A dendronised polymer for bulk heterojunction solar cells
Justin
Yu
a,
Kwan H.
Lee
a,
Yuliang
Zhang
a,
Michael F. G.
Klein
b,
Alexander
Colsmann
b,
Uli
Lemmer
b,
Paul L.
Burn
*a,
Shih-Chun
Lo
a and
Paul
Meredith
*a
aCentre for Organic Photonics & Electronics, The University of Queensland, Brisbane, Queensland, Australia 4072. E-mail: p.burn2@uq.edu.au; meredith@physics.uq.edu.au
bLight Technology Institute, Karlsruhe Institute of Technology (KIT), 76131, Karlsruhe, Germany
First published on 2nd September 2011
Abstract
A Gilch polymerisation was used to form a high molecular weight poly[1,4-phenylenevinylene] with pendent first generation biphenyl dendrons attached to every monomer unit of the backbone. The attachment of the bulky side-chain was found not to disrupt the conjugation of the polymer backbone and the polymer formed an ordered intramolecular structure in the solid-state. Bulk heterojunction (BHJ) organic solar cells were manufactured by blending the polymer at different ratios with [6,6]-phenyl-C61-butyric acid methyl ester (PCBM). Under AM1.5 testing conditions the device with the polymer:PCBM ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 by weight was found to have an optimum Voc = 0.86 V, Jsc = 1.4 mA cm−2, FF = 37% and PCE of 0.44%, with the PCE 30 times higher than the only other report on a dendronised polymer used in a BHJ device.
:3 by weight was found to have an optimum Voc = 0.86 V, Jsc = 1.4 mA cm−2, FF = 37% and PCE of 0.44%, with the PCE 30 times higher than the only other report on a dendronised polymer used in a BHJ device.
Introduction
Dendronised polymers are an emerging class of synthetic macromolecules. As with many new material systems much of the early effort has focused on methodologies for their synthesis1 and understanding the effect of polymer and/or dendron type and generation on topology.2,3 One of the key characteristics of dendronised polymers is that as the generation of the side-chain dendron or dendrimer increases, the order in the polymer backbone changes. For example, a flexible randomly oriented backbone will become more elongated and rigid with an increase of side-chain generation. By changing the type of dendron, its generation and the polymer backbone it has been found that the macromolecular architecture and bulk packing can be varied to give materials that can form columns or tubes,4 a range of liquid crystalline phases,5,6 micelles and well-defined colloidal/microsphere structures.7 Dendronised polymers have been studied for oxygen permselective membranes,8 hierarchically structured fibers,9 holographic data storage,10 as mechanical response materials,11 liquid crystal polymers,12 and biofunctional surfaces.13In contrast there has been less focus on dendronised polymers with optoelectronic properties. There have been only a few reports on the photophysical properties of such materials and even fewer on their use in devices such as an organic light-emitting diodes or solar cells.14–18 Apart from the recent work on phosphorescent poly(dendrimers)16–18 much of the effort on optoelectronic materials has concentrated on dendronised conjugated polymers, and in particular using the dendrons to control adverse interchain interactions that can lead to the quenching of luminescence. Flexible dendrons have been mainly studied,19–21 although there have been a small number of reports of conjugated dendrons attached to a poly(fluorene) backbone.22
In this manuscript, we describe the synthesis and properties of a poly[1,4-phenylenevinylene] (PPV) dendronised polymer with rigid conjugated biphenyl based first generation dendron side-chains, and its performance as the active material in a bulk-heterojunction (BHJ) organic photovoltaic (OPV) device.
Results and discussion
Synthesis and physical properties
The synthetic route to polymer 6 is shown in Scheme 1. A macromonomer approach was adopted to ensure each ‘monomer unit’ in the polymer backbone had a dendron attached. The Gilch double-dehydrohalogenation polymerisation23 was chosen for the polymer formation as it typically gives high-molecular weight polymers. The first key step in the synthesis was the coupling of the benzylbromide focused first generation dendron 1 to phenol 2 using conditions typically used for the building of Fréchet dendrons.24 Under these conditions dendronised 3 was formed in a 94% yield. The ester groups were then reduced with lithium aluminium hydride to give the di-alcohol 4 in a good yield of 81%. Finally, an Appel reaction was used to convert the alcohols to the bromides ready for polymerisation and monomer 5 was isolated in a 53% yield. The polymerisation was carried out by rapidly adding a solution of monomer 5 to a tetrahydrofuran solution containing an excess of potassium tert-butoxide. A problematic occurrence often seen in Gilch polymerisations is the formation of a gel,25 and this was encountered in the synthesis of 6. However, a combination of stirring and sonication was found to be sufficient to break up the gel and give a visually homogenous, highly viscous solution. After purification by precipitation, the light orange coloured 6 was obtained in a ∼68% yield. Polymer 6 was found to have good solubility (up to ∼5 mg cm−3) in chloroform, tetrahydrofuran, and hot (≈60 °C) chlorobenzene.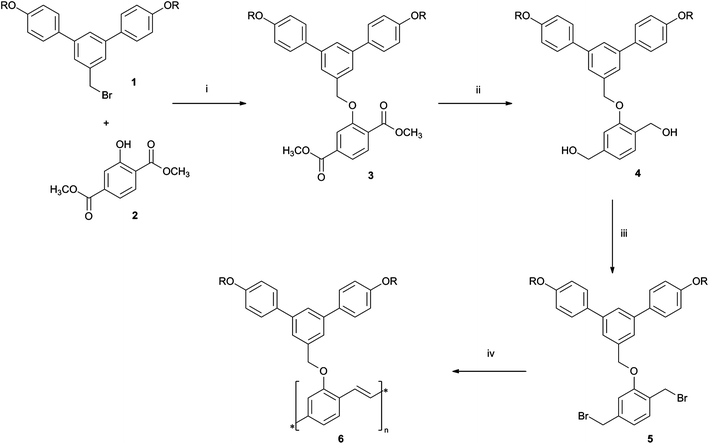 | ||
| Scheme 1 Reagents and conditions: (i) K2CO3, 18-crown-6, acetone, reflux 25 h; (ii) LiAlH4, THF, reflux, 4.75 h; then H+(aq); (iii) PPh3, CBr4, CH2Cl2, r.t., 1 h; (iv) t-BuOK, THF, r.t. 20.5 h. R = 2-ethylhexyl. | ||
The first step in the characterisation of 6 was the 1H NMR spectrum. The 1H NMR spectrum of 6 in deuterated chloroform shown in Fig. 1 is broad, which is indicative of a polymer being formed, and there is no evidence for remaining monomer or short chain oligomers. Infrared spectroscopy of polymer 6 showed an absorption at 964 cm−1, which corresponds to the trans-vinylene CH out-of-plane bend. It is important to have transvinylene linkages as the presence of cis-linkages shortens the effective conjugation length due to twisting of the polymer backbone.26–29Gel permeation chromatography of purified 6 showed that it had an ![[M with combining macron]](https://www.rsc.org/images/entities/i_char_004d_0304.gif) w of 260 kDa and polydispersity index of 2.4. Finally, the thermal stability of 6 was investigated by thermogravimetric analysis under nitrogen and a 5% weight loss was observed at 344 °C.
w of 260 kDa and polydispersity index of 2.4. Finally, the thermal stability of 6 was investigated by thermogravimetric analysis under nitrogen and a 5% weight loss was observed at 344 °C.
 | ||
| Fig. 1 1H NMR spectrum of polymer 6 in CDCl3. | ||
Photophysical and electronic properties
The solution (chlorobenzene) and film (spin-coated from a chlorobenzene solution) UV-visible absorption and photoluminescence (PL) spectra of 6 are shown in Fig. 2. The absorption spectra of PPVs are generally divided into two key wavelength regions. The localised π–π* transitions are found at short wavelengths (≈200 nm) while the absorptions due to the delocalised π-system are observed in the visible region. The ratio between these two transitions is often used as a guide in understanding the level of delocalisation in a particular PPV.30 In solution the long wavelength absorption due to delocalised π–π* transitions is relatively broad and featureless. The chlorobenzene solvent masks the short wavelength absorptions of the dendrons and localised transitions. The solution PL spectrum shows a peak at 514 nm ([0,0] transition) and the emission has poorly defined shoulders corresponding to the [0,1] and [0,2] transitions. In moving from solution to solid-state several important changes are observed: first, the absorption spectrum of the film has peaks at 206 nm, 273 nm, and 473 nm. The peaks at 206 nm and 473 nm correspond to the localised (polymer backbone and dendrons) and delocalised (polymer) π–π* transitions, respectively. The extra absorption peak at 273 nm corresponds to the chromophores in the first generation dendrons. Second, there is a red shift in the of absorption maxima in the visible region and there is a well-defined shoulder. Third, the red shift in the absorption is mirrored in the PL spectrum with an emission maximum at 540 nm. Finally, vibrational modes are clearly resolvable in the PL spectrum. These results demonstrate that the polymer in the film is significantly more ordered than in the solution state. Importantly, the peak maxima for the absorption and emission are similar to other mono-alkoxylated PPVs31,32 indicating that the bulky dendrons are not causing substantial twisting of the polymer backbone. Interestingly, excitation at either 265 nm or 465 nm resulted in only emission being observed from the polymer backbone showing that energy transfer from the dendron to the polymer backbone was efficient. | ||
| Fig. 2 Solution (chlorobenzene) and film (cast from chlorobenzene) absorption (Abs) and photoluminescence (PL) spectra of polymer 6. The excitation wavelength used to obtain the emission spectra for the film and solution were 442 nm and 430 nm, respectively. | ||
The final step in characterising the optoelectronic properties of polymer 6 was to determine whether the energy levels would be compatible with the electron acceptor, [6,6]-phenyl-C61-butyric acid methyl ester (PCBM), for use in photovoltaic devices. We first determined the ionisation potential of a film of polymer 6 spin-coated from tetrahydrofuran onto glass using Photoelectron Spectroscopy in Air (PESA). The ionization potential was 5.4 eV. We then estimated the electron affinity of polymer 6 to be ≈3.0 eV by adding the energy from the optical gap (2.4 eV) to the ionisation potential. The optical gap was calculated from the film absorption and PL data shown in Fig. 2 by the standard method: the corrected absorbance and PL spectra were obtained by dividing the absorbance by eV and PL by eV3; the spectra were then normalised and plotted against eV;33 and finally the optical gap was taken as the intersect of the absorption and PL spectra. An electron affinity of ≈3.0 eV for 6 means that PCBM will be a suitable electron acceptor when blended together in a bulk heterojunction solar cell.
OPV device performance
Photovoltaic cells with a Glass/ITO/PEDOT:PSS (75 nm)/‘Active Layer’/Ca (30 nm)/Al (200 nm) architecture were fabricated (ITO = indium-tin-oxide; PEDOT:PSS = poly[ethylenedioxythiophene]:poly[styrenesulphonic acid]). The ‘Active Layer’ consisted of polymer 6 blended with the soluble fullerene derivative PCBM in three different weight ratios of polymer 6:PCBM (1:2, 1:3, and 1:4). Fig. 3 shows the current density-voltage (J–V) responses of the best performing device from each of the blend ratios, and Table 1 summarises the best performance characteristics along with average values in each group. Under air mass 1.5 (AM1.5) illumination, a ratio of polymer 6:PCBM of 1:3 achieved the best overall photon conversion efficiency (PCE) of 0.44% with an open circuit voltage (Voc) of 0.86 V, short circuit current density (Jsc) of 1.40 mA cm−2, and fill-factor (FF) of 37%. The largest change in efficiency occurs in moving from the 1:2 to the 1:3 blend with the latter and 1:4 the blend having similar performance. All three blend ratios have similar Vocs at around 0.86 V (Table 1) with the cause of the lower PCE for the 1:2 blend being primarily due to the lower short circuit current density. This is related directly to the light harvesting and free carrier extraction efficiency. The decrease in Jsc seen for the 1:2 blend is likely to be due to decreased hole mobility and/or a relative imbalance of electron and hole transport caused by the reduced polymer fraction. It is also worthy of note that the active layers in all devices are thick for polymer-based BHJ devices and this leads to relatively high series resistances as indicated by the J–V slope around short circuit. The incident photon-to-current conversion efficiency (IPCE) response of the 1:3 blend shown in the inset of Fig. 3 shows a peak efficiency at 350 nm. This wavelength corresponds to a minimum in the absorption spectrum of 6 (Fig. 2) and shows that light absorption by the PCBM also plays an important role in device performance. Between 400 nm and 500 nm there is a clear contribution to the IPCE from light absorption by polymer 6. While the efficiency of the devices does not yet rival the state-of-the-art ‘low bandgap polymers’ the incorporation of conjugated dendrons onto the polymer backbone is a major step forward in the design of dendronised polymers for organic solar cells. A PCE of 0.44% is a thirty-fold improvement compared to the only other report of a dendronised PPV, which is comprised of flexible first generation dendrons.31
 | ||
| Fig. 3 Best J–V characteristics of BHJ devices made from each blend ratio under AM1.5 illumination and in the dark. The inset shows the IPCE response curve for the 1:3 device. | ||
| Device | PCE (%) | FF (%) | J sc (mA cm−2) | V oc (V) | Thickness (nm) |
|---|---|---|---|---|---|
| a Average of 4 devices. | |||||
| 1:2 |
0.30 | 34 | 1.01 | 0.86 | — |
| Average | 0.26 ± 0.03 | 33.2 ± 0.6 | 0.93 ± 0.07 | 0.85 ± 0.01 | 578 ± 213 |
| 1:3 |
0.44 | 37 | 1.40 | 0.86 | — |
| Average | 0.40 ± 0.03 | 36.5 ± 0.3 | 1.29 ± 0.08 | 0.85 ± 0.01 | 625 ± 143 |
| 1:4 |
0.42 | 37 | 1.29 | 0.87 | — |
| Averagea | 0.39 ± 0.02 | 36.5 ± 0.7 | 1.26 ± 0.04 | 0.86 ± 0.01 | 498 ± 65 |
Conclusion
In summary, we have developed the first of a new class of dendronised soluble PPV derivatives. In spite of the rigidity and steric bulk of the dendron side-chains the polymer had a high molecular weight, and was comprised primarily of trans-vinylene linkages. The polymer was thermally stable and could form films characterised by structured absorption and emission spectra. Bulk-heterojunction solar cells at blend ratios of 1:2, 1:3 and 1:4 were fabricated and tested and found to be more efficient when the molar ratio of PCBM to polymer ‘monomer’ unit was significantly above 1:1. The best performance was for a 1:3 blend by weight that had a 0.44% PCE, representing the highest efficiency reported to date for a dendronised polymer in OPV devices.
Experimental
1H and 13C NMR spectra were recorded on an Oxford Instruments 400 MHz spectrometer. Chemical shifts are reported in parts per million (ppm) and referenced to the residual solvent peak for deuterated chloroform at 7.26 ppm for 1H, and 77.16 ppm for 13C.34G1-bp H = dendron branching phenyl H, spH = surface phenyl H, PBH = protons on the phenyl ring that become the polymer backbone. MALDI-TOF spectra were recorded on an Applied Biosystems Voyager MALDI-TOF mass spectrometer in positive reflectron mode, the matrix used was 2,5-dihydroxybenzoic acid. Elemental analyses were carried out by Mr. George Blazak of the Elemental Microanalysis Research Laboratory at the University of Queensland. Infrared spectra of neat samples were recorded on a Perkin-Elmer Spectrum 100 FT-IR Spectrometer. UV-Visible absorption measurements were recorded on a Cary Varian 5000 UV-Vis-NIR spectrophotometer. Steady-state photoluminescence spectra were recorded on a Horiba Jobin-Yvon Fluorolog Tau3. Thermogravimetric analyses (TGA) were performed with a Perkin-Elmer STA 6000 under nitrogen, the decomposition temperature (Td(5%)) is reported as the 5% weight loss temperature. Gel permeation chromatography was carried out using Waters Styragel HT 6E and Styragel HT 3 columns running in series, in tetrahydrofuran at 1 cm3 min−1 at 40 °C, calibrated to Waters poly(styrene) standards. Centrifugation was performed using a Sigma 2–5 centrifuge.Flash column chromatography was performed using Merck silica gel, 230–400 mesh. Columns with length less than 5 cm are referred to as a silica plug. When the eluent solvent is a mixture, the composition is given as a volume ratio of the respective solvents. Solvents were dried using the following techniques: tetrahydrofuran was distilled from sodium/benzophenone; acetone was heated at reflux over anhydrous potassium carbonate, then distilled and stored over activated 4 Å molecular sieves under argon prior to use; anhydrous dichloromethane was obtained by distillation over calcium hydride and then stored over activated 4 Å molecular sieves under argon prior to use. Ethyl acetate, n-hexane, dichloromethane, and light petroleum used in column chromatography and extractions were all distilled prior to use. All other reagents were used as received from chemical suppliers without further purification.
Device preparation and characterisation
All process steps were carried out in a clean room. Bulk heterojunction solar cells were fabricated on patterned ITO-glass slides (16 mm × 16 mm, 150 nm ITO, 12 Ω/□, VisionTek) with the anode pattern formed by a lithography process and etched with hydrochloric acid. The substrates were cleaned with detergent (Hellmanex® II), followed by successive sonication in water, acetone, and 2-propanol for 10 min each, then blown dry with nitrogen. After exposing the substrates to an oxygen plasma for 120 s in order to remove remaining organic residues on the surfaces, they were transferred to a glovebox (<1 ppm oxygen and water) and kept under a nitrogen atmosphere for the entire deposition process. A 75 nm layer of PEDOT:PSS (Clevios™ P VP AI4083, Heraeus) was deposited by spin-coating at 4000 rpm for 30 s, then residual water was removed by annealing the substrates in a vacuum oven for 30 min at 150 °C.Solutions of 6 and PCBM were also prepared under a nitrogen atmosphere by adding the required amount of PCBM to a 5 mg cm−3 solution of 6 in chlorobenzene to give weight ratios of 1:2, 1:3, and 1:4 and stirring the mixture at 80 °C. No filtration was performed due to the high viscosity of the solutions. The solutions were stirred at 80 °C until immediately prior to spin-coating, as gelation generally occurred after around 5 min after cooling. A 120 μL aliquot of solution was deposited onto the PEDOT:PSS coated ITO-glass substrate. The 1:3 and 1:4 solutions were spin-coated at 1500 rpm for 60 s, with 3 s acceleration time, while the 1:2 solution spin-time was slightly shorter at 30 s. The films were then placed into a thermal evaporation chamber under a vacuum of (8 × 10−7 mbar) and the devices completed by depositing 30 nm of calcium (at a rate of 3 Å s−1), followed by 200 nm of aluminium (at a rate of 3 Å s−1). The devices had active device areas of 4 mm2, and each substrate contained 6 devices. The film thicknesses were determined using a Dektak 150 surface profilometer.
A spectrally monitored Oriel 300 W solar simulator was used to simulate sunlight according to the ASTM-G173-03e1 standard for hemispherical solar irradiance.35 The J–V characteristics of the solar cells were recorded with a source measure unit (Keithley 238). To avoid degradation during the measurement of the IPCE the solar cells were encapsulated. The homebuilt IPCE setup consisted of a 450 W Xenon light source, a chopper wheel (280 Hz), a 300 mm monochromator (LOT-Oriel), a custom designed current amplifier (DLPCA-S Femto Messtechnik) and a digital lock-in (eLockinin203 Anfatec). A modified photoreceiver (OE-200-S Femto Messtechnik) with a Si/InGaAs sandwich diode was used to monitor the stability of the monochromatic light beam. Initial calibration was achieved with a reference silicon diode (NIST traceable calibration, Thorlabs).
Dimethyl 2-(3,5-bis[4-(2-ethylhexyloxy)phenyl]benzyloxy)terephthalate 3
A mixture of 136 (100 mg, 0.48 mmol), 237 (262 mg, 0.45 mmol), potassium carbonate (99 mg, 0.71 mmol), and 18-crown-6 (12.6 mg, 0.0476 mmol) in anhydrous acetone (10 cm3) was heated at reflux under argon for 25 h. Water (5 cm3) and hydrochloric acid (3 M, 0.5 cm3) were added and the solution was allowed to cool to r.t. The mixture was extracted with dichloromethane (3 × 5 cm3) and the combined extracts were dried over anhydrous magnesium sulphate, filtered and the solvent removed. The residue was purified by flash column chromatography over silica, using ethyl acetate:n-hexane (1:6 to 1:4) as eluent, to afford 3 as a light yellow oil (318 mg, 94%). Found: C, 76.1%; H, 8.15%; C45H56O7 requires C, 76.2%; H, 8.0%; νmax(film)/cm−1 1725 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O); λmax (CH2Cl2)/nm 268 [log (ε/dm3 mol−1 cm−1) (4.82)], 324sh (3.60); 1H NMR (400 MHz, CDCl3) δ: 0.90–0.97 (12 H, m, CH3), 1.31–1.57 (16 H, m, CH2), 1.71–1.81 (2 H, m, CH), 3.88–3.92 (7 H, m, OCH2 and OCH3), 3.94 (3 H, s, OCH3), 5.33 (2 H, s, OCH2Ar), 7.00 and 7.59 (8 H, AA′BB′, spH), 7.64 (2 H, d, J = 1.5, G1-bpH), 7.68 (1 H, dd, J = 8, 1.5, PBH), 7.68 (1 H, dd, J = 1.5, 1.5, G1-bpH), 7.78 (1 H, d, J = 1.5, PBH), 7.85 (1 H, d, J = 8, PBH); 13C NMR (100 MHz, CDCl3) δ: 11.3, 14.2, 23.2, 24.0, 29.2, 30.7, 39.6, 52.5, 52.7, 70.8, 71.0, 114.7, 115.0, 121.8, 123.8, 125.0, 125.2, 128.3, 131.8, 133.4, 134.5, 137.3, 141.9, 157.8, 159.3, 166.3, 166.6; m/z (MALDI) found: 731.3 (100%), 732.3 (53%), 733.3 (12%); C45H56O7Na requires 731.4 (100%), 732.4 (51%), 733.4 (14%).
O); λmax (CH2Cl2)/nm 268 [log (ε/dm3 mol−1 cm−1) (4.82)], 324sh (3.60); 1H NMR (400 MHz, CDCl3) δ: 0.90–0.97 (12 H, m, CH3), 1.31–1.57 (16 H, m, CH2), 1.71–1.81 (2 H, m, CH), 3.88–3.92 (7 H, m, OCH2 and OCH3), 3.94 (3 H, s, OCH3), 5.33 (2 H, s, OCH2Ar), 7.00 and 7.59 (8 H, AA′BB′, spH), 7.64 (2 H, d, J = 1.5, G1-bpH), 7.68 (1 H, dd, J = 8, 1.5, PBH), 7.68 (1 H, dd, J = 1.5, 1.5, G1-bpH), 7.78 (1 H, d, J = 1.5, PBH), 7.85 (1 H, d, J = 8, PBH); 13C NMR (100 MHz, CDCl3) δ: 11.3, 14.2, 23.2, 24.0, 29.2, 30.7, 39.6, 52.5, 52.7, 70.8, 71.0, 114.7, 115.0, 121.8, 123.8, 125.0, 125.2, 128.3, 131.8, 133.4, 134.5, 137.3, 141.9, 157.8, 159.3, 166.3, 166.6; m/z (MALDI) found: 731.3 (100%), 732.3 (53%), 733.3 (12%); C45H56O7Na requires 731.4 (100%), 732.4 (51%), 733.4 (14%).
2,5-Bis(hydroxymethyl)-(3,5-bis[4-(2-ethylhexyloxy)phenyl]benzyloxy)benzene 4
A solution of 3 (131 mg, 0.18 mmol) in anhydrous tetrahydrofuran (2 cm3) was added dropwise to a stirred suspension of lithium aluminium hydride (49 mg, 1.3 mmol) in anhydrous tetrahydrofuran (3 cm3) at 0 °C under argon. The reaction was heated at reflux for 4.75 h, before being cooled to r.t. and placed in an ice-bath. Ethyl acetate (7 cm3) was added and the white gelatinous mixture was poured onto ice (20 cm3). Sulphuric acid (10%, 1.5 cm3) was added until the solution was at pH = 1. Brine was added and then the mixture was extracted with ethyl acetate (2 × 20 cm3). The combined extracts were dried over anhydrous magnesium sulphate, filtered, and the solvent removed. The residue was passed through a silica plug using a solvent mixture of ethyl acetate:n-hexane (1:4) as eluent to give 4 as a light yellow viscous oil (98 mg, 81%). Found: C, 78.8%; H, 8.7%; C43H56O5 requires C, 79.1%; H, 8.7%; νmax(film)/cm−1 3261, 3346 (OH); λmax (CH2Cl2)/nm 270 [log (ε/dm3 mol−1 cm−1) (4.67)]; 1H NMR (400 MHz, CDCl3) δ: 0.91–0.98 (12 H, m, CH3), 1.32–1.60 (16 H, m, CH2), 1.72–1.81 (2 H, m, CH), 1.90 (1 H, br, OH), 2.37 (1 H, br, OH), 3.89–3.91 (4 H, m, OCH2), 4.68 (2 H, s, ArCH2OH), 4.75 (2 H, s, ArCH2OH), 5.20 (2 H, s, OCH2Ar), 6.94 (1 H, brd, J = 7.5, PBH), 7.00 and 7.57 (8 H, AA′BB′, spH), 7.07 (1 H, brs, PBH), 7.30 (1 H, brd, J = 7.5, PBH), 7.53 (2 H, d, J = 1.5, G1-bpH), 7.70 (1 H, dd, J = 1.5, 1.5, G1-bpH); 13C NMR (100 MHz, CDCl3) δ: 11.3, 14.2, 23.2, 24.0, 29.2, 29.8, 30.7, 39.5, 62.0, 65.3, 70.4, 70.7, 110.3, 115.0, 119.4, 124.2, 125.2, 128.3, 129.0, 133.2, 137.7, 142.1, 142.2, 157.0, 159.4; m/z (MALDI) found: 675.3 (100%), 676.3 (37%), 677.3 (9%); C43H56O5Na requires 675.4 (100%), 676.4 (48%), 677.4 (12%).
2,5-Bis(bromomethyl)-(3,5-bis[4-(2-ethylhexyloxy)phenyl]benzyloxy)benzene 5
A solution of triphenylphosphine (1.06 g, 4.04 mmol) in anhydrous dichloromethane (32 cm3) was added dropwise to a solution of 4 (978 mg, 1.5 mmol) and carbon tetrabromide (1.34 g, 4.04 mmol) at r.t. under argon over 23 s. The reaction was stirred for 1 h before water (∼3 cm3) was added. The organic was phase collected, and the aqueous layer was extracted with dichloromethane (3 × 4 cm3). The combined organic phases were dried over anhydrous magnesium sulphate, filtered and the solvent removed. The residue was purified in two steps. First, the mixture was filtered through a silica plug using n-hexane to remove excess carbon tetrabromide, and then ethyl acetate:n-hexane (1:4) was used as eluent to obtain the product containing fraction. The product containing fraction was concentrated and then purified by flash column chromatography using ethyl acetate:n-hexane (1:9 to 1:4) as eluent to give 5 as a light yellow oil (621 mg, 53%). Found: C, 66.6%; H, 6.9%; C43H54Br2O3 requires C, 66.3%; H, 7.0%; λmax (CH2Cl2)/nm 267 [log (ε/dm3 mol−1 cm−1) (4.77)]; 1H NMR (400 MHz, CDCl3) δ: 0.90–0.97 (12 H, m, CH3), 1.31–1.61 (16 H, m, CH2), 1.71–1.81 (2 H, m, CH), 3.89–3.91 (4 H, m, OCH2), 4.46 (2 H, s, ArCH2Br), 4.61 (2 H, s, ArCH2Br), 5.27 (2 H, s, OCH2Ar), 6.97–7.02 (5 H, m, PBH and spH), 7.04 (1 H, d, J = 1.5, PBH), 7.33 (1 H, d, J = 7.5, PBH), 7.60 (4 H, 1/2AA′BB′, spH), 7.64 (2 H, d, J = 1.5, G1-bpH), 7.70 (1 H, dd, J = 1.5, 1.5, G1-bpH); 13C NMR (100 MHz, CDCl3) δ: 11.3, 14.2, 23.2, 24.0, 28.8, 29.2, 30.7, 33.4, 39.6, 70.4, 70.7, 113.0, 115.0, 121.7, 124.0, 125.1, 127.0, 128.4, 131.3, 133.3, 137.5, 140.1, 141.9, 156.9, 159.4; m/z (MALDI) found: 776.2 (48%), 777.2 (23%), 778.2 (100%), 779.2 (49%), 780.2 (55%), 781.2 (24%), 782.2 (6%); C43H54Br2O3 requires 776.2 (49%), 777.2 (24%), 778.2 (100%), 779.2 (47%), 780.2 (58%), 781.2, (24%), 782.2 (6%).
Poly[2-(3,5-bis{4-[2-ethylhexyloxy]phenyl}benzyloxy)-1,4-phenylenevinylene] 6
A solution of 5 (199.8 mg, 0.257 mmol) in anhydrous tetrahydrofuran (1.5 cm3) was rapidly injected into a tetrahydrofuran solution of potassium tert-butoxide (0.175 M, 8.8 cm3) at r.t. under argon. A yellow colour was observed to quickly evolve in the reaction mixture, and ∼15 s after reaction initiation a gel was observed to form. Tetrahydrofuran (10 cm3) was added 2 min after monomer addition and the sealed reaction mixture was sonicated for 1.5 h until the gel was dispersed. The viscous solution was then stirred in the dark for a further 19 h at r.t. Tetrahydrofuran (30 cm3) was added to dilute the polymer solution which was then slowly pipetted into a 1:2 water:methanol mixture (195 cm3) resulting in a fibrous orange polymer precipitate. The impure precipitate was collected by centrifuging the suspension at 2000 rpm in ∼10 cm3 portions, decanting the supernatant each time. The combined residual solids were triturated with methanol (3 × ∼10 cm3) then dried under reduced pressure. The dried impure polymer was then dissolved in tetrahydrofuran (∼50 cm3) with sonication, and then precipitated by slow addition into methanol (∼250 cm3). The polymer precipitate was collected by centrifuging aliquots of the mixture and removing the supernatant before the solids were combined and triturated with methanol. Finally, the solid was dried under reduced pressure to yield polymer 6 as a light orange fibrous solid (107.9 mg, ∼68%). Td(5%) = 344 °C; w = 2.6 × 105, n = 1.1 × 105, PDI = 2.4; found: C, 83.2%; H, 8.6%; C43H52O3 requires C, 83.7%; H, 8.5%; νmax(film)/cm−1 964 (HCC–H); λmax (chlorobenzene)/nm 462; λmax (film)/nm 206, 273, 473, 503sh; 1H NMR (400 MHz, CDCl3) δ: 0.75–0.95 (12 H, br, CH3), 1.15–1.60 (16 H, br, CH2), 1.60–1.75 (2 H, br, CH), 3.55–3.90 (4 H, br, OCH2), 4.80–5.40 (2 H, brm, OCH2Ar), 6.60–7.70 (16 H, br, PBH, G1-bpH, spH, and vinyl-H).
Acknowledgements
Professor Paul Burn is the recipient of an Australian Research Council Federation Fellowship (Project FF0668728) and Professor Paul Meredith the recipient of a Queensland Smart State Senior Fellowship and University of Queensland Vice Chancellor's Senior Research Fellowship. We thank the Australian Federal Government and the Federal Minister for Education and Research (BMBF, Germany) for support under the Australia-Germany Solar Photovoltaics Research Call 2010/2011, the University of Queensland (Strategic Initiative - Centre for Organic Photonics & Electronics), the Queensland Government National and International Research Alliance Program [Queensland Organic Solar Cell Alliance (OSCA)]. We also thank Dr S. Watkins at the Commonwealth Science and Industrial Research Organisation (Clayton VIC) for the PESA measurement. Dr Kwan H. Lee is a University of Queensland Postdoctoral Research Fellow. A UQ GSITA travel scholarship was awarded to Justin Yu to enable aspects of the work to be undertaken.References
- H. Frauenrath, Prog. Polym. Sci., 2005, 30, 325 CrossRef CAS.
- B. M. Rosen, C. J. Wilson, D. A. Wilson, M. Peterca, M. R. Imam and V. Persec, Chem. Rev., 2009, 109, 6275 CrossRef CAS.
- Y. Chen and X. Xiong, Chem. Commun., 2010, 46, 5049 RSC.
- T. Kaneko, T. Horie, S. Matsumoto, M. Teraguchi and T. Aoki, Macromol. Chem. Phys., 2009, 210, 22 CrossRef CAS.
- A. J. Soininen, E. Kasëmi, A. D. Schlüter, O. Ikkala, J. Ruokolainen and R. Mezzenga, J. Am. Chem. Soc., 2010, 132, 10882 CrossRef CAS.
- C. Li, A. D. Schlüter, A. Zhang and R. Mezzenga, Adv. Mater., 2008, 20, 4530 CrossRef CAS.
- S. Achelle, Á. Blanco, M. López-García, R. Sapienza, M. Ibisate, C. López and J. Rodríguez-López, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 2659 CrossRef CAS.
- T. Kaneko, K. Yamamoto, M. Asano, M. Teraguchi and T. Aoki, J. Membr. Sci., 2006, 278, 365 CrossRef CAS.
- S. Feng, X. Xiong, G. Zhang, N. Xia, Y. Chen and W. Wang, Macromolecules, 2009, 42, 281 CrossRef CAS.
- A. Khan, A. E. Daugaard, A. Bayles, S. Koga, Y. Miki, K. Sato, J. Enda, S. Hvilsted, G. D. Stucky and C. J. Hawker, Chem. Commun., 2009, 425 RSC.
- I. Popa, B. Zhang, P. Maroni, A. D. Schlüter and M. Borkovec, Angew. Chem., Int. Ed., 2010, 49, 4250 CrossRef CAS.
- C.-A. Yang, G. Wang, H. Xie, Q. Wang, H. Zhang, E. Chen and Q. Zhou, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1149 CrossRef CAS.
- M. Pohl, N. Michaelis, F. Meister and T. Heinze, Biomacromolecules, 2009, 10, 382 CrossRef CAS.
- Q. Peng, J. Xu, M. Li and W. Zheng, Macromolecules, 2009, 42, 5478 CrossRef CAS.
- Z. Tan, R. P. Tang, E. J. Zhou, Y. He, C. Yang, F. Xi and Y. Li, J. Appl. Polym. Sci., 2008, 107, 514 CrossRef CAS.
- J. P. Gunning, J. W. Levell, M. F. Wyatt, P. L. Burn, J. Robertson and I. D. W. Samuel, Polym. Chem., 2010, 1, 730 RSC.
- W.-Y. Lai, J. W. Levell, A. C. Jackson, S.-C. Lo, P. V. Bernhardt, I. D. W. Samuel and P. L. Burn, Macromolecules, 2010, 43, 6986 CrossRef CAS.
- J. W. Levell, J. P. Gunning, P. L. Burn, J. Robertson and I. D. W. Samuel, Org. Electron., 2010, 11, 1561 CrossRef CAS.
- Y. Fu, Y. Li, J. Li, S. Yan and Z. Bo, Macromolecules, 2004, 37, 6395 CrossRef CAS.
- C.-W. Wu, C.-M. Tsai and H.-C. Lin, Macromolecules, 2006, 39, 4298 CrossRef CAS.
- C.-W. Wu, H.-H. Sung and H.-C. Lin, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 6765 CrossRef CAS.
- S. Setayesh, A. C. Grimsdale, T. Weil, V. Enkelmann, K. Müllen, F. Meghdadi, E. J. W. List and G. Leising, J. Am. Chem. Soc., 2001, 123, 946 CrossRef CAS.
- H. G. Gilch and W. L. Wheelwright, J. Polym. Sci., Part A-1, 1966, 4, 1337 CrossRef CAS.
- C. J. Hawker and J. M. J. Fréchet, J. Am. Chem. Soc., 1990, 112, 7638 CrossRef CAS.
- T. Schwalm, J. Wiesecke, S. Immel and M. Rehahn, Macromol. Rapid Commun., 2009, 30, 1295 CrossRef CAS.
- A. M. Spring, C.-Y. Yu, M. Horie and M. L. Turner, Chem. Commun., 2009, 2676 RSC.
- S. Son, A. Dodabalapur, A. J. Lovinger and M. E. Galvin, Science, 1995, 269, 376 CAS.
- Y. Pang, J. Li, B. Hu and F. E. Karasz, Macromolecules, 1999, 32, 3946 CrossRef CAS.
- C. Huang, W. Huang, J. Guo, C.-Z. Yang and E.-T. Kang, Polymer, 2001, 42, 3929 CrossRef CAS.
- G. R. Webster and P. L. Burn, Synth. Met., 2004, 145, 159 CrossRef CAS.
- R. Tang, Z. Tan, C. Cheng, Y. Li and F. Xi, Polymer, 2005, 46, 5341 CrossRef CAS.
- S.-C. Lo, L.-O. Pålsson, M. Kilitziraki, P. L. Burn and I. D. W. Samuel, J. Mater. Chem., 2001, 11, 2228 RSC.
- J. R. Lakowicz, “Principles of Fluorescence Spectroscopy”, 3rd Ed., Springer, 2006 Search PubMed.
- H. E. Gottlieb, V. Kotlyar and A. Nudelman, J. Org. Chem., 1997, 62, 7512 CrossRef CAS.
- American Society of Testing & Materials (ASTM), in Annual book of ASTM standards 2006, Vol. 14.04, ASTM International, West Conshohocken, USA 2006, Ch. 03e1 Standard Tables for Reference Solar Spectral Irradiances: Direct Normal and Hemispherical on 37° Tilted Surface, DOI:10.1520/G0173-03E01.
- K. Fujimoto, Y. Tokuda, H. Maekawa, Y. Matsubara, T. Mizuno and I. Nishiguchi, Tetrahedron, 1996, 52, 3889 CrossRef CAS.
- S.-C. Lo, R. N. Bera, R. E. Harding, P. L. Burn and I. D. W. Samuel, Adv. Funct. Mater., 2008, 18, 3080 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |