Synthesis of glycerin carbonate-based intermediates using thiol–ene chemistry and isocyanate free polyhydroxyurethanes therefrom†
Sofia
Benyahya
a,
Myriam
Desroches
a,
Rémi
Auvergne
*a,
Stéphane
Carlotti
b,
Sylvain
Caillol
a and
Bernard
Boutevin
a
aInstitut Charles Gerhardt Montpellier UMR 5253 - Equipe Ingénierie et Architectures Macromoléculaires, Ecole Nationale Supérieure de Chimie de Montpellier, 8 rue de l'Ecole Normale, 34296, Montpellier Cedex 5, France. E-mail: remi.auvergne@enscm.fr; Fax: +33467147220; Tel: +33467144305
bLaboratoire de Chimie des Polymères Organiques UMR 5629 Université Bordeaux 1/CNRS, Ecole Nationale Supérieure dev Chimie, de Biologie & de Physique, 16 Avenue Pey-Berland, 33607, Pessac Cedex, France. E-mail: carlotti@enscbp.fr; Tel: 05 40 00 27 34
First published on 6th September 2011
Abstract
A new synthesis of 4-[(prop-2-en-1-yloxy)methyl]-1,3-dioxolan-2-one (AGC) was performed by Williamson ether synthesis from 4-(hydroxymethyl)-1,3-dioxolan-2-one. Dicyclocarbonates were synthesized by UV thiol–ene coupling of allyl-cyclocarbonate with a 2,2′-oxydiethanethiol. This photochemical thiol–ene reaction was carried out under air, with neither solvent nor photoinitiator. The products, obtained with high yield, were characterized by 1H NMR and FTIR analysis. The synthesized dicyclocarbonates were used without purification to synthesize polyhydroxyurethanes without isocyanate by step growth polyaddition with 1,10-diaminodecane. The synthesized polyhydroxyurethanes were characterized by 1H NMR, FTIR, ATG and DSC analysis. These polyhydroxyurethanes exhibited glass transition temperatures from −31 °C to −14 °C, molecular weight from 7,000 g mol−1 to 9000 g mol−1 and degradation temperature for 5% of weight loss (Td 5%) between 227 °C and 250 °C.
Introduction
Polyurethanes (PUs) are one of the most dynamic groups of polymers, exhibiting versatile properties suitable for use in practically all fields of polymer applications—foams, elastomers, thermoplastics, thermorigids, adhesives, coatings, sealants, fibers and so on. Generally, PUs are obtained from the reaction of an oligomeric polyol (low molar mass polymer with terminal hydroxyl groups) and a diisocyanate (or polyisocyanate). However, the use of diisocyanate should be avoided for several reasons. Isocyanate reactants are generally very harmful for human health, particularly for people exposed during polyurethane synthesis, and could entail adverse health effects such as asthma, dermatitis, conjunctivitis and acute poisoning.1 Moreover, isocyanates are synthesized from phosgene which is a very toxic chemical that raises questions in industry. Therefore the synthesis of PUs from step growth polyaddition of dicyclocarbonates and diamines should be favored.2,3 Thus, this old reaction is currently gaining a great deal of attention as a substitution route for the synthesis of PUs. This method is particularly interesting since no hazardous isocyanates are used. Moreover dicyclocarbonate reactants could be provided from renewable resources such as glycerin. This route allows polyhydroxyurethanes (PHUs) with hydrogen bonds to be obtained, presenting higher chemical and hydrolysis resistances.The synthesis of PHUs from step growth polyaddition of dicyclocarbonates and diamines was extensively reported in the literature, particularly by Endo.4 Indeed, several cyclocarbonates were synthesized and some polyhydroxyurethanes were thereof characterized.5–8
Several methods are used to synthesize five-membered cyclic carbonates (Scheme 1).9–18 Most of these methods are based on epoxide or diol reactants. This is also the case of dicyclocarbonate syntheses (Scheme 2 (1–5)).6,17,19–21 The production of methyl esters from vegetable oil leads to glycerin as a by-product. Thus, the use of glycerin carbonate (4-(hydroxymethyl)-1,3-dioxolan-2-one) presents a great interest for the synthesis of dicyclocarbonate. Generally, syntheses of PHUs from glycerin carbonate are based on esterification reactions from glycerin carbonate and dicarboxylic acid or derivatives (Scheme 2 (6)). In a previous study,22 a five-membered dicyclocarbonate bis[(2-oxo-1,3-dioxolan-4-yl)methyl] benzene-1,4-dicarboxylate DCterter was prepared by esterification of carboxylic acid groups of benzene-1,4-dicarboxylic acid with alcohol function of commercial 4-(hydroxymethyl)-1,3-dioxolan-2-one. This method was also reported by other authors for the synthesis of various polydicyclocarbonates, either symmetric or asymmetric.8,23 Dicyclocarbonates synthesized present ester bonds on the carbon chain, thus the resulting materials are more sensitive to hydrolysis reaction.
 | ||
| Scheme 1 Various synthesis routes to obtain cyclic carbonates molecules. | ||
 | ||
| Scheme 2 Various dicyclocarbonate synthesis routes. | ||
Furthermore, the synthesis of thioether five- and six-membered dicyclocarbonates by radical addition of ethane-1,2-dithiol with 4-(3-butenyl)-1,3-dioxolan-2-one(Scheme 2 (7))14 or by reaction between 4-ethenyl-1,3-dioxolan-2-one (AC) and several thiols (Scheme 2 (8))24 has been reported. These dicyclocarbonates allow polyhydroxyurethanes to be obtained without ester bonds. However, these monomers were not synthesized from glycerol derivatives and were very expensive.
Another cyclocarbonate monomer is reported in the literature, the 4-[(prop-2-en-1-yloxy)methyl]-1,3-dioxolan-2-one (AGC). AGC was obtained by different methods (Scheme 3) without the use of glycerin carbonate derivatives.
![Different methods for 4-[(prop-2-en-1-yloxy)methyl]-1,3-dioxolan-2-one (AGC) synthesis.](/image/article/2011/PY/c1py00289a/c1py00289a-s3.gif) | ||
| Scheme 3 Different methods for 4-[(prop-2-en-1-yloxy)methyl]-1,3-dioxolan-2-one (AGC) synthesis. | ||
According to the literature, AGC molecule was generally synthesized by reaction between carbon dioxide and allyl glycidyl ether by homogeneous25 or heterogeneous26 catalysis (Scheme 3 (9)). Indeed, Schmidt reported the synthesis of 4-((allyloxy)methyl)-1,3-dioxolan-2-one by carbonylation of oxiran-2-ylmethyl prop-2-en-1-yl carbonate in the presence of chromium (III) octaethylporphyrinato tetracarbonylcobaltate [(OEP)-Cr(THF)2][Co(CO)4] (Scheme 3 (9)).27 This carbonate was also synthesized from reaction between 4-methyloxetan-2-one and 2-[(prop-2-en-1-yloxy)methyl]oxirane with tetrabutylazanium bromide TBAB (Scheme 3 (10))9 or by transcarbonatation between an allyl-diol and an organic carbonate (Scheme 3 (11)).28 Finally, AGC was synthesized from reaction between propane-1,2,3-triol and 4-(chlorooxy)-4-oxobut-1-ene with palladium (Scheme 3 (12)).29
In the literature, thiol–ene coupling was initially reported on 4-(3-butenyl)-1,3-dioxolan-2-one thermally initiated with a radical initiator and in solvent. Purifications were needed and yields varied between 50 and 60%.14 UV thiol–ene coupling with photoinitiator was also reported.24 However, the use of a photoinitiator represents a drawback since residual fragments of photoinitiator remain in the polymer and could result in accelerated ageing or yellowing of materials. In a previous study,30 UV thiol–ene coupling on vegetable oils without any photoinitiator has been described.
Therefore, in this study, the 4-[(prop-2-en-1-yloxy)methyl]-1,3-dioxolan-2-one AGC has been synthesized by a new, easy and inexpensive method, based on the use of the Williamson reaction. Synthesized AGC and commercial AC have been used to synthesize bis-AGC and bis-AC from thiol ene addition (click chemistry), and bis-AGC and bis-AC were polymerized with 1,10-diaminedecane (DA10) to obtain polyhydroxyurethane materials.
Results and discussion
Two different cyclocarbonates, 4-ethenyl-1,3-dioxolan-2-one AC and 4-[(prop-2-en-1-yloxy)methyl]-1,3-dioxolan-2-one AGC, were used for the synthesis of dicyclocarbonates bis-AC and bis-AGC by thiol–ene coupling. AC cyclocarbonate is a commercial chemical, whereas the AGC cyclocarbonate was synthesized by Williamson ether reaction. As AC cyclocarbonate is a commercial product, its characterization is not reported here. The AGC cyclocarbonate synthesis is reported herein.Synthesis of dicyclocarbonates: bis-AGC and bis-AC
Bis-AGC and bis-AC were synthesized by reaction between a dithiol and AGC or AC, respectively. This reaction was carried out by UV thiol–ene coupling without a photoinitiator (Scheme 4). | ||
| Scheme 4 Synthesis of dicyclocarbonate by thiol–ene coupling (ratio thiol/double bond of 1/1, irradiated at 10 W cm−2). | ||
This reaction was realized without any solvent, with a thiol/double bonds ratio of 1/1 and an irradiation between 250 and 450 nm, at 10 W cm−2. Firstly, dicyclocarbonate bis-AC was synthesized in one step by UV thiol–ene coupling of 2,2′-oxydiethanethiol on commercial cyclocarbonate AC in 2 hours. The 1H NMR spectrum of the product allows clear identification of the expected product (Fig. 1).
 | ||
| Fig. 1 1H NMR spectrum of commercial cyclocarbonate (AC) after thiol–ene coupling (ratio thiol/double bond of 1/1, irradiated 2 hours at 10 W cm−2). | ||
Addition of thiol was confirmed by the disappearance of signals corresponding to the vinylic protons at 5.43 ppm (CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2) and 5.88 ppm (CHCH2), and by the appearance of signals corresponding to the thioether protons between 2.65 ppm and 2.78 ppm (CH2–S, signal d, d′) and between 1.94 ppm and 2.08 ppm (CH2–CH2–S, signal c, c′) (Fig. 1). These signals corresponding to protons in α and β positions of sulfur appeared as a multiplet dedoubled due to the vicinity of the asymmetric carbon b. The product of thiol addition on the more substituted carbon is not observed in this spectrum, which confirms the selectivity of thiol–ene coupling.
CH2) and 5.88 ppm (CHCH2), and by the appearance of signals corresponding to the thioether protons between 2.65 ppm and 2.78 ppm (CH2–S, signal d, d′) and between 1.94 ppm and 2.08 ppm (CH2–CH2–S, signal c, c′) (Fig. 1). These signals corresponding to protons in α and β positions of sulfur appeared as a multiplet dedoubled due to the vicinity of the asymmetric carbon b. The product of thiol addition on the more substituted carbon is not observed in this spectrum, which confirms the selectivity of thiol–ene coupling.
The UV thiol–ene coupling was then successfully used for the synthesis of dicyclocarbonate AGC and was carried out in 5 hours. This lower reactivity of this reaction is in agreement with allylic ether reactivity. As observed previously, 1H NMR analysis confirmed thiol addition (Fig. 2). Indeed, the disappearance of signals corresponding to the protons of double bonds at 5.26 ppm (CHCH2) and at 5.85 ppm (CHCH2) and the appearance of signals corresponding to the protons of thioether in the α position of sulfur at 3.58 ppm (CH2–S, signal f; Fig. 2) and in the β position at 1.81 ppm (CH2–CH2–S, signal e; Fig. 2) were observed.
 | ||
| Fig. 2 1H NMR spectrum of synthesized cyclocarbonate (AGC) after thiol–ene coupling (ratio thiol/double bond of 1/1, irradiated 5 hours at 10 W cm−2). | ||
In this case, thiol–ene coupling for the synthesis of dicyclocarbonates fits perfectly with the Sharpless principles of “click chemistry”31 stoichiometry of reactant 1/1, no solvent, no catalyst, high yield and no by-product.
Polyhydroxyurethanes synthesis
Then, step growth polyaddition of synthesized building blocks bis-AC and bis-AGC with commercial decane-1,10-diamine DA10 was carried out and yielded respectively PHU-bis-AC and PHU-bis-AGC polyhydroxyurethanes (Scheme 5). Scheme 5 presents idealized structures (isomer primary–secondary alcohol); most isomers such as primary–primary alcohol, primary–secondary alcohol and secondary–secondary alcohol were obtained. | ||
| Scheme 5 Step growth polyaddition of DA10 and AC or AGC. | ||
Both synthesized PHUs were characterized by 1H and 13C NMR spectrometry and by FTIR analysis. The 1H NMR spectrum of synthesized PHU-bis-AGC is reported in Fig. 3.

This spectrum confirms the formation of the carbamate group with the signal of the proton on nitrogen at 8 ppm.32 The shifts of characteristic signals of carbonate function (aa', b, cc' in Fig. 2) in a, b, c and x, y, z (Fig. 3) are observed. These signals represent the different isomers of cyclocarbonate ring opening. Indeed, ring opening of the cyclocarbonate function by the primary amine function is not regioselective and leads to three PHU isomers, with primary and secondary alcohols.4–8,33 The percent of primary and secondary alcohols was determined thanks to y and b proton integration according to eqn (1):
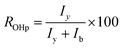 | (1) |
The ratio of primary and secondary alcohols was 25/75, which is in agreement with Endo studies.34
The FTIR spectrum (Fig. 4) confirms formation of PHU-bis-AGC polyhydroxyurethane with the three characteristic bands of the carbamate function: N–H bond stretching vibration, hydrogen bonded CO stretching and N–H bond deformation are respectively observed at 3078 cm−1, 1686 cm−1 and 1530 cm−1.
 | ||
| Fig. 4 Infrared spectra of bis-AGC and PHU-bis-AGC. | ||
The large absorption band of the hydroxyl group OH at 3326 cm−1 also appeared.
On the PHU-bis-AGCFTIR spectrum, the absence of the absorption band of CO carbonyl of the carbonate group at 1785 cm−1 reveals a total conversion of initial bis-AGC.
Table 1 summarizes the main characteristics of both synthesized PHUs, Mn (SEC), Tg (DSC) and Td 5% (TGA). The corresponding figures are given in the ESI†.
As expected, the carbon chain length of dicyclocarbonate influences the Tg value of polymer obtained. Thus, the AGC monomer presents two methyl groups and one ether bond in the chain, leading to a more flexible chain than the AC monomer. This assumption was confirmed by Tg values −31 °C and −14 °C for PHU-bis-AGC and PHU-bis-AC, respectively (see the ESI, Fig. S1†).
The molecular weight values obtained were similar, 7000 and 9000 g mol−1, with polydispersity indices (PI) of 1.5 and 3.2 for PHU-bis-AC and PHU-bis-AGC, respectively. These values are low but still comparable to those reported in the literature. The SEC chromatogram is presented in the ESI, Fig. S2†. Indeed, Endo et al. reported the step growth polyaddition of 4,4′-[ethane-1,2-diylbis(sulfanediylbutane-4,1-diyl)]bis(1,3-dioxolan-2-one) in DMAc at 50 °C in 48 h. This reaction led to a PHU with a yield of 67% and a molecular weight of 7500 g mol−1. After 14 days of reaction under the same conditions, a conversion of 95% is reached with a molecular weight of 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1 (Scheme 6).14
000 g mol−1 (Scheme 6).14
![Step growth polyaddition of 1,2-bis[4-(1,3-dioxolan-2-one-4-yl)-butylthio)ethane with 3,3′-[butane-1,4-diylbis(oxy)]dipropan-1-amine.11](/image/article/2011/PY/c1py00289a/c1py00289a-s6.gif) | ||
| Scheme 6 Step growth polyaddition of 1,2-bis[4-(1,3-dioxolan-2-one-4-yl)-butylthio)ethane with 3,3′-[butane-1,4-diylbis(oxy)]dipropan-1-amine.11 | ||
Finally, concerning thermal degradation measured by ATG, synthesized PHU-bis-AGC and PHU-bis-AC exhibit a degradation temperature (Td 5%) of 249 °C and 227 °C, for 5% of weight loss, respectively. These values confirm the stability of these polymers above 200 °C, as other PUs. The thermal degradation of PHU polymers proceeds in three steps: the first step, between 250 and 350 °C (for PHU-bis-AC) and between 250 and 380 °C (for PHU-bis-AGC) for a weight loss corresponding to 75%; a second degradation step until 480 °C with a weight loss of 10%; and the last step until 580 °C for total degradation (see the ESI, Fig. S3†).
Concerning PHU-bis-AC, thermal degradation under air or nitrogen conditions for 5% of degradation occurred at 227 °C and 225 °C, respectively. We did not observe any difference during thermal degradation under air or nitrogen conditions until 350 °C. Between 350 °C and 450 °C, a difference of 10% was observed, oxidation phenomena under air explain this difference. After 450 °C, the polymer reaches total degradation at 580 °C (see the ESI, Fig. S4†).
Experiment section
Materials
Sodium hydride (95%), 1,10-diaminodecane (DA10) (95%), 3-bromo-1-propene (99%), sodium iodide (99.99%), 2,2′-oxydiethanethiol, 4-ethenyl-1,3-dioxolan-2-one (99%) were purchased from Sigma Aldrich and used as received. 4-(Hydroxymethyl)-1,3-dioxolan-2-one (JEFFSOL®) was supplied by Huntsman (Saint-Mihiel, France). Sodium chloride, magnesium sulfate, tertrahydrofuran (THF) and N,N-dimethylmethanamide (DMF) were purchased from SDS Carlo Erba (Val de Reuil, France). Before use, THF and DMF were dried according to current methods, then distilled and stored under argon atmosphere. Deuterated solvents (CDCl3 and DMSO) were purchased from Eurisotop (Saint-Aubin, France).Analytical techniques
All nuclear magnetic resonance (1H, 13C NMR) measurements were recorded on a Bruker AC-400 MHz spectrometer at room temperature in deuterated chloroform (CDCl3) or dimethylsulfoxide (DMSO). The chemical shifts were reported in parts per million relative to tetramethylsilane.IR spectra were recorded with a Nicolet 210 FT-IR spectrometer. Size exclusion chromatography (SEC) was performed on a Varian ProStar Model 210 equipped with an RI refractive index detector. Two PLgel 5 μm MIXED-C 600 mm were used at 70 °C with a 0.8 mL·min−1 flow rate of DMF, calibrated using PMMA standards. Differential scanning calorimetry (DSC) analyses were performed under inert atmosphere with a calorimeter DSC1 from Mettler Toledo. The polymer was weighted in an aluminium pan and consecutively placed in the measurement heating cell. An empty pan was used as reference. All the samples were heated under inert atmosphere from −120 to 100 °C at a heating rate of 20 °C min−1.
Three runs were recorded and the glass transition temperature (Tg) values were measured during the second run and confirmed by a third run. Tgs were calculated at the inflexion point of the heat capacity jump.
Thermogravimetric analyses (TGA) were performed using a TGA Q50 W/MFC apparatus of TA Instruments under air flow (25 ml min−1) from room temperature to 580 °C at a heating rate of 20 °C min−1. The analysis consisted of registering the weight loss of the sample as a function of temperature.
Synthesis
RMN
1H/CDCl3 d6: δ (ppm) = 5.86–5.75 (1H, m, CH2CH–), 5.24–5.14 (2H, dd, CH2CH–), 4.83–4.77 (1H, m, CH2–CH–O–), 4.49–4.45 (2H, m, O–CH–CH2), 4.37–4.32 (2H, d, CH–CH2–O), 3.67–3.54 (2H, m, CH2–O–CH2).
RMN
13C/CDCl3 d6: δ (ppm) = 155.4 (O–CO–O), 115.45 (CH2CH–), 132.8 (CH2CH–), 75.77 (CH2–O–CH2), 72 (O–CH–CH2), 71.8 (CH–CH2–O), 61.7 (O–CH–CH2).
RMN 1H of Bis AG/CDCl3 d6: δ (ppm) = 4.90 (1H, m), 4.57 (1H, t), 4.12 (1H, t), 3.62 (2H, t), 2.77 (2H, m), 2.70 (2H, t), 1.95–2.10 (2H, m).
RMN 13C of Bis AG/CDCl3 d6: δ (ppm) = 155.0 (O–CO–O), 75.8 (O–CH–CH2), 70.9 (O–CH2–CH2–S), 69.4 (CO–O–CH2–CH), 34.1 (O–CH2–CH2–S), 32.0 (CH–CH2–CH2–S), 27.8 (CH–CH2–CH2–S).
RMN 1H of Bis AGC/CDCl3 d6: δ (ppm) = 4.80 (1H, m), 4.50 (1H, t), 4.38 (1H, t) 3.70 (2H, m), 3.60 (2H, t), 3.58 (2H, t), 2.68 (2H, t), 2.60 (2H, t), 1.82 (2H, m).
RMN 13C of Bis AGC/CDCl3 d6: δ (ppm) = 154.6 (O–CO–O), 74.6 (O–CH–CH2), 70.0 (O–CH2–CH2–S), 69.7 (CO–O–CH–CH2–O), 69.4 (CO–O–CH2–CH), 65.5 (O–CH2–CH2–CH2–S), 31.1 (O–CH2–CH2–S), 29.0 (O–CH2–CH2–CH2–S), 28.5 (O–CH2–CH2–CH2–S).
Polyhydroxyurethane (PHU-bis-AC and PHU-bis-AGC) syntheses
Step growth polyaddition of dicyclocarbonates bis-AC and bis-AGC was realized with DA10 under stirring in distilled DMF and under argon atmosphere at 75 °C during 48 h. At the end of reaction, DMF is evaporated and PHU-bis-AC and PHU-bis-AGC polymers were precipitated in methanol. After drying, PHUs were quantitatively obtained.Both synthesized PHUs were characterized by 1H NMR spectrometry and by FTIR analysis. Only 1H NMR and FTIR spectra of bis-AGC dicyclocarbonate and PHU-bis-AGC corresponding polyhydroxyurethane are reported.
Conclusion
A new one-step synthesis of 4-[(prop-2-en-1-yloxy)methyl]-1,3-dioxolan-2-one AGC, from 4-(hydroxymethyl)-1,3-dioxolan-2-one (glycerol derivatives), has been reported. This route is particularly interesting since it is an inexpensive, one-step synthesis from biobased glycerin carbonate. Then precursors of PHU have been synthesized by thiol–ene coupling of dithiol with AGC and AC. This “click-chemistry” reaction is particularly advantageous since it requires neither solvent nor photoinitiator and is quantitative in stoichiometric conditions. Finally, polyhydroxyurethanes have been synthesized by step growth polyaddition reaction of bis-AC and bis-AGC with 1,10-diaminedecane. This reaction, carried out without isocyanates, is a less hazardous synthesis. The synthesized polyhydroxyurethanes contain neither hydrolysis sensitive ester bonds nor UV sensitive aromatic groups and exhibit low Tg, therefore they could profitably be used in exterior coatings.Acknowledgements
This work was supported by ANR project MatetPro, Resipoly Chrysor and SEG Dielectriques companies, the authors thank Pr. Alain Fruchier for fruitful discussion.References
- M. H. Karol and J. A. Kramarik, Toxicol. Lett., 1996, 89, 139–146 CrossRef CAS.
- W. Ried and W. Merkel, Angew. Chem., Int. Ed. Engl., 1969, 8, 379–380 CrossRef CAS.
- H. Tomita, F. Sanda and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 3678–3685 CrossRef CAS.
- A. Steblyanko, W. Choi, F. Sanda and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 2375–2380 CrossRef CAS.
- N. Kihara and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 1993, 31, 2765–2773 CrossRef CAS.
- M.-R. Kim, H.-S. Kim, C.-S. Ha, D.-W. Park and J.-K. Lee, J. Appl. Polym. Sci., 2001, 81, 2735–2743 CrossRef CAS.
- L. Ubaghs, N. Fricke, H. Keul and H. Höcker, Macromol. Rapid Commun., 2004, 25, 517–521 CrossRef CAS.
- Q. Li, W. Zhang, N. Zhao, W. Wei and Y. Sun, Catal. Today, 2006, 115, 111–116 CrossRef CAS.
- CN101376632A, 2009.
- T. Nishikubo, T. Iizawa, M. Iida and N. Isobe, Tetrahedron Lett., 1986, 27, 3741–3744 CrossRef CAS.
- M. Aresta, A. Dibenedetto, C. Dileo, I. Tommasi and E. Amodio, J. Supercrit. Fluids, 2003, 25, 177–182 CrossRef CAS.
- B. M. Bhanage, S.-i. Fujita, Y. Ikushima and M. Arai, Green Chem., 2003, 5, 429–432 RSC.
- H. Tomita, F. Sanda and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 860–867 CrossRef CAS.
- H. Tomita, F. Sanda and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 4091–4100 CrossRef CAS.
- H. Tomita, F. Sanda and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 162–168 CrossRef CAS.
- H. Komura, T. Yoshino and Y. Ishido, Bull. Chem. Soc. Jpn., 1973, 46, 550–553 CrossRef CAS.
- K. Weissermel, Industrial Organic Chemistry, Wiley-VCH, 3rd edn, 1997 Search PubMed.
- M. Aresta, A. Dibenedetto, C. Dileo, I. Tommasi and E. Amodio, J. Supercrit. Fluids, 2003, 25, 177–182 CrossRef CAS.
- G. Proempers, H. Keul and H. Hoecker, Des. Monomers Polym., 2005, 8, 547–569 CrossRef CAS.
- S. P. Rannard and N. J. Davis, Org. Lett., 1999, 1, 933–936 CrossRef CAS.
- K. T. Sprott and E. J. Corey, Org. Lett., 2003, 5, 2465–2467 CrossRef CAS.
- S. Benyahya, B. Boutevin, S. Caillol, V. Lapinte, J.-P. Habas, 2011. submitted.
- DE3937116A1, 1991.
- C. N. Tang, H. B. Nulwala, K. Damodaran, P. Kaur and D. R. Luebke, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 2024–2032 CrossRef CAS.
- C. Qi, H. Jiang, Z. Wang, B. Zou and S. Yang, Synlett, 2007, 255–258 CAS.
- Y. Xiong, H. Wang, R. Wang, Y. Yan, B. Zheng and Y. Wang, Chem. Commun., 2010, 46, 3399–3401 RSC.
- J. A. R. Schmidt, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2005, 127, 11426–11435 CrossRef CAS.
- Z. Zhang, L. J. Lyons, R. West, K. Amine and R. West, Silicon Chem., 2007, 3, 259–266 CrossRef CAS.
- F. Guibe and M. L. Y. Saint, Tetrahedron Lett., 1981, 22, 3591–3594 CrossRef CAS.
- M. Desroches, S. Caillol, V. Lapinte, R. Auvergne and B. Boutevin, Macromolecules (Washington, DC, U.S.), 2011, 44, 2489–2500 CAS.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- B. Boutevin, J. P. Hugon and Y. Pietrasanta, Eur. Polym. J., 1981, 17, 723–727 CrossRef CAS.
- H. Tomita, F. Sanda and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 851–859 CrossRef CAS.
- B. Ochiai, S.-I. Sato and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 3408–3414 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1py00289a |
| This journal is © The Royal Society of Chemistry 2011 |