DOI:
10.1039/C1PY00333J
(Paper)
Polym. Chem., 2011,
2, 2674-2682
Hyperbranched glycoconjugated polymer from natural small molecule kanamycin as a safe and efficient gene vector†
Received
23rd July 2011
, Accepted 21st August 2011
First published on 19th September 2011
Abstract
The exploration of safe and efficient polycationic gene vectors from natural small molecules such as kanamycin was proposed. Cationic hyperbranched glycoconjugated polymer was synthesized by the Michael-addition polymerization of kanamycin and N,N′-methylenebisacrylamide, and the resultant product was well characterized by FTIR, 1H NMR, 13C NMR, SEC-MALLS and ζ-potential analyses. The nitrogen content (7.3%) of this kanamycin-based hyperbranched glycoconjugated polymer was much lower than that (32.6%) of polyethylenimine (PEI) control. Moreover, this resultant polymer could be degraded in acidic conditions. Therefore, the hyperbranched glycoconjugated polymer showed low cytotoxicity, even lower than that of natural biomacromolecule chitosan. Due to the existence of various primary, secondary and tertiary amines in the polymer backbone, hyperbranched glycoconjugated polymer exhibited high buffering capacity and strong pDNA condensation ability. In vitro transfection showed that the luciferase expression of hyperbranched glycoconjugated polymer was about 4.4 × 108 RLU per mg protein, approximately 33-fold greater than that of chitosan transfection. These results demonstrate that the construction of highly branched polycations from natural small molecules provides a new opportunity for developing safe and efficient gene vectors.
Introduction
One of the main targets in the Human Genome Project (HGP) which was accomplished in June 2000 was to understand and treat human diseases.1 With the completion of the HGP and the development of molecular biology, the function of numerous genes, such as p53 gene, p16 gene and Rb gene, has been identified, which lays a solid foundation for gene therapy.2–5 To deliver the exogenous treatment genes into the nucleus of the targeted cells, gene vectors are indispensable. Viruses are quite effective gene delivery vectors. However, the safety concerns of viral vectors limit their clinical applications, which promotes the investigation of nonviral vectors.6–9
Among various nonviral vectors, cationic polymers are particularly interesting because of their several advantages, such as high compound stability, low/absent immunogenicity, and convenience of synthesis and modification. Especially, many reports have confirmed that the gene transportation and transfection activity can be readily regulated by branched architecture of polycations.10–12 By introduction of a branching structure into the polymer backbone, the cationic polymers exhibit compact and globular structures in combination with a great number of various amine groups, which facilitates the DNA condensation, gene delivery and transfection improvement.12 Unfortunately, highly branched polycations usually show undesirable cytotoxicity.13–21 Considering that cytotoxicity is closely related to the high molecular weight of polycations, different degradable linkages are used to cross-link the low molecular weight polycations.22,23 However, the degradation products might be harmful in a toxicological perspective. On the other hand, many natural biomacromolecules/biopolymers with low cytotoxicity such as chitosan and polylysine have been used as gene vectors, but their transfection efficiency is frequently disappointing.24–28 Additionally, natural biomacromolecules/biopolymers with high molecular weight are usually hard to be optimized.29,30 Therefore, safe and efficient polycations are still required for developing gene vectors.
The preparation of cationic polymers from natural small molecules provides a new strategy for the exploration of satisfactory gene vectors. Kanamycin is a kind of natural aminoglycoside, which comes from the metabolism of bacteria and is harmless to eukaryotes. Clinically, it is commonly used as an antiseptic to prevent bacteria infections, so it is usually much less cytotoxic than synthetic molecules. If kanamycin with plenty of amine groups is used as a monomer to prepare polymer, a safe and efficient polycation could be expected. Considering that kanamycin contains four active NH2 groups, it can be readily polymerized into highly branched glycoconjugated polymer through Michael-addition reaction with divinyl monomers.31–33 In the present work, the cationic hyperbranched glycoconjugated polymer was prepared by Michael-addition polymerization of kanamycin and N,N′-methylenebisacrylamide via a green chemistry method. In vitro cytotoxicity and transfection efficiency of kanamycin-based hyperbranched glycoconjugated polymer were evaluated. The experimental data showed that the cytotoxicity was low while the transfection efficiency was very high. These results confirm that natural small molecules with plenty of amines such as kanamycin can be used to construct the safe and efficient gene vectors.
Experimental section
Materials
N,N′-Methylenebisacrylamide (MBA, 98%) was purchased from J&K China Chemical Ltd. Polyethylenimine (PEI, water free, Mw = 25 kDa, Mn = 10 kDa), low molecular weight chitosan (approximately 50–190 kDa based on viscosity) and 3-(4,5-dimethyl-thiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) were purchased from Sigma-Aldrich. Kanamycin sulfate (potency > 750) was purchased from Amresco. Dialysis bag (3.5 kDa) was purchased from Shanghai Green Bird Development Co. Ltd., China. BCA protein assay kit was purchased from Beyotime (China). Luciferase assay kit was obtained from Promega.
Synthesis of hyperbranched poly(kanamycin-MBA) by Michael-addition polymerization
The hyperbranched poly(kanamycin-MBA) (HPKM) was synthesized via a conventional Michael-addition polymerization. Briefly, kanamycin sulfate (2.9 g, 0.005 mol) and MBA (1.18 g, 0.0075 mol) were dissolved in 100 mL distilled water and stirred at 60 °C for 80 h. Crude products were concentrated by rotary evaporation. Exhaustive dialysis against exchanged distilled water was performed for 48 h with a dialysis bag (molecular weight cutoff 3500). The residues were evaporated and dried in a vacuum oven overnight and saved in the dryer. Size exclusion chromatography-multiangle laser light scattering (SEC-MALLS) measurements gave the weight-average molecular weight (Mw), number-average molecular weight (Mn) and the polydispersity index (PDI), as summarized in Table 1, (yield: 1.28 g, 36%). 1H NMR (400 MHz, D2O, 298 K) δ: 1.0–2.2 (CHCH2CH); 2.2–2.6 (O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CCH2CH2); 2.8–3.3 (OCCH2CH2); 2.8–3.3 (CHCH2NH2, CHCH2NH, CHCH2N); 2.6–3.3 (NCH, CHNH, CHNH2); 4.4 (NHCH2NH); 3.7 (CH2OH). 13C NMR (400 MHz, D2O, 298 K) δ: 26.0–30.8 (NH2CHCH2CHNH–, –NHCHCH2CHNH2, –NHCHCH2CHNH–); 31.2–32 (NH2CHCH2CHNH2); 32–33.5 (OCCH2CH2N–); 33.6–35.2 (OCCH2CH2NH); 39.6–41 (NH2CH2CH); 41–42.4 (–NHCH2CH, –NCH2CH); 42–44,44.8 (OCCH2CH2NH); 44.5 (NHCH2NH); 48 (NH2CHCHO–); 48.2–49.2 (OCCH2CH2N–); 49.6–51.2 (NH2CHCHO–); 53.2–56.0 (NH2CHCHOH–); 56.0–56.8 (–NHCHCHOH–, –NCHCHOH); 56.8–59.4 (–NHCHCH–O–, –NCHCH–O–); 60.0 (–OCHCH2OH); 175.0 (CO). IR (cm−1): 3350 (νas OH, νas NH2), 2926 (νs CH2), 1665 (νas carbonyl), 1533 (δNH, δNH2, νC–N), 1045 (νC–O–C).
CCH2CH2); 2.8–3.3 (OCCH2CH2); 2.8–3.3 (CHCH2NH2, CHCH2NH, CHCH2N); 2.6–3.3 (NCH, CHNH, CHNH2); 4.4 (NHCH2NH); 3.7 (CH2OH). 13C NMR (400 MHz, D2O, 298 K) δ: 26.0–30.8 (NH2CHCH2CHNH–, –NHCHCH2CHNH2, –NHCHCH2CHNH–); 31.2–32 (NH2CHCH2CHNH2); 32–33.5 (OCCH2CH2N–); 33.6–35.2 (OCCH2CH2NH); 39.6–41 (NH2CH2CH); 41–42.4 (–NHCH2CH, –NCH2CH); 42–44,44.8 (OCCH2CH2NH); 44.5 (NHCH2NH); 48 (NH2CHCHO–); 48.2–49.2 (OCCH2CH2N–); 49.6–51.2 (NH2CHCHO–); 53.2–56.0 (NH2CHCHOH–); 56.0–56.8 (–NHCHCHOH–, –NCHCHOH); 56.8–59.4 (–NHCHCH–O–, –NCHCH–O–); 60.0 (–OCHCH2OH); 175.0 (CO). IR (cm−1): 3350 (νas OH, νas NH2), 2926 (νs CH2), 1665 (νas carbonyl), 1533 (δNH, δNH2, νC–N), 1045 (νC–O–C).
| Sample |
MBA/mol |
Kanamycin/mol |
M
w (×103) |
M
n (×103) |
dn/dc |
PDI |
Yield |
ζ-Potential/mV |
D (%) |
T (%) |
L (%) |
DB |
|
D, T, and L represent the fractions of the dendritic, terminal, and linear units, respectively.
|
| HPKM |
0.0075 |
0.005 |
6.4 |
4.8 |
0.151 |
1.3 |
36% |
12 ± 0.5 |
23 |
26 |
51 |
0.49 |
Polymer characterization
1H NMR, and 13C NMR spectra were recorded with a Bruker AVANCEIII 400 spectrometer in DMSO-d6 or D2O at 298 K. An inverse gated 1H decoupling method was used in quantitative 13C NMR spectra measurement. Fourier transform infrared (FTIR) measurements were performed on a Bruker Equinox-55 FTIR spectrometer with KBr pellets. The molecular weight and molecular weight distribution of HPKM were determined by a SEC-MALLS system consisting of a separation module (Waters 2690D), a refractive index detector (RI, Waters 2414), and a MALLS detector (Wyatt DAWN EOS). Two chromatographic columns of Shodex OHpak SB-803 and SB-802.5 (Showa Denko, Japan) with a Shodex SB-G precolumn were used. A mixed solution of NaAc and acetonitrile was used as eluent, and the flow rate of eluent was 0.6 mL min−1. Here, the eluent was prepared as described by Jiang and co-workers.22 The data were analyzed by Astra software (Wyatt Technology). The ζ-potential was measured with Zetasizer 2000 (Malvern, U.K.). The polymer was dissolved in ultra-pure water at a concentration of 5 mg mL−1, and the ζ-potential values were averaged over three repeated measurements.
Cell cultures
COS-7 cells (a cell line derived from kidney cells of the African green monkey) were cultured according to the general method. Briefly, Dulbecco's Modified Eagle Medium (DMEM) with 10% fetal bovine serum (FBS) was used, and penicillin (100 units per mL) and streptomycin (100 μg mL−1) were used to prevent anti-microbial contamination. Cells were grown at 37 °C with 5% CO2 environment. Cells were subcultured every 3 days.
In vitro degradation of HPKM was performed at 37 °C in the different PBS solutions (phosphate buffered saline, pH = 7.4, 5.5, and 2.5). The concentrations of HPKM aqueous solutions were set at 10 mg mL−1. Some HPKM aqueous solutions were taken out at the predetermined intervals, freeze-dried and analyzed by 1H NMR in D2O.
Cell viability assay
A standard MTT method was used in cell viability assay. Briefly, 96-well plates were used and COS-7 cells were seeded at a density of 6000 cells per well in 200 μL medium. The cells were cultured for 24 h and then DMEM was replaced with fresh medium including HPKM at different concentrations (0.01, 0.1, 0.2, 0.5, 1, 2 mg mL−1). PEI and chitosan were set as controls. 200 μL solution was added into the correspondent well and cells were incubated for 48 h, and then 20 μL MTT (5 mg mL−1) was added into every well. After cells were incubated for 4 h, unreacted dyes were removed carefully. After that, 200 μL DMSO was added in every well, and the optical density was recorded at the wavelength of 490 nm (VarioskanFlash). The cell viability was calculated by the formula as follows:
| Cell viability = ([OD]sample/[OD]control) × 100% |
Evaluation of buffering capacity
The pH titrations of polycation solutions were carried out with a Sartorius PB-10 pH meter and a Sartorius pH/ATC electrode, and the method referred to the literature.34 HPKM and chitosan were diluted to 1 mg mL−1 with 0.15 M NaCl solution, using 0.15 M NaCl solution as a negative control. The starting pH of solutions was set to 11 with 0.1 M NaOH solution. Then, 50 μL 0.1 M HCl solution was dropped into 10 mL assessed solution every time. The pH values were recorded.
Biophysical properties of the HPKM/pDNA complexes
The pGL3-control vector as a model plasmid DNA (pDNA) was used. The pDNA purification was carried out according to endofree plasmid purification handbook (QIAGEN). A certain amount of pDNA was dissolved in deionized water. The HPKM solution of various concentrations was added separately at different molar ratios of polymer nitrogen to pDNA phosphorus (N/P ratios: 0, 1, 5, 10, 20, 30, 60, 80). 1% agarose gel was produced by melting 1 g agarose into 100 mL 0.5 × TAE (Tris–acetate–EDTA) buffer, and then the appropriate amount of ethidium bromide (EB) was added before solidification. The total volume of complex solution was regulated to 18 μL with deionized water, and the appropriate amount of 6 × DNA loading buffer was added. Agarose gel electrophoresis was performed for about 1 h at 100 voltage. The profile was recorded with a UV transilluminator (Bio-RAD Gel-phase system).
The morphology of HPKM/pDNA complexes was observed by atomic force microscopy (AFM, Nanoscope IIIa microscope operating, tapping mode). Samples were prepared as follows. Both 15 μL pDNA solution containing 1 μg pDNA and 15 μL HPKM solutions with different concentrations were prepared in Hepes buffer (NaCl 10 mM, MgCl2 4 mM, Hepes 4 mM, pH = 7.4). The HPKM/pDNA complexes were obtained by mixing two solutions with various N/P ratios (0, 5, 10, 60) at room temperature for 5 min. Then, 90 μL Hepes buffer was added. 5 μL complex solution was deposited onto freshly cleaved ruby mica for 5 min at room temperature. The samples were washed out by distilled water several times, and the surface was dried naturally in air. The scan size was 2 × 2 μm and images were processed by Nanoscope software.
In vitro transfection assay
For transfection studies, COS-7 cells were seeded at a density of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells per well in 100 μL medium in 96-well plates and incubated for 24 h at 37 °C and 5% CO2. Immediately prior to transfection, the medium was removed. Then, cells were washed and replaced with fresh and prewarmed DMEM in the absence of 10% FBS. HPKM/pDNA complexes at the N/P ratios of 0, 5, 10, 20, 30, and 60 (corresponding to 0.2 μg plasmid per well) were added to each well, and the cells were incubated at 37 °C for 4 h. The medium was then replaced with fresh DMEM supplemented with 10% FBS and the cells were lysed afterward. Chitosan/pDNA and branched PEI/pDNA complexes at the N/P ratio of 10, which exhibited the highest transfection efficiency, were set as control. The luciferase assay was carried out according to manufacturer's protocol (Promega, Madison, WI). Relative light units (RLUs) were measured with GloMaxTM 96 microplate luminometer (Promega). The obtained RLUs were normalized with respect to protein concentration in the cell extract determined using the BCA protein assay kit (Beyotime, China).
000 cells per well in 100 μL medium in 96-well plates and incubated for 24 h at 37 °C and 5% CO2. Immediately prior to transfection, the medium was removed. Then, cells were washed and replaced with fresh and prewarmed DMEM in the absence of 10% FBS. HPKM/pDNA complexes at the N/P ratios of 0, 5, 10, 20, 30, and 60 (corresponding to 0.2 μg plasmid per well) were added to each well, and the cells were incubated at 37 °C for 4 h. The medium was then replaced with fresh DMEM supplemented with 10% FBS and the cells were lysed afterward. Chitosan/pDNA and branched PEI/pDNA complexes at the N/P ratio of 10, which exhibited the highest transfection efficiency, were set as control. The luciferase assay was carried out according to manufacturer's protocol (Promega, Madison, WI). Relative light units (RLUs) were measured with GloMaxTM 96 microplate luminometer (Promega). The obtained RLUs were normalized with respect to protein concentration in the cell extract determined using the BCA protein assay kit (Beyotime, China).
Results and discussion
Synthesis and characterization of HPKM
Kanamycin-based hyperbranched polymer HPKM was synthesized via Michael-addition polymerization, and the synthetic route is given in Scheme 1. N,N′-Methylenebisacrylamide (MBA) with two vinyl groups reacts as an A2 monomer, while kanamycin with four NH2 groups as a B8 monomer. By mixing MBA and kanamycin sulfate at a molar ratio of 1:1.5 in aqueous solution, polycondensation-addition occurs. It has been reported that the Michael-addition of monomers with multi-amines tends to form hyperbranched polymers in the solvent of water.12,31,32 After polymerization for 80 h at 60 °C, highly branched polycation HPKM forms through an A2 + B8 reaction. Here, the reaction is carried out in water and base catalysts are unnecessary due to the presence of amines,31–33 which makes it a convenient and green chemistry.
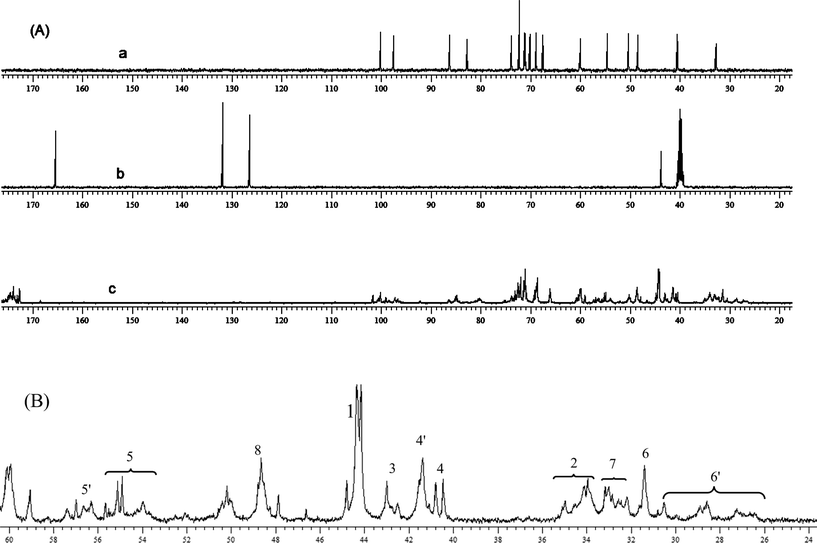
The resultant HPKM was characterized by FTIR, 1H NMR, 13C NMR, SEC-MALLS and ζ-potential techniques. Fig. 1 shows the FTIR spectra of kanamycin sulfate, MBA, and HPKM. For kanamycin sulfate, the bands at 1045 and 1533 cm−1 are assigned to the C–O–C stretching vibration and amino bending vibration, respectively. In the spectrum of MBA, the 1655 cm−1 band comes from the carbonyl stretching vibration. All of these three characteristic bands can be observed in the curve of HPKM. In the meantime, the bands at 2926 and 2860 cm−1 can be assigned to the asymmetric and symmetric CH2 stretching vibrations, respectively. The FTIR data suggest that HPKM is polymerized from kanamycin and MBA monomers.
Fig. 2a shows the 1H NMR of kanamycin sulfate, in which the sharp oligosaccharide signals can be distinguished clearly. The protons at 5.6–6.6 ppm in Fig. 2b correspond to the double-bond of MBA. After the Michael-addition polymerization, these vinyl signals disappear and all proton signals become broad in Fig. 2c, suggesting the formation of polymerized product HPKM. Fig. 3A shows the 13C NMR spectra of kanamycin sulfate, MBA, and HPKM. The signals at 126.5 and 132.0 ppm come from the double-bond of MBA, which disappear completely after polymerization. As expected, two new signals around 43.0 and 34.0 ppm assigning to the methylene groups from MBA units appear in the 13C NMR spectrum of HPKM.
Careful examination of the HPKM structure shows that eleven different types of structural units may be present, including four linear units, four dendritic units and three terminal units. All of these structural units are listed in Scheme 2. With the help of 1H NMR and 13C NMR spectra of kanamycin sulfate and MBA, the detailed assignment of HPKM could be obtained12,21 and the results are given in Fig. 3B. Correspondingly, the degree of branching (DB) of HPKM can be calculated from quantitative 13C NMR analysis based on the following equation:35
where
D,
T, and
L represent the fractions of the dendritic, terminal, and linear units, respectively. As listed in
Table 1, the content of dendritic, terminal, and linear units is 23%, 26% and 51%, respectively. Therefore, the DB of HPKM is 0.49, suggesting the formation of
hyperbranched polymer.
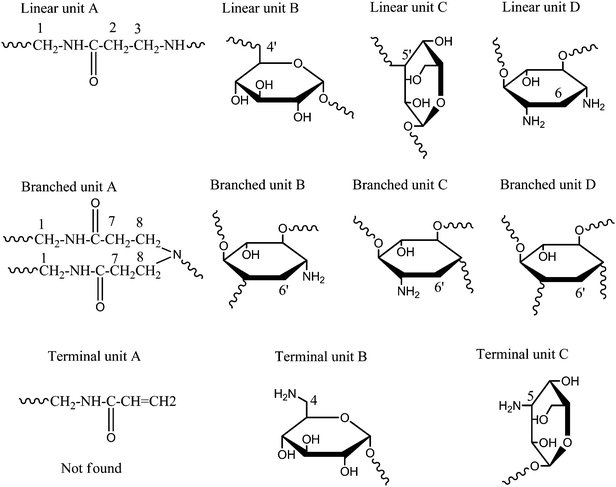 |
| | Scheme 2 Schematic illustration of the molecular structure of HPKM. | |
The molecular weights and their polydispersity index (PDI) of HPKM were measured by SEC-MALLS, and the data are summarized in Table 1. In detail, the number- and weight-average molecular weights (Mn and Mw) are 4.8 × 103 and 6.4 × 103 g mol−1, respectively, with PDI about 1.3. ζ-Potential analysis demonstrates that HPKM is positively charged, attributing to the presence of various amino groups.
HPKM contains many amide and glycosidic linkages, which can be hydrolysed in an acidic environment.12,36 The in vitro degradation behavior of HPKM was evaluated in PBS at 37 °C under neutral (pH 7.4) and acidic (pH 5.5, 2.5) conditions, respectively. The degradation products were lyophilized for 1H NMR analysis in D2O. The 1H NMR spectra of the original HPKM and the degradation products at different times are shown in Fig. 4. For HPKM, the signal at 3.32 ppm is associated with glycosidic linkages, the signals at 2.85 and 2.44 ppm are related to amide, while the signal at 1.32 ppm comes from cyclohexane unit in kanamycin. It is very clear that the proton signals at 3.32, 2.85, 2.44 and 1.32 ppm shift downfield with time, suggesting the degradation of glycosidic and amide bonds. Moreover, the 1H NMR spectra of HPKM are almost unchanged at a neutral condition (pH 7.4) in 17 days, while the degradation rate of HPKM is accelerated at pH 2.5 (Fig. S3 and S4†). This indicates that the hydrolysis of the amide and glycosidic bond in HPKM can be catalyzed by acid.
 |
| | Fig. 4
1H NMR spectra of HPKM in D2O at different degradation time cultured at 37 °C under acidic conditions (pH 5.5): (a) 0 day; (b) 5 days; (c) 9 days, and (d) 17 days. | |
In vitro cytotoxicity of HPKM
The cytotoxicity of HPKM in comparison with PEI and chitosan was evaluated by the MTT assay in COS-7 cell line after 48 h of incubation with polymers. Fig. 5 shows that the HPKM samples with different concentrations exhibit much less toxicity in COS-7 cells than the PEI control. The cell viability after 48 h incubation with HPKM up to 2 mg mL−1 remains nearly 87%, which is even higher than that of chitosan (80%). The low cytotoxicity of HPKM might be related to three reasons. First, kanamycin is one kind of natural and degradable product coming from the metabolism of Streptomyces kanamyceticus and is harmless to eukaryotes. Second, the polymerization and purification of HPKM are carried out in water so that the cytotoxicity originated from organic solvents can be avoided. At last, HPKM presents a low charge density. The nitrogen content of PEI, chitosan and HPKM is 32.6%, 8.8%, and 7.3%, respectively, which is consistent with cytotoxicity of PEI, chitosan and HPKM. The low cytotoxicity makes HPKM a potential vector for gene transfection.
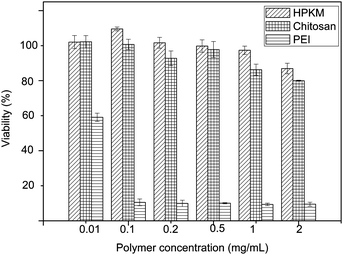 |
| | Fig. 5
In vitro cytotoxicity of HPKM against COS-7 cells after 48 h incubation (mean ± SD, n = 3). | |
Buffering capacity of HPKM
A strong buffering capacity of cationic polymer is frequently related to a high transfection activity due to the low nuclease degradation and fast endosomal escape of polymer/pDNA complexes.37 Therefore, the buffering capacity of HPKM was assessed by acid–base titration before in vitro transfection. As shown in Fig. 6, the pH of NaCl solution (negative control) changes rapidly with addition of HCl solution, while HPKM exhibits an excellent acid-buffering capacity. Compared to natural polysaccharide chitosan which is widely used in gene transfection, HPKM shows a higher buffering capacity. This demonstrates that HPKM with some primary, secondary, and tertiary amines might be a potential gene vector for high transfection activity.
Biophysical properties of HPKM/pDNA complexes
Stable HPKM/pDNA complexes can protect pDNA from enzymatic degradation and promote the gene transfection efficiency through passive targeting to the tumor tissues via the enhanced permeability and retention (EPR) effect. Therefore, the pDNA condensation capacity of HPKM is an important parameter to reflect its gene transfection efficiency. Fig. 7 shows the agarose gel electrophoresis images of the HPKM/pDNA complexes at different N/P ratios. It can be found that the migration of pDNA is obviously retarded at N/P = 5. Further increasing the N/P ratio, the pDNA is completely blocked. The agarose gel electrophoresis results suggest that HPKM can complex with pDNA very efficiently.
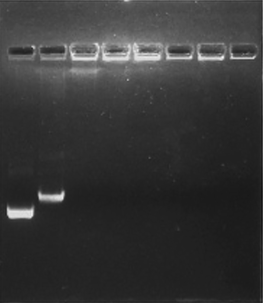 |
| | Fig. 7 Agarose gel electrophoretic images of the HPKM/pDNA complexes: N/P ratios of 0, 1, 5, 10, 20, 30, 60, and 80 from left to right in each well. | |
The DNA condensation capability of HPKM was also studied by AFM. Fig. 8 shows the morphology of pure pDNA and HPKM/pDNA complexes with different N/P ratios. For pure pDNA, the typical plectonemic conformation can be observed in Fig. 8a. For the HPKM/pDNA complexes with an N/P of 5, Fig. 8b shows pDNA is condensed into particles with wide size distributions. Further increasing N/P ratio results in the complete complexation of pDNA into nanoparticles with an average diameter less than 100 nm, as shown in Fig. 8c and d. The AFM observations confirm that pDNA can be efficiently condensed by HPKM.
 |
| | Fig. 8 AFM images of pDNA condensation by HPKM: (a) pure pDNA as control; (b–d) HPKM/pDNA complexes with N/P = 5, 10, 60. Each image represents a 2 × 2 μm scan. | |
In vitro transfection assay
In vitro transfection activities of HPKM/pDNA complexes at various N/P ratios were evaluated in COS-7 cells in comparison with chitosan by the luciferase assay (Fig. 9). With the increase of N/P ratios from 5 to 60, the transfection efficiency increases first and then reaches a plateau. The transfection activity of the HPKM/pDNA complexes with an N/P ratio of 60 reaches 4.4 × 108 RLU per mg protein, which is 33 times higher than the control chitosan at an optical N/P ratio of 10.
 |
| | Fig. 9 Luciferase expression of the HPKM/pDNA at various N/P ratios in COS-7 cells, (the expression levels were measured 48 h later, mean ± SD, n = 3). | |
The high transfection efficiencies of HPKM could be attributed to three reasons. First, highly branched molecular structure benefits HPKM for enhanced condensing capacity. Branched polymers have many advantages, such as having more functional terminals and higher DNA condensation capability,10–12 which can steadily enhance the transfection ability of polymeric vectors. Second, HPKM presents better water solubility than most natural polysaccharides. For example, the low molecular weight chitosan (approximately 50–190 kDa based on viscosity) is poorly water-soluble and can be only dissolved in dilute acid.26,38 In the physiological environment, the aggregation and precipitation of chitosan/pDNA complexes result in the low transfection efficiency. However, HPKM shows a satisfying solubility in a wide range of pH and consequently avoids the precipitation and any other morphological changes. Third, HPKM possesses a balance between plasmid condensation and release. HPKM successfully condenses pDNA into nanoparticles for efficient cell internalizations. On the other hand, numerous secondary amines and tertiary amines are incorporated into HPKM backbones for sufficient plasmid release together with the endosomal escape. For natural polymers like chitosan and polylysine, only primary amines exist. The novel polycation HPKM provides us a new perspective that natural small molecules can be used to synthesize low toxic and highly efficient gene vectors.
Conclusions
In the present work, low toxic and highly efficient hyperbranched glycoconjugated polymer HPKM as a degradable non-viral gene vector was synthesized from natural small molecule kanamycin. The results showed that pDNA can be effectively condensed by HPKM, and the transfection activity in COS-7 reached 4.4 × 108 RLU per mg protein, which was significantly higher than that of chitosan. At the same time, the in vitro cytotoxicity of HPKM was significantly lower than that of PEI, even lower than that of natural polysaccharide chitosan. These results demonstrate that the combination of natural small molecules and hyperbranched polymers can provide low toxic and highly efficient gene vectors.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (20974062, 30700175) and National Basic Research Program 2009CB930400, Shanghai Natural Science Foundation of the Science and Technology Commission of Shanghai Municipal Government (No. 09ZR1415100), a Fundamental Key Project (No. YG2010MS92) of Shanghai Jiaotong University, Shanghai Leading Academic Discipline Project (No. B202), and China National Funds for Distinguished Young Scientists (21025417).
References
-
International human genome sequencing consortium, Nature, 2001, 409, 860–921 Search PubMed.
- A. W. MacInnes, A. Amsterdam, C. A. Whittaker, N. Hopkins and J. A. Lees, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 10408–10413 CrossRef CAS.
- T. Ueba, T. Nosaka, J. A. Takahashi, F. Shibata, R. Z. Florkiewicz, B. Vogelstein, Y. Oda, H. Kikuchi and M. Hatanaka, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 9009–9013 CrossRef CAS.
- K. Strati and P. F. Lambert, Cancer Res., 2007, 67, 11585–11593 CrossRef CAS.
- N. Gonzalez-Paz, W. J. Cheng, R. F. McClure, E. Blood, M. M. Oken, B. V. Ness, C. D. James, P. J. Kurtin, K. Henderson, G. J. Ahmann, M. Gertz, M. Lacy, A. Dispenzieri, P. R. Greipp and R. Fonseca, Blood, 2007, 109, 1228–1232 CrossRef CAS.
- J. Grisham, Nat. Biotechnol., 2000, 18, 254–255 CrossRef CAS.
- J. Fox, Nat. Biotechnol., 1999, 17, 1153 CrossRef CAS.
- T. Niidome and L. Huang, Gene Ther., 2000, 9, 1647–1652 CrossRef.
- M. A. Mintzer and E. E. Simanek, Chem. Rev., 2009, 109, 259–302 CrossRef CAS.
- M. Kramer, J. F. Stumbe, G. Grimm, B. Kaufmann, U. Kruger, M. Weber and R. Haag, ChemBioChem, 2004, 5, 1081–1087 CrossRef.
- Y. F. Zhou, W. Huang, J. Y. Liu, X. Y. Zhu and D. Y. Yan, Adv. Mater., 2010, 22, 4567–4590 CrossRef CAS.
- R. B. Wang, L. Z. Zhou, Y. F. Zhou, G. L. Li, X. Y. Zhu, H. C. Gu, X. L. Jiang, H. Q. Li, J. L. Wu, L. He, X. Q. Guo, B. S. Zhu and D. Y. Yan, Biomacromolecules, 2010, 11, 489–495 CrossRef CAS.
- J. Chen, C. Wu and D. Oupicky, Biomacromolecules, 2009, 10, 2921–2927 CrossRef CAS.
- Y. Lim, S. Kim, Y. Lee, W. Lee, T. Yang, M. Lee, H. Suh and J. Park, J. Am. Chem. Soc., 2001, 123, 2460–2461 CrossRef CAS.
- R. B. Arote, E. Lee, H. Jiang, Y. Kim, Y. Choi, M. Cho and C. Cho, Bioconjugate Chem., 2009, 20, 2231–2241 CrossRef CAS.
- D. W. Pack, A. S. Hoffman, S. Pun and P. S. Stayton, Adv. Drug Delivery Rev., 2005, 4, 581–593 CAS.
- J. B. Kim, J. S. Choi, K. Nam, M. Lee, J. S. Park and J. K. Lee, J. Controlled Release, 2006, 114, 110–117 CrossRef CAS.
- H. Y. Nam, K. Nam, H. J. Hahn, B. H. Kim, H. J. Lim, H. J. Kim, J. S. Choi and J. S. Park, Biomaterials, 2009, 30, 665–673 CrossRef CAS.
- A. Beyerle, M. Irmler, J. Beckers, T. Kissel and T. Stoeger, Mol. Pharmaceutics, 2010, 7, 727–737 CrossRef CAS.
- Y. Liu, D. C. Wu, W. D. Zhang, X. Jiang, C. B. He, T. S. Chung, S. H. Goh and K. W. Leong, Angew. Chem., Int. Ed., 2005, 44, 4782–4785 CrossRef CAS.
- Y. Chen, L. Zhou, Y. Pang, W. Huang, F. Qiu, X. Jiang, X. Zhu, D. Yan and Q. Chen, Bioconjugate Chem., 2011, 22, 1162–1170 CrossRef.
- J. Liu, X. Jiang, L. Xu, X. Wang, W. E. Hennink and R. Zhuo, Bioconjugate Chem., 2010, 21, 1827–1835 CrossRef CAS.
- M. Breunig, U. Lungwitz, R. Liebl and A. Goepferich, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 14454–14459 CrossRef CAS.
- M. S. Shim and Y. J. Kwon, Biomaterials, 2010, 31, 3404–3413 CrossRef CAS.
- X. Zhang, M. Oulad-Abdelghani, A. N. Zelkin, Y. Wang, Y. Hakel, D. Mainard, J. C. Voegel, F. Caruso and N. Benkirane-Jessel, Biomaterials, 2010, 31, 1699–1706 CrossRef CAS.
- T. G. Park, J. H. Jeong and S. W. Kim, Adv. Drug Delivery Rev., 2006, 58, 467–486 CrossRef CAS.
- Y. Huang, H. Yu, L. Guo and Q. Huang, J. Phys. Chem. B, 2010, 114, 7719–7726 CrossRef CAS.
- K. L. Chang, Y. Higuchi, S. Kawakami, F. Yamashita and M. Hashida, Bioconjugate Chem., 2010, 21, 1087–1095 CrossRef CAS.
- M. N. V. R. Kumar, R. A. A. Muzzarelli, C. Muzzarelli, H. Sashiwa and A. J. Domb, Chem. Rev., 2004, 104, 6017–6084 CrossRef.
- A. Watthanaphanit, P. Supaphol, T. Furuike, S. Tokura, H. Tamura and R. Rujiravanit, Biomacromolecules, 2009, 10, 320–327 CrossRef CAS.
- L. Chen, X. Y. Zhu, D. Y. Yan, Y. Chen, Q. Chen and Y. F. Yao, Angew. Chem., Int. Ed., 2006, 45, 87–90 CrossRef CAS.
- H. S. Wan, Y. Chen, L. Chen, X. Y. Zhu, D. Y. Yan, B. Li, T. Liu, L. Zhao, X. L. Jiang and G. Z. Zhang, Macromolecules, 2008, 41, 465–470 CrossRef CAS.
- B. D. Mather, K. Viswanathan, K. M. Miller and T. E. Long, Prog. Polym. Sci., 2006, 31, 487–531 CrossRef CAS.
- J. M. Benns, R. I. Mahato and S. W. Kim, J. Controlled Release, 2002, 79, 255–269 CrossRef CAS.
- C. J. Hawker, R. Lee and J. M. J. Fréchet, J. Am. Chem. Soc., 1991, 113, 4583 CrossRef CAS.
- S. Holtan, Q. Zhang, W. I. Strand and G. Skjåk-Bræk, Biomacromolecules, 2006, 7, 2108–2121 CrossRef CAS.
- O. Boussif, F. Lezoualch, M. A. Zanta, M. D. Mergny, D. Scherman, B. Demeneix and J. P. Behr, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 7297–7301 CrossRef CAS.
- O. Germershaus, S. Mao, J. Sitterberg, U. Bakowsky and T. Kissel, J. Controlled Release, 2008, 125, 145–154 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here.