S. P. Armes, Chem. Commun., 2007, 3021–3035 RSC.
- R. K. O'Reilly, M. J. Joralemon, C. J. Hawker and K. L. Wooley, New J. Chem., 2007, 31, 718–724 RSC.
- R. Fu and G.-D. Fu, Polym. Chem., 2011, 2, 465–475 RSC.
- C. J. Hawker, A. W. Bosman and E. Harth, Chem. Rev., 2001, 101, 3661–3688 CrossRef CAS.
- W. A. Braunecker and K. Matyjaszewski, Prog. Polym. Sci., 2007, 32, 93–146 CrossRef CAS.
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
-
Handbook of RAFT Polymerization, ed. C. Barner-Kowollik, Wiley-VCH, Weinheim, 2008 Search PubMed.
- H. G. Schild, Prog. Polym. Sci., 1992, 17, 163–249 CrossRef CAS.
- A. E. Smith, X. W. Xu and C. L. McCormick, Prog. Polym. Sci., 2010, 35, 45–93 CrossRef CAS.
- E. N. Savariar and S. Thayumanavan, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 6340–6345 CrossRef CAS.
- Y. Li, B. S. Lokitz and C. L. McCormick, Macromolecules, 2006, 39, 81–89 CrossRef CAS.
- Y. Li, B. S. Lokitz, S. P. Armes and C. L. McCormick, Macromolecules, 2006, 39, 2726–2728 CrossRef CAS.
- J. Zhang, X. Jiang, Y. Zhang, Y. Li and S. Liu, Macromolecules, 2007, 40, 9125–9132 CrossRef CAS.
- S. Pascual and M. J. Monteiro, Eur. Polym. J., 2009, 45, 2513–2519 CrossRef CAS.
- M. Enerhardt and P. Théato, Macromol. Rapid Commun., 2005, 26, 1488–1493 CrossRef.
- S. M. Heilmann, J. K. Rasmussen and L. R. Krepski, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 3655–3677 CrossRef CAS.
- A. Guyomard, D. Fournier, S. Pascual, L. Fontaine and J.-F. Bardeau, Eur. Polym. J., 2004, 40, 2343–2348 CrossRef CAS.
- D. Fournier, S. Pascual, V. Montembault, D. M. Haddleton and L. Fontaine, J. Comb. Chem., 2006, 8, 522–530 CrossRef CAS.
- C. Lucchesi, S. Pascual, G. Dujardin and L. Fontaine, React. Funct. Polym., 2008, 68, 97–102 CrossRef CAS.
- G. J. Drtina, S. M. Heilmann, D. M. Moren, J. K. Rasmussen, L. R. Krespski, H. K. Smith, R. A. Pranis and T. C. Turek, Macromolecules, 1996, 29, 4486–4489 CrossRef CAS.
- J. A. Tripp, J. A. Stein, F. Svec and J. M. J. Fréchet, Org. Lett., 2000, 2, 195–198 CrossRef CAS.
- J. A. Tripp, F. Svec and J. M. J. Fréchet, J. Comb. Chem., 2001, 3, 216–223 CrossRef CAS.
- L. Fontaine, T. Lemêle, J.-C. Brosse, G. Sennyey, J.-P. Senet and D. Wattiez, Macromol. Chem. Phys., 2002, 203, 1377–1384 CrossRef CAS.
- G. J. Drtina, L. C. Haddad, J. K. Rasmussen, B. N. Gaddam, M. G. Williams, S. J. Moeller, R. T. Fitzsimons, D. D. Fansler, T. L. Buhl, Y. N. Yang, V. A. Weller, J. M. Lee, T. J. Beauchamp and S. M. Heilmann, React. Funct. Polym., 2005, 64, 13–24 CrossRef CAS.
- S. P. Cullen, I. C. Mandel and P. Gopalan, Langmuir, 2008, 24, 13701–13709 CrossRef CAS.
- J. E. Barringer, J. M. Messman, A. L. Banaszek, H. M. Meyer, III and S. M. Kilbey, II, Langmuir, 2009, 25, 262–268 CrossRef CAS.
- J. M. Messman, B. S. Lokitz, J. M. Pickel and S. M. Kilbey, II, Macromolecules, 2009, 42, 3933–3941 CrossRef CAS.
- M. E. Buck, A. S. Breitbach, S. K. Belgrade, H. E. Blackwell and D. M. Lynn, Biomacromolecules, 2009, 10, 1564–1574 CrossRef CAS.
- T. H. Ho, M. E. Levere, J.-C. Soutif, V. Montembault, S. Pascual and L. Fontaine, Polym. Chem., 2011, 2, 1258–1260 RSC.
- A. J. Convertine, B. S. Lokitz, Y. Vasileva, L. J. Myrick, C. W. Scales, A. B. Lowe and C. L. McCormick, Macromolecules, 2006, 39, 1724–1730 CrossRef CAS.
- Y. Ya Cao, X. X. Zhu, J. Luo and H. Liu, Macromolecules, 2007, 40, 6481–6488 CrossRef.
- B. S. Lokitz, J. M. Messman, J. P. Hinestrosa, J. Alonzo, R. Verduzco, R. H. Brown, M. Osa, J. F. Ankner and S. M. Kilbey, II, Macromolecules, 2009, 42, 9018–9026 CrossRef CAS.
- S. Pascual, T. Blin, P. J. Saikia, M. Thomas, P. Gosselin and L. Fontaine, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 5053–5062 CrossRef CAS.
-
C. M. Schilli, A. H. E. Müller, E. Rizzardo, S. H. Thang and Y. K. Chong, Advances in Controlled/Living Radical Polymerization, in ACS Symposium Series 854, American Chemical Society, Washington, DC, 2003, pp. 603–618, ch. 41 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterization of PNIPAM-CTA and determination of LCST values by UV-Vis spectroscopy. See DOI: 10.1039/c1py00320h |
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here.