Organic-metalloblock copolymersvia photocontrolled living anionic ring-opening polymerization†
Laurent
Chabanne
,
Inmaculada
Matas
,
Sanjib K.
Patra
and
Ian
Manners
*
School of Chemistry, University of Bristol, BS8 1TS, U.K. E-mail: ian.manners@bristol.ac.uk; Fax: +44 (0)117 929 0509; Tel: +44 (0)117 928 7650
First published on 12th September 2011
Abstract
A new method for the preparation of organic-organometallic diblock copolymers including a polyferrocenylsilane (PFS) metalloblock through photocontrolled ring-opening polymerization (ROP) is reported. Polystyrene (PS) homopolymers end-capped with a cyclopentadienyl group (1) were used as macroinitiators for the photocontrolled ROP of sila[1]ferrocenophanes [Fe(η-C5H4)2Si{C≡CtBu}2] 3a and [Fe(η-C5H4)2Si(Me)(C≡CSiMe3)] 3b to afford diblock copolymers with controlled molecular weights and block ratios, as well as low polydispersities (PDI < 1.2). Block copolymer PSm-b-[Fe(η-C5H4)2Si{C≡C(t-Bu)}2]n4 was clusterized with [Co2(CO)8], forming the highly metallized PSm-b-[Fe(η-C5H4)2Si{Co2(CO)6C2(t-Bu)}2]n (PS-bb-(Co-PFS), 7). The diblock PSm-b-[Fe(η-C5H4)2Si(Me)(C≡CH)]n6 was prepared by selective desilylation of PSm-b-[Fe(η-C5H4)2Si(Me)(C≡CSiMe3)]n5 was then reacted with ClAuP(n-Bu)3 in the presence of an amine as HCl acceptor to afford PSm-b-[Fe(η-C5H4)2Si(Me){C≡CAuP(n-Bu)3}]n (PS-bbb-(Au-PFS), 8). Preliminary studies on the self-assembly of these materials in thin films showed phase separation with metal-rich nanodomains within an organic matrix.
Introduction
Block copolymers self-assemble in the solid-state into patterns with nanometre size domains provided that the miscibility of the blocks is low and the molecular weight is sufficiently high.1 Due to this ability to spontaneously organize into periodic nanostructures, the use of block copolymers is promising for the development of a range of applications.2–7 Diblocks that contain a metalloblock are especially attractive as their self-assembly results in materials with phase-separated metal-containing domains.8–19 For example, thin films of polyferrocenylsilane (PFS) block copolymers with organic coblocks have been explored as nanotemplates.20–22 As a result of the much larger resistance to reactive ion etching of the metalloblock, the pattern obtained by self-assembly of a PS-b-PFS (PS = polystyrene) diblock could be transferred directly into layers of materials onto which the film was deposited, to finally afford a dense array of magnetic cobalt dots.21 An analogous approach was used to obtain a Ag nanotextured surface, which showed high and uniform enhancement in Raman spectroscopy.23 The presence of metals in only one block has also allowed the direct preparation of spatially defined, nanoscopic regions of well-dispersed nanoparticles following appropriate treatment.8–10,24 This can result in nanopatterned ceramic materials with useful catalytic or magnetic properties. For example, diblocks copolymers containing a PFS block were used as precursors to well-defined arrays of catalytically active nanostructures: when cast as a thin film, these pyrolysed diblock copolymers were used to prepare suspended single- or multi-walled carbon nanotubes under a hydrocarbon atmosphere.25–27 Patterned arrays of magnetic nanocylinders were also obtained from the pyrolysis of UV-crosslinked PS-b-PFS films.24 PFS block copolymer films have also attracted attention because of the redox-activity of the iron center28 and the associated conductivity of the metal-rich nanodomains.29Among metallopolymers,30–37 PFSs represent a versatile class of materials: a wide range of polymers have been prepared, with low to high glass transition temperature, and which includes hydrophilic polyelectrolytes and hydrophobic or even fluorinated materials.38 However, very few types of functional PFS block copolymers have been reported, as these materials have been prepared by organolithium-mediated sequential living polymerization. This process takes place by Si-Cp (Cp = cyclopentadienyl) bond cleavage and involves strongly basic initiating and propagating sites, which are incompatible with the presence of base-sensitive side-groups.11,39,40 Recently, our group has developed an alternative, living ROP method involving photoactivation of the metallocenophane monomer.41,42 This method, termed photocontrolled ROP, involves irradiation of the monomer with Pyrex-filtered emission from a mercury lamp (λ > 310 nm) in a donor solvent, such as THF (Scheme 1). A solvated ring-slipped species is believed to form, that is subsequently attacked by a Cp anion initiator. This results in the cleavage of the weakened Fe-Cp bond, leading to a ring-opened species possessing a free Cp propagating anion that can attack another ring-slipped monomer. Crucially, the initiator and propagating sites, which are delocalized, pendent Cp anions, are much less basic than for classical anionic ROP. Hence, monomers with functionalities incompatible with the latter method have been successfully polymerized using this photocontrolled ROP.43–46 Phosphorus-bridged ferrocenophanes have also been shown to undergo photocontrolled ROP.47 This process has allowed for the polymerization of iron-48 and cobalt-based49 metallocenophanes with hydrocarbon bridges that are resistant to anionic polymerization using organolithium reagents. Furthermore, by using labile initiators that can be displaced from the metal center, cyclic polymetallocenes can be prepared.50 However, to date, the photocontrolled ROP has been limited to the preparation of materials containing only metalloblocks.
![Mechanism for the photocontrolled ROP of silicon-bridged [1]ferrocenophanes (* = photoexcited; S = donor solvent, e.g.THF).](/image/article/2011/PY/c1py00298h/c1py00298h-s1.gif) | ||
| Scheme 1 Mechanism for the photocontrolled ROP of silicon-bridged [1]ferrocenophanes (* = photoexcited; S = donor solvent, e.g.THF). | ||
Many applications require the presence of a metal-free block in addition to the metalloblock. For example, an organic block is essential to afford contrast in etch resistance for use in nanolithography.51 Herein we report the first preparation of organic-organometallic block copolymers using photocontrolled ROP. Specifically, block copolymers with base-sensitive C≡C triple bonds have been prepared. They were then clusterised to afford organic/highly metallized block copolymers, which constitute promising precursors to periodic arrays of functional nanoparticles.24,52
Results and discussions
Preparation of Cp-capped polystyrenes 1a–d
In order to prepare the desired organic-organometallic block copolymers we chose to synthesize organic macroinitiators for the photocontrolled ROP of metallocenophanes. As PS has proven to be a useful block for nanolithography or nanopatterning (elevated glass transition temperature, UV-crosslinkability, lack of etch resistance, immiscibility with PFS), we chose this as a complementary block for our studies. A simple one-pot route was designed to afford the cyclopentadienyl-capped PS (PS-Cp), that was used as macroinitiator (Scheme 2).53,54 First, living polystyryllithium PS-Li in THF, generated by initiation with s-BuLi, was quenched with a large excess (20 equivalents) of dichlorodimethylsilane to afford chlorosilane-capped PS-Cl, and, following the removal of all volatiles (solvent and excess silane), was reacted with a large excess (4 equivalents) of magnesocene (MgCp2) in THF. Chlorotrimethylsilane was then added to quench the excess MgCp2, and all volatiles were removed under vacuum. Successive precipitations in hexanes and methanol (in air) and drying afforded PS-Cp (1) as a white powder with isolated yields ranging from 53% to 96%, depending on the molecular weight.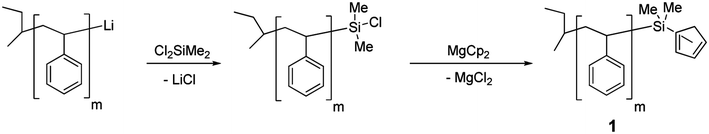 | ||
| Scheme 2 Preparation of PS-Cp macroinitiator 1. | ||
The 1H NMR spectrum of polymer 1d was consistent with a Cp-capped polystyrene (ESI†, Fig. S1). The resonance for the two methyl groups attached to the silicon was found to be split into three signals (δ 0.15 to −0.10 ppm, −0.11 to −0.27 ppm and −0.30 to −0.49 ppm). This was attributed to three different environments for the SiMe2 end-group, depending on the position of the silicon on the terminal uncoordinated Cp ring. Furthermore, the integration of this signal matched the integration of that associated with the two methyl groups inherited from the s-BuLi initiator (δ 0.80 to 0.55 ppm), confirming a virtually complete end-capping of the polymer. Matrix-Assisted Laser Desorption-Ionization Time-Of-Flight (MALDI TOF) analysis of a low molecular weight polymer confirmed the presence of the terminal group (Fig. S2, ESI†). Gel-permeation chromatography (GPC) in THF (e.g.1a shown in Fig. 1) revealed monomodal traces with low polydispersities (Table 1). Little to no shoulder was observed at low retention times, which could have arisen from chain-chain coupling between PS-Cl and PS-Li during the reaction of the latter with dichlorodimethylsilane.

| Polymer | Styrene(g) | s-BuLi (mmol) | M:I ratio | Expected Mn (g mol−1) | Observed Mn (g mol−1)a | PDIa | Isolated yield(%) | |
|---|---|---|---|---|---|---|---|---|
| a Determined using triple-detection GPC in THF. | ||||||||
| 1a | PS365-Cp | 9.09 | 0.28 | 311 | 32,500 | 38,000 | 1.01 | 96 |
| 1b | PS216-Cp | 18.18 | 0.85 | 205 | 21,400 | 22,500 | 1.02 | 90 |
| 1c | PS76-Cp | 4.09 | 0.63 | 62 | 6,600 | 7,900 | 1.04 | 86 |
| 1d | PS28-Cp | 4.55 | 1.96 | 22 | 2,400 | 2,900 | 1.03 | 53 |
Photocontrolled ROP of ferrocenophanes 3a and 3b initiated by PS macroinitiators (2)
To prepare the actual macroinitiator for a photocontrolled ROP, the Cp end group of the PS 1 prepared as above must be deprotonated. An ideal base for this purpose would deprotonate the Cp group without reacting with sila[1]ferrocenophanes (by nucleophilic attack on the silicon), so that it could be used in excess to ensure complete deprotonation of the Cp end-groups. Sodium hexamethyldisilazane NaN(SiMe3)2 (NaHMDS), a moderate, sterically encumbered base, was initially explored. To probe its reactivity towards sila[1]ferrocenophanes, a small amount of NaHMDS solution in THF was added to ethylmethylsila[1]ferrocenophane in THF, with the target that the base would be too sterically hindered to react with the monomer. After an overnight stirring (ca. 16 h), the red solution was found to have turned amber, a signature of ring-opening, and after precipitation in methanol, poly(ferrocenylethylmethylsilane) was isolated as an orange polymer. The mechanism involved is presumably nucleophilic attack of the amide base at the bridging silicon, resulting in the opening of the ring and a chain propagation as for organolithium-induced anionic ROP. Indeed, GPC analysis showed a monomodal, broad trace with high molecular weight (Mn = 190,000, PDI = 1.25) and tailing at low molecular weight, a characteristic feature of slow initiation.55 However, when an equimolar amount of NaCp and NaHMDS were added to a solution of ethylmethylsila[1]ferrocenophane and irradiated overnight (ca. 16 h) at 5 °C, the resulting polymer showed a monomodal, narrow molecular weight distribution by GPC with a molecular weight (Mn = 43 000, PDI = 1.09) close to the expected molecular weight for this ratio of monomer to Cp initiator (Mn = 40 000). This suggested that the rate of initiation of NaHMDS is negligibly slow under the conditions of photocontrolled ROP and that, as a consequence, an excess of NaHMDS can still be used to deprotonate 1 without preventing the controlled polymerisation of ferrocenophanes.We therefore proceeded to the preparation of diblock copolymers (Scheme 3) using monomers 3a and 3b with alkyne substituents (Fig. 2). Such species were previously shown to undergo side-reactions during organolithium-initiated ROP, preventing their incorporation into block copolymers. However, we have found that the formation of PFSs with narrow and controlled size distributions are accessible using photocontrolled ROP.43 In addition, the resulting polymers were shown to be versatile precursors to highly metallized polymers containing various metals.56 Hence, these monomers constitute ideal candidates to demonstrate the new opportunities enabled by the photocontrolled ROP technique.
 | ||
| Scheme 3 Photocontrolled ROP of ferrocenophanes from PS-Cp macroinitiator 2. | ||
![Alkyne-containing sila[1]ferrocenophanes 3a and 3b.](/image/article/2011/PY/c1py00298h/c1py00298h-f2.gif) | ||
| Fig. 2 Alkyne-containing sila[1]ferrocenophanes 3a and 3b. | ||
An excess of NaHMDS (1.5 to 2 equivalents) in THF was used to deprotonate the Cp-terminated PS 1. To the resulting macroinitiator 2 was added monomer 3a (Fig. 2). The red solution was then photoirradiated at 5 °C until it turned light amber (typically a few hours), quenched with a few drops of ClSiMe3 and dried. Several precipitations from THF in large amounts of methanol afforded block copolymer 4. Even when 2 equivalents of NaHMDS were employed, no evidence for the formation of amide-initiated PFS homopolymer was observed by GPC analysis. A small amount of left-over PS was detected, presumably resulting from polystyrene chains not bearing the desired terminal Cp group. Nevertheless, the amount of PS homopolymer was similar to that commonly observed for PFS block copolymers prepared from sequential addition of monomers.11 Characterization using 1H, 13C and 29Si NMR spectroscopy confirmed the expected structures. The spectra clearly consisted of the superimposition of the spectra of polystyrene and the corresponding PFS reported earlier:43 Notably, the protons associated with the tert-butyl group of the alkyne side-group were found at δ = 1.24 ppm, while the aromatic protons of both polystyrene and PFS were observed in their respective regions (δ = 7.3–6.4 ppm and δ = 4.92 and 4.68 ppm, respectively). The signals associated with the carbons belonging to the triple bond in the PFS block were observed in the 13C NMR spectrum at δ = 116.6 and 79.4 ppm, and 29Si NMR revealed a single signal at δ = −46.0 ppm.
In order to expand the range of metals that can be incorporated onto a PFS block, a new monomer 3b was designed, which contained a TMS-protected alkyne group (TMS = trimethylsilyl). The preparation of monomer 3b was adapted from the synthesis of similar alkyne-containing monomers reported previously.43 Characterization by 1H, 13C and 29Si NMR spectroscopy, single-crystal X-ray crystallography, mass spectrometry and elemental analysis confirmed the expected structure. As observed with 3a, the photocontrolled ROP of 3b initiated by the macroinitiator 2 afforded diblock copolymers 5 (Table 2, Scheme 3) after quenching and workup. Monomodal, narrow size distributions were observed by GPC, and a small amount of PS homopolymer but no trace of PFS homopolymer was detected. The 29Si NMR spectrum in CDCl3 showed the presence of two different signals for the silicon in the main chain (δ = −24.8 ppm) and that in the TMS group (δ = −18.7 ppm). The presence of TMS group was also apparent in the 1H NMR spectrum, with a resonance accounting for 9 protons (δ = 0.27 ppm) when compared to the 8 Cp protons of the ferrocene group. The alkynyl carbons were detected by 13C NMR spectroscopy at δ = 115.1 and 112.8 ppm.
| Macrointiator | Mn of PS (g mol−1)a | m/nb | DPn of PFS blockb | Calculated total Mn (g mol−1)c | PDId | |
|---|---|---|---|---|---|---|
| a Measured using triple-detection GPC. b Block ratio determined using 1H NMR. c Calculated from block ratio and Mn of PS. d Determined using conventional calibration GPC in THF. e Monomer = 3a. f Monomer = 3b. | ||||||
| 4a e | PS216-Cp(1b) | 22,500 | 22 | 10 | 26,200 | 1.11 |
| 4b e | PS216-Cp(1b) | 22,500 | 10 | 22 | 30,600 | 1.11 |
| 4c e | PS216-Cp(1b) | 22,500 | 6.5 | 33 | 34,900 | 1.10 |
| 5a f | PS216-Cp(1b) | 22,500 | 3.5 | 62 | 42,500 | 1.14 |
| 5b f | PS216-Cp(1b) | 22,500 | 6.5 | 33 | 33,300 | 1.13 |
| 5c f | PS216-Cp(1b) | 22,500 | 14 | 15 | 27,500 | 1.11 |
| 5d f | PS365-Cp(1a) | 38,000 | 10 | 36 | 49,800 | 1.11 |
| 5e f | PS365-Cp(1a) | 38,000 | 3 | 122 | 77,400 | 1.19 |
Remarkably, the steric hindrance of the internal Si-alkyne bond of diblock copolymers 5 was found to be sufficiently high to allow the selective cleavage of the external Si-alkyne bond using sodium methoxide in a mixture of THF and methanol at 0 °C (Scheme 4). 1H NMR spectroscopy in CDCl3 confirmed the disappearance of the signals associated with the TMS group, and a new singlet attributed to the alkynyl proton was detected at δ = 2.61 ppm. The 13C NMR signals for the alkyne group (δ = 94.4 and 88.5 ppm) were upfield to the corresponding signals of the precursors (δ = 115.1 and 112.8 ppm). Size distributions of the resulting polymer 6 remained narrow according to GPC (PDI < 1.2).
 | ||
| Scheme 4 Selective desilylation of diblock copolymers 5. | ||
Synthesis and characterization of cobalt- and gold-containing diblock copolymers 7 and 8
To prepare diblock copolymers containing a metal-free and a highly metallized block, the precursors described above were reacted with metallic moieties. First, a diblock copolymer 4 was clusterized with [Co2(CO)8], as previously described with analogous homopolymers,56 to obtain 7 (Scheme 5). Multinuclear NMR analysis confirmed the inclusion of cobalt clusters in the polymer, with notably a shift of the signal of the tert-butyl protons, the presence of new 13C signals associated with the COs in the cluster and the broadening of signals previously reported to occur upon clusterization.56 Determination of a percentage of clusterisation by NMR proved difficult, not only because of broad resonances, but also because the signals of the methyl groups overlap with the resonance corresponding to the CH2 group of the polystyrene block. In addition, observation of resonance from the t-butyl group of the precursors (δ = 1.23 ppm) was difficult but suggested that most of the repeat units were clusterized at least once. New resonances were detected between δ 1.5 and δ 1.35 ppm and were attributed to the tert-butyl groups of mono- and disubstituted units. As this type of material was previously found to interact strongly with the styrene-divinylbenzene GPC columns,56 no attempt was made to characterize the molecular weight distribution of this polymer.![Clusterization of 4a–c with [Co2(CO)8] to obtain cobalt-containing polymer 7.](/image/article/2011/PY/c1py00298h/c1py00298h-s5.gif) | ||
| Scheme 5 Clusterization of 4a–c with [Co2(CO)8] to obtain cobalt-containing polymer 7. | ||
Diblock copolymer 6, bearing terminal alkyne side groups, constitutes an ideal scaffold for a range of functional diblock copolymers that could be obtained after alkyne-azide Huisgen cycloaddition, the flagship of click chemistry.57,58 However, in the context of this article, we chose to use this functionality as an opportunity to incorporate new metals into PFS via formation of a σ-bonded complexes.59Diblock copolymer 6 was reacted in THF with an excess (1.4 equivalents) of ClAuP(n-Bu)3 in the presence of an amine as an HCl acceptor using an adapted procedure reported for small molecules (Scheme 6).60 After filtration, precipitations in pentane and drying, the resulting air- and moisture-stable diblock copolymer 8 was obtained as a light orange solid, that was soluble in toluene, chloroform and THF. 1H NMR in CDCl3 showed a nearly complete consumption of the terminal alkyne moieities (94%), with the nearly total disappearance of the resonances associated with the alkynyl proton, and the presence of new peaks corresponding to the butyl groups of the phosphine ligand. Only one very small resonance corresponding to nonsubstituted units could be observed by 29Si NMR spectroscopy (δ = −23.8 ppm), while the major peak was found at δ = −29.7 ppm. 31P NMR showed a single resonance at δ = 28.9 ppm. GPC analysis revealed an expected shift to lower retention times and a slight broadening of the distribution (Fig. S6, ESI†).
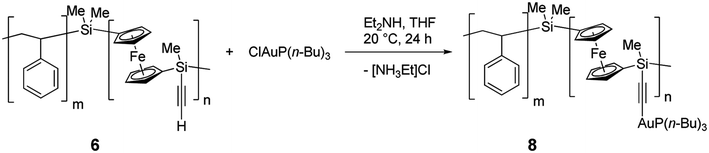 | ||
| Scheme 6 Preparation of gold-containing polymer 8. | ||
Self-assembly of organic-highly metallized diblock copolymers 7 and 8
In order to confirm the ability of the diblock copolymers 7 and 8 to self-assemble into phase-separated nanodomains, thin films were prepared by simply drop-casting solutions of the materials, in toluene and THF, respectively, on carbon-coated copper grids. Transmission Electron Microscopy (TEM) revealed the presence of phase-separated domains in both cases (Fig. 3). Dark domains were attributed to the electron-rich block, i.e.Co-PFS and Au-PFS, while lighter regions were assigned to the polystyrene domains. EDX spectroscopy confirmed the presence of cobalt and gold in the respective films, in addition to the silicon and iron found in the PFS backbone (Fig. S7, ESI†). | ||
| Fig. 3 Bright field TEM images of thin films prepared by drop-casting: (a) and (b) a filtered toluene solution (10 mg ml−1) of PS216216-bb-(Co-PFS)33337 and (c) and (d) a THF solution (10 mg ml−1) of PS365365-bb-(Au-PFS)1221228. | ||
Summary
Diblock copolymers containing an organic block and a PFS block with base-sensitive alkyne substituents were successfully prepared via photocontrolled anionic ROP. In a further step, diblock copolymers containing a metal-free and a highly metallized block were obtained by reaction with appropriate metal complexes. PS-bb-(CoCo-PFS)7 was prepared by clusterisation of an internal alkyne functionality, while PS-bb-(Au-PFS)8 was obtained via formation of a gold alkynyl side group. Most promisingly, the latter material was shown to be stable in air and in solution. Further investigations on the self-assembly of these diblock copolymers will be performed in the near future, with systematic analysis of phase-separated materials with different block ratios. The resulting arrays of metallic nanodomains from these and related block copolymers may be useful for the fabrication of nanogranular in-gap spintronic devices,61 as templates for nanolithography using reactive ion etching, for the direct preparation of surfaces exhibiting surface-enhanced Raman scattering, or for various catalytic processes62 in water-gas shift reactors,63fuel-cell devices64 or for the preparation of carbon nanotubesviachemical vapour deposition (CVD).65 Organic-metalloblock copolymers such as 4 and 5 are also of interest for functionalization by click reactions that may also provide access to interesting new materials.Experimental section
Equipment and materials
Unless otherwise stated, all reactions were carried out under a dry dinitrogen atmosphere using standard Schlenk line techniques or an Ar-filled glove-box. All air- or moisture-sensitive solids and solutions, as well as PS-Cp macroinitiators 1, were stored in a glovebox. Deuterated benzene used for analysis of water-sensitive compounds was distilled from Na/benzophenone and stored in an Ar-filled glove-box. Dichlorodimethylsilane and chlorotrimethylsilane were purchased from Fisher and distilled over CaH2. Chloromethylsila[1]ferrocenophane56 and bis(3,3-dimethylbut-1-ynyl)sila[1]ferrocenophane 3a66 were prepared according to previously reported literature procedures and purified by repeated recrystallizations from hexanes and sublimations. Ethynyltrimethylsilane was purchased from Aldrich, degassed by freeze-pump-thaw cycles and stored over molecular sieves. Styrene, obtained from Sigma-Aldrich, was dried over CaH2 for 24 h and subsequently distilled twice under reduced pressure prior to polymerizations. Cyclohexane was washed several times with 95 mol % H2SO4, until the acid remained uncolored. It was then washed with a 10 M NaOH solution, rinsed with distilled water until neutral, dried over anhydrous MgSO4 and stirred over CaH2 for 24 h. It was finally distilled under reduced pressure immediately before use. Tetrahydrofuran (THF) for polymerizations was distilled under reduced pressure from Na/benzophenone. sec-BuLi (1.4 M solution in cyclohexane) was purchased from Acros Organics and used as received. NaCp (2 M, THF) and sodium hexamethyldisilazide (NaHMDS, 1 M, THF) were purchased from Sigma-Aldrich and used as received. Magnesocene (MgCp2) was prepared according to a literature-known procedure67 and repeatedly sublimed until pure by 1H NMR. [Co2(CO)8] was purchased from Strem Chemicals Inc. and sublimed immediately before use. Dry N2-saturated diethyl ether was collected from a Grubbs system using filtration through an alumina column impregnated with deoxygenated catalysts in vacuo-dried glassware. HAuCl4·3H2O (49.5% Au) was purchased from Alfa Aesar and used as received. Thiodiglycol was purchased from Aldrich and degassed prior to use. Diethylamine (HNEt2) was purchased from Fisher and distilled over CaH2. All solvents for precipitations were used as supplied.The mercury lamp used for polymerization experiments was purchased from Photochemical Reactors Ltd. The emission lines were as follows: 577–579, 546, 436, 408–405, 366–365, 334, 313, 302, 297, 289, 280, 270, 265, 254 nm. Polymerizations were conducted in Pyrex tubes which filter out λ < 310 nm. The tubes were immersed in an ethylene glycol/water bath maintained at 5 °C through a thermocouple that reads the bath temperature into a feedback loop connected to a Julabo FP 50 refrigerator/heater circulator, which circulated ethanol through a stainless steel coil placed within the bath.
1H, 13C, 31P and 29Si NMR spectra were obtained from JEOL ECP300 and ECP400, Varian400 and Varian500 spectrometers. 1H resonances were referenced internally to residual protonated solvent resonances (C6H6: 7.16 ppm; CDCl3: 7.26 ppm) and 13C resonances were referenced internally to deuterated solvent resonances (C6D6: 128.06 ppm; CDCl3: 77.16 ppm).6829Si and 31P resonances were referenced externally to tetramethylsilane and H3PO4 respectively. Prior to drop-casting, the C6D6 solution of cobalt–containing polymer was filtered through glass wool topped with small magnetic bars to remove any magnetic impurity. Matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectra were collected on a 4700 Proteomics Analyser (Applied Biosystems) equipped with a Nd:Yag laser, operating at 335 nm. Positive ion mass spectra were obtained in reflector mode over a range of m/z values. Each spectrum was an accumulation of 10000 laser shots over 50 points on the sample (200 shots/point). Laser intensity was varied. Samples were prepared in THF using dithranol as a matrix and either silver or sodium trifluoroacetate. 10 mg mL−1 of matrix in THF, 1 mg mL−1 of salt in THF and 10 mg mL−1 solution of polymer in THF were mixed in volume ratio 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 matrix:polymer:salt and drop-cast by micropipette into sample wells. The electron ionization mass spectrometry (EI MS) data was collected on a VG Instruments Autospec (70 eV).
:1:1 matrix:polymer:salt and drop-cast by micropipette into sample wells. The electron ionization mass spectrometry (EI MS) data was collected on a VG Instruments Autospec (70 eV).
The polymers were analyzed with a Viscotek VE 2001 Triple-Detector Gel Permeation Chromatograph equipped with automatic sampler, pump, injector, in-line degasser, column oven (30 °C), styrene/divinylbenzene columns with pore sizes of 500 Å and 100 000 Å, VE 3580 refractometer, four-capillary differential viscometer and 90° angle laser and low angle laser (7°) light scattering detector (VE 3210 & VE270). A 0.025% Butylated Hydroxytoluene stabilized THF (Fisher) was used as the eluent, with a flow rate of 1.0 mL/min. Samples were dissolved in the eluent (2 mg mL−1) and filtered (Acrodisc, PTFE membrane, 0.45 μm) before analysis. The calibrations were made from a polystyrene standard from Viscotek (triple-detection) and from polystyrene standards from Aldrich (conventional calibration). This equipment allows the exact measure of homopolymers molecular weights and polydispersity indexes (PDIs) using triple-detection calibration, or molecular weights relative to polystyrene for block copolymers using conventional calibration.
Transmission electron microscopy was performed on JEOL 1200EX Mk2, using a standard tungsten filament at a 120 kV acceleration voltage. The pictures were taken from a MegaViewII digital camera. Energy-dispersive X-ray analyses (EDX) were performed using an Oxford Instruments ISIS 300 system with silicon detector and beryllium window. Copper grids, mesh 400, were coated with an Agar TEM Turbo Carbon Coater, where carbon was sputtered on mica sheet before deposition on the copper grids via flotation. TEM samples were prepared by drop-casting: using a Pasteur pipette, one drop of polymer solution was placed on a carbon-coated copper grid atop a piece of filter paper to remove excess solvent. Prior to drop-casting, the 10 mg mL−1toluene solution of cobalt–containing polymer was filtered through glass wool topped with small magnetic bars and then through a 0.2 μm PTFE membrane to remove any solid impurity.
Synthesis of chlorodimethylsilane-capped polystyrene
A representative polymerization is described. Styrene (5 mL, 43.6 mmol) was dissolved in cyclohexane (ca. 30 mL) and initiated with 1.4 mL of sec-BuLi (1.4 M solution in cyclohexane, 2.0 mmol) at ambient temperature in an inert atmosphere glovebox. The colourless solution rapidly turned orange, indicative of the presence of living polystyrenyl anions. After a 1 h stirring, a large excess of dichlorodimethylsilane (5 mL, 41.5 mmol) was rapidly added and the mixture gradually (a few minutes to a few hours) turned colourless. After an overnight stirring, solvent and left-over silane were removed under vacuum. The small molecular weight polymer prepared for NMR analysis was precipitated in hexane, isolated and dried under vacuum for 24 h, leaving a white powder, with a yield of 3.6 g (79%). In order to minimize handling of this moisture-sensitive polymer, polymers destined for the preparation of macroinitiator were simply dried under high dynamic vacuum (<10−2 mbar) for 24 h, to remove the maximum amount of unreacted silane, before further reaction in the same flask.1H NMR (399.8 MHz, C6D6, 25 °C): δ = 7.25–6.35 (Ph), 2.7–1.9 (CH), 1.9–1.35 (CH2), 0.8–0.55 (CH), 0.15–(−0.10) (m, 6H, Si(CH3)2).
Synthesis of Cp-capped polystyrene 1
In a typical reaction, MgCp2 (130 mg, 0.84 mmol) in tetrahydrofuran (THF, ca. 20 mL) was added into the flask containing PS-Cl prepared as above (1.0 g, 0.43 mmol). The solution was stirred overnight. 1 mL of ClSiMe3 was then added to the mixture and the solution was stirred for about one hour. Solvent and excess silane were removed under vacuum and the white solid was dried under vacuum overnight. The polymer was redissolved in THF, quickly precipitated into rapidly stirring hexanes and filtered. It was then precipitated twice from THF into methanol and finally dried in a vacuum oven (60 °C, 3 days), leaving the white powder 1d, with a yield of 530 mg (53%). Low molecular weight polystyrenes were slightly soluble in hexanes. The preparation of polymers with higher molecular weight resulted in up to nearly quantitative isolated yields.1H NMR (399.8 MHz, C6D6): δ = 7.25–6.35 (Ph), 3.21 (Cp), 2.7–1.9 (CH), 1.9–1.35 (CH2), 0.8–0.55 (CH), 0.15–(−0.49) (m, 6H, Si(CH3)2); GPC analysis (THF, triple-detection): Mn = 2 900, PDI = 1.03.
MALDI (see ESI†): m/z: 2598.9 for C4H9(C8H8)23Si(CH3)2C5H5·Na+.
Test reaction of a sila[1]ferrocenophane with NaHMDS
Ethylmethylsila[1]ferrocenophane (200 mg, 0.78 mol) was dissolved into ca. 2 mL of THF. NaHMDS (10 μl, 10 mmol, THF, 1M) was added along with ca. 1 mL of THF. The solution was stirred overnight (ca. 16 h), by which time the solution turned light amber. A few drops of degassed methanol were added to quench the reaction, the polymer was stirred for 30 min, then precipitated three times from THF into methanol and dried under vacuum (ca. 10−3 mbar) overnight, leaving an orange gummy polymer, with a yield of 196 mg (98%). GPC analysis showed a high molecular weight distribution with a tailing at low molecular weight, which are characteristic of slow initiation.GPC analysis (THF, triple-detection): Mn = 190 000, PDI = 1.25
Evaluation of initiator efficiency of NaHMDS vs.NaCp
In an attempt to evaluate the polymerisation efficiency of NaHMDS in competition with NaCp, an equimolar amount (10 μmol) of NaHMDS (THF, 1M, 10 μL) and NaCp (THF, 2M, 5 μL) was introduced in a tube containing a solution of monomer (ethylmethylsila[1]ferrocenophane, 400 mg) in THF. The resulting mixture was photo-irradiated at 5 °C overnight and quenched with chlorotrimethylsilane. A few drops of degassed methanol were added to quench the reaction, the polymer was stirred for 30 min, then precipitated three times from THF into methanol and dried under vacuum (ca. 10−3 mbar) overnight, leaving an orange gummy polymer, with a yield of 399 mg (100%).GPC analysis (THF, triple-detection): Mn = 43 000, PDI = 1.09
Synthesis of precursor PSm-b-[Fe(η-C5H4)2Si{C≡C(t-Bu)}2]n4
In a typical reaction, NaHMDS (1 M, 45 μL, 0.045 mmol) was added to a THF (3 mL) solution of Cp-capped PS2161b (Mn = 22 500, 500 mg, 0.022 mmol). The solution was stirred at room temperature for 1 h. Bis(3,3-dimethylbut-1-ynyl)sila[1]ferrocenophane 3a (300 mg, 0.80 mmol) was added along with THF (3 mL) and the flask was quickly placed into a 5 °C bath and photo-irradiated overnight (ca. 16 h), after which the solution was light amber. A few drops of ClSiMe3 were added to quench the polymerization. All volatiles were removed under vacuum. The polymer was dissolved in a small amount of THF and precipitated in a large amount of methanol containing a few drops of diethylamine. After two additional precipitations in pure methanol, the polymer was dried at 40 °C in a vacuum oven for 3 d, leaving the light yellow powder 4c, with a yield of 709 mg (89%).1H NMR (399.8 MHz, C6D6, 25 °C): δ 7.3–6.4 (Ph), 4.92 (Cp), 4.68 (Cp), 2.55–1.95 (br, CH), 1.85–1.4 (br, CH2), 1.24 (s, CH3); 13C NMR (125.7 MHz, C6D6, 25 °C): δ 147.0–145.0 (Ph, quaternary C), 126.5–125.8 (Ph) 116.6 (SiC≡C), 79.4 (SiC≡C), 74.95, 74.85, 74.19, 74.04 (Cp) 68.05 (ipso-C), 47.6–41.6 (CHCH2CH), 41.0 (CHPh), 31.1 (CH(CH3)3), 28.5 (CH(CH3)3); 29Si DEPT NMR (99.3 MHz, C6D6, 25 °C): δ −46.0; GPC analysis (THF, conventional calibration): Mn = 31 900, PDI = 1.10.
Synthesis of polymer PSm-b-[Fe(η-C5H4)2Si(CH3){C≡CSi(CH3)3}]n5
In a typical reaction, NaHMDS (1 M, 36 μL, 0.036 mmol) was added to a solution of Cp-capped PS2161b (Mn = 22 500, 600 mg, 0.027 mmol) in ca. 3 mL THF. The solution was stirred at room temperature for 1 h. [Fe(η-C5H4)2Si(CH3){C≡CSi(CH3)3}] 3b (150 mg, 0.66 mmol) was added along with ca. 2 mL THF and the flask was quickly put into a 5 °C bath and photo-irradiated for 8 h). The solution became light amber. A few drops of ClSiMe3 were added to quench the polymerization. All volatiles were removed under vacuum. The polymer was dissolved in a small amount of THF and precipitated in a large amount of methanol containing a few drops of diethylamine. After three more precipitations from THF to methanol, the polymer was dried in a vacuum oven (40 °C, ca.10−2 mbar) for 3 days, leaving the light yellow powder 5c, with a yield of 726 mg (97%).1H NMR (499.9 MHz, C6D6, 25 °C): δ = 7.3–6.4 (Ph), 4.60, 4.58, 4.56 (br, 4H, Cp), 4.48 (br, 1H, Cp), 4.45 (br, 1H, Cp), 4.34 (br, 1H, Cp), 4.29 (br, 1H, Cp), 2.55–1.95 (br, CH), 1.85–1.4 (br, CH2), 0.77, 0.76, 0.75 (br, 3H, CH3), 0.27 (Si(CH3)3); 13C{1H} NMR (75.6 MHz, C6D6, 25 °C): δ 147.0–144.5 (Ph, quaternary C), 126.5–125.8 (Ph), 115.1 (C≡CSi(CH3)3), 112.8 (C≡CSi(CH3)3), 74.69, 74.59, 74.27, 74.19, 73.37, 73.26 73.22, 73.14 (Cp), 68.69, 68.67, 68.65, 68.62 (ipso-Cp), 47.6–41.6 (CHCH2CH), 41.0 (CHPh) 0.2 (Si(CH3)3). −0.56, −0.63, −0.70 (CH3); 29Si DEPT NMR (99.3 MHz, C6D6, 25 °C): δ = −19.3 (C≡CSi(CH3)3), −25.3 (Si(CH3)).
GPC (THF, conventional calibration) Mn = 24 800, PDI = 1.11.
Synthesis of precursor PSm-b-[Fe(η-C5H4)2Si(CH3)(C≡CH)]n6
In a typical experiment, PS216-b-[Fe(η-C5H4)2Si(CH3){C≡CSi(CH3)3}]155c (27 500 g mol−1, 600 mg) was dissolved in ca. 30 mL THF, to which ca. 10 mL methanol were added. The flask was placed in an ice/water bath and stirred for ca. 5 min, after which ca. 60 mg (1.11 mmol) of NaOMe were added. The solution was stirred for 3 h at 0 °C. A large excess of NH4Cl was added with a spatula and the solution was stirred for a further 5 min. It was then poured into a large amount (ca. 450 mL) of methanol to precipitate the polymer. After three more precipitations from THF to methanol, the polymer was dried in a vacuum oven (40 °C, ca.10−2 mbar) for 3 days, leaving the light orange powder 6c, with a yield of 473 mg (88%).1H NMR (300.5 MHz, CDCl3, 25 °C): δ = 7.3–6.3 (Ph), 4.45–4.3, 4.22, 4.21, 4.14, 4.11 (Cp), 2.61 (C≡CH) 2.35–1.7 (br, CH), 1.70–1.2 (br, CH2), 0.64 (CH3); 13C{1H} NMR (75.6 MHz, CDCl3, 25 °C): δ = 147.0–144.5 (Ph, quaternary C), 128.6–127.2 (Ph, Cortho and Cmeta), 126.0–125.3 (Ph, Cpara), 94.4 (C≡CH), 88.5 (C≡CH), 74.5–73.5, 72.7 (Cp), 68.0 (ipso-Cp)), 46.0–41.0 (CHCH2CH), 40.5 (CHPh), −1.04 (CH3);; 29Si DEPT NMR (99.3 MHz, CDCl3, 25 °C): δ = −23.7; GPC (THF, conventional calibration) Mn = 23 800, PDI = 1.12.
Synthesis of PSm-b-(Co-PFS)n7
PS216-b-[Fe(η-C5H4)2Si{C≡C(t-Bu)}2]334c (500 mg, 1.0 mmol alkyne) was reacted with [Co2(CO)8] (700 mg, 2.0 mmol) in toluene at room temperature for 4 h. A Teflon cannula equipped with a filter paper was used to transfer the solution into a flask containing rapidly stirring, dry hexanes. The liquid was removed with a cannula equipped with a filter paper and the solid was dried under vacuum. Dry toluene was added to redissolve the solid and the solution was transferred into a flask containing rapidly stirring hexanes using a cannula equipped with filter paper. After another precipitation from toluene to hexanes, the polymer was dried under vacuum (ca. 10−3 mbar) for 3 days, leaving the dark brown solid 7, with a yield of 369 mg (52% based on 75% clusterization, i.e. 1.5 per unit). The degree of clusterization was difficult to determine due to overlapping of the resonances of the t-butyl groups of the PFS block with the resonance of the methylene protons of the PS block. Only one silicon peak was observed by 29Si NMR using the NNE technique (inverse gate decoupling to suppress negative nuclear overhauser effect). As this type of polymer is known to interact strongly with GPC columns, no attempt was made to perform GPC analysis on these polymers.1H NMR (399.8 MHz, C6D6, 25 °C): δ 7.3–6.4 (Ph), 5.5–4.7 (Cp), 2.55–1.95 (br, CH), 1.85–1.35 (br, CH2 and CH3), 1.24 (s, CH3); 29Si NNE NMR (59.6 MHz, C6D6, 25 °C): δ −4.5; FTIR (25 °C, CH2Cl2) νmax/cm−1 = 2083m, 2065w, 2046s and 2020s cm−1 (CO).
Synthesis of chloro(tri-n-butylphosphine)gold(I) [ClAuP(n-Bu3)]
[ClAuP(n-Bu3)] was prepared following a method adapted from the literature.69 A solution of thiodiglycol (0.180 g, 1.912 mmol) in 1 mL of ethanol was added to a solution of HAuCl4·3H2O (0.275 g, 0.698 mmol) in 2 mL of distilled water. When the orange solution became colourless, it was cooled down to −5 °C and a solution of n-Bu3P (0.18 mL, 0.720 mmol) in 1 mL of ethanol was added dropwise to the solution while stirring. Stirring was continued for 30 min. to get a white turbid solution. All the volatiles were removed in vacuum to obtain an oily colourless product which was washed with a cold water-ethanol (2:1) solution (3 mL × 2). It was further purified by flash chromatography on silica gel using 3:7 ethyl acetate/hexanes as eluent. [ClAuP(n-Bu3)], and after drying in vacuo, was obtained as a colourless oil, with a yield of 0.215 g (72%).
1H NMR (299.9 MHz, CDCl3): δ 1.8–1.3 (m, CH2), 0.86 (t, CH3); 31P{1H} (121 MHz, CDCl3, 25 °C): δ 22.3;
Synthesis of PSm-b-(Au-PFS)n8
In a typical reaction, PS365-b-[Fe(η-C5H4)2Si(CH3)(C≡CH]1226e (30 mg, 0.05 mmol PFS units) and ClAuP(nBu)3 (50 mg, 0.115 mmol) were dissolved in a 4 mL of a 1:1 mixture of THF/diethylamine. After a few minutes, the solution turned cloudy, due to precipitation of insoluble (NH2Et2)Cl as a fine white power. The vial containing the solution was covered with aluminium foil and the mixture was stirred for 24 h. The solution was filtered on glass wool to remove insoluble (NH2Et2)Cl and the polymer was precipitated 3 times in rapidly stirring pentane. The product was then dried at 60 °C for 24 h, leaving the orange solid 8, with a yield of ca. 90%. Comparison of the alkyne resonance with the Cp resonances in the 1H NMR spectrum indicated that ca. 94% of the triple bonds were substituted. GPC indicated a slight broadening of the distribution attributed to aggregation and/or interaction with the column (see Fig. S6).
1H NMR (399.8 MHz, CDCl3): δ = 7.3–6.3 (br, Ph). 4.6–4.0 (Cp), 2.63 (C≡CH), 2.30–1.65 (CH and PCH2CH2), 1.65–1.25 (CHCH2 and CH2CH2CH2CH3), 0.90 (CH2CH3), 0.63 (Si(CH3)); 13C{1H} NMR (100.5 MHz, CDCl3, 25 °C): δ 155.0 (CAu), 147.0–144.5 (Ph, quaternary C), 128.6–127.2 (Ph, Cortho and Cmeta), 126.0–125.3 (Ph, Cpara) 106.6 (CSi), 74.5–68.5 (Cp), 46.0–41.0 (CHCH2CH), 40.5 (CHPh) 27.2 (s, CH2CH3), 25.6 (d, PCH2, 2JCP = 31.9 Hz), 24.4 (d, PCH2CH2, 1JCP = 14.0 Hz), 13.8 (s, CH2CH3), 0.4 (s, Si(CH3)); 31P{1H} NMR (121.7 MHz, CDCl3, 25 °C): δ 28.9; 29Si NMR (59.7 MHz, CDCl3, 25 °C): δ −29.7; GPC (THF, conventional calibration): Mn = 75 600.
Acknowledgements
I. Manners wishes to thank EPSRC for funding this research. I. Matas would like to thank the Fundación Ramón Areces for a postdoctoral fellowship. S. K. Patra thanks the EU for a Marie Curie Postdoctoral Fellowship.Notes and references
- F. S. Bates, Science, 1991, 251, 898–905 CAS.
- J. Y. Cheng, C. A. Ross, H. I. Smith and E. L. Thomas, Adv. Mater., 2006, 18, 2505–2521 CrossRef CAS.
- I. W. Hamley, Angew. Chem., Int. Ed., 2003, 42, 1692–1712 CrossRef CAS.
- C. De Rosa, F. Auriemma, R. Di Girolamo, G. P. Pepe, T. Napolitano and R. Scaldaferri, Adv. Mater., 2010, 22, 5414–5419 CrossRef CAS.
- Y. A. Elabd and M. A. Hickner, Macromolecules, 2011, 44, 1–11 CrossRef CAS.
- L. Chen, W. A. Phillip, E. L. Cussler and M. A. Hillmyer, J. Am. Chem. Soc., 2007, 129, 13786–13787 CrossRef CAS.
- P. R. L. Malenfant, J. L. Wan, S. T. Taylor and M. Manoharan, Nat. Nanotechnol., 2007, 2, 43–46 CrossRef CAS.
- Y. N. C. Chan, G. S. W. Craig, R. R. Schrock and R. E. Cohen, Chem. Mater., 1992, 4, 885–894 CrossRef CAS.
- Y. N. C. Chan and R. R. Schrock, Chem. Mater., 1993, 5, 566–570 CrossRef.
- L. A. Miinea, L. B. Sessions, K. D. Ericson, D. S. Glueck and R. B. Grubbs, Macromolecules, 2004, 37, 8967–8972 CrossRef CAS.
- D. A. Rider, K. A. Cavicchi, K. N. Power-Billard, T. P. Russell and I. Manners, Macromolecules, 2005, 38, 6931–6938 CrossRef CAS.
- P. Chadha and P. J. Ragogna, Chem. Commun., 2011, 47, 5301–5303 RSC.
- C. Cui, E. M. Bonder and F. Jäkle, J. Am. Chem. Soc., 2010, 132, 1810–1812 CrossRef CAS.
- L. Ren, C. G. Hardy and C. Tang, J. Am. Chem. Soc., 2010, 132, 8874–8875 CrossRef CAS.
- M. Roerdink, M. A. Hempenius and G. J. Vancso, Chem. Mater., 2005, 17, 1275–1278 CrossRef CAS.
- M. Gallei, B. V. K. J. Schmidt, R. Klein and M. Rehahn, Macromol. Rapid Commun., 2009, 30, 1463–1469 CrossRef CAS.
- C. Kloninger, D. Knecht and M. Rehahn, Polymer, 2004, 45, 8323–8332 CrossRef CAS.
- M. Ramanathan and S. B. Darling, Soft Matter, 2009, 5, 4665–4671 RSC.
- J.-F. Gohy, B. G. G. Lohmeijer and U. S. Schubert, Chem.–Eur. J., 2003, 9, 3472–3479 CrossRef CAS.
- M. Ramanathan, E. Nettleton and S. B. Darling, Thin Solid Films, 2009, 517, 4474–4478 CrossRef CAS.
- J. Y. Cheng, C. A. Ross, V. Z. H. Chan, E. L. Thomas, R. G. H. Lammertink and G. J. Vancso, Adv. Mater., 2001, 13, 1174–1178 CrossRef CAS.
- D. A. Rider and I. Manners, Polym. Rev., 2007, 47, 165–195 CrossRef CAS.
- J. Lu, D. Chamberlin, D. A. Rider, M. Liu, I. Manners and T. P. Russell, Nanotechnology, 2006, 17, 5792 CrossRef CAS.
- D. A. Rider, K. Liu, J.-C. Eloi, L. Vanderark, L. Yang, J.-Y. Wang, D. Grozea, Z.-H. Lu, T. P. Russell and I. Manners, ACS Nano, 2008, 2, 263–270 CrossRef CAS . 8.
- S. Lastella, Y. J. Jung, H. Yang, R. Vajtai, P. M. Ajayan, C. Y. Ryu, D. A. Rider and I. Manners, J. Mater. Chem., 2004, 14, 1791–1794 RSC.
- C. Hinderling, Y. Keles, T. Stöckli, H. F. Knapp, T.d. l. Arcos, P. Oelhafen, I. Korczagin, M. A. Hempenius, G. J. Vancso, R. Pugin and H. Heinzelmann, Adv. Mater., 2004, 16, 876–879 CrossRef CAS.
- J. Q. Lu, T. E. Kopley, N. Moll, D. Roitman, D. Chamberlin, Q. Fu, J. Liu, T. P. Russell, D. A. Rider, I. Manners and M. A. Winnik, Chem. Mater., 2005, 17, 2227–2231 CrossRef CAS.
- D. A. Rider, M. A. Winnik and I. Manners, Chem. Commun., 2007, 4483–4485 RSC.
- J. K. Li, S. Zou, D. A. Rider, I. Manners and G. C. Walker, Adv. Mater., 2008, 20, 1989–1993 CrossRef CAS.
- G. R. Whittell, M. D. Hager, U. S. Schubert and I. Manners, Nat. Mater., 2011, 10, 176–188 CrossRef CAS.
- K. A. Williams, A. J. Boydston and C. W. Bielawski, Chem. Soc. Rev., 2007, 36, 729–744 RSC.
- I. Manners, Frontiers in Transition Metal-Containing Polymers, Wiley-VCH, Weinheim, 2007 Search PubMed.
- V. Marin, E. Holder, R. Hoogenboom and U. S. Schubert, Chem. Soc. Rev., 2007, 36, 618–635 RSC.
- P. R. Andres and U. S. Schubert, Adv. Mater., 2004, 16, 1043–1068 CrossRef CAS.
- W.-Y. Wong, Macromol. Chem. Phys., 2008, 209, 14–24 CrossRef CAS.
- I. Dragutan, V. Dragutan and H. Fischer, J. Inorg. Organomet. Polym. Mater., 2008, 18, 311–324 CrossRef CAS.
- A. O. Moughton and R. K. O'Reilly, Macromol. Rapid Commun., 2010, 31, 37–52 CrossRef CAS.
- V. Bellas and M. Rehahn, Angew. Chem., Int. Ed., 2007, 46, 5082–5104 CrossRef CAS.
- C. Kloninger and M. Rehahn, Macromolecules, 2004, 37, 1720–1727 CrossRef CAS.
- H. Wang, M. A. Winnik and I. Manners, Macromolecules, 2007, 40, 3784–3789 CrossRef CAS.
- M. Tanabe and I. Manners, J. Am. Chem. Soc., 2004, 126, 11434–11435 CrossRef CAS.
- M. Tanabe, G. W. M. Vandermeulen, W. Y. Chan, P. W. Cyr, L. Vanderark, D. A. Rider and I. Manners, Nat. Mater., 2006, 5, 467–470 CrossRef CAS.
- W. Y. Chan, A. J. Lough and I. Manners, Organometallics, 2007, 26, 1217–1225 CrossRef CAS.
- Z. Wang, G. Masson, F. C. Peiris, G. A. Ozin and I. Manners, Chem.–Eur. J., 2007, 13, 9372–9383 CrossRef CAS.
- S. Hilf, P. W. Cyr, D. A. Rider, I. Manners, T. Ishida and Y. Chujo, Macromol. Rapid Commun., 2005, 26, 950–954 CrossRef CAS.
- G. S. Smith, S. K. Patra, L. Vanderark, S. Saithong, J. P. H. Charmant and I. Manners, Macromol. Chem. Phys., 2010, 211, 303–312 CrossRef CAS.
- S. K. Patra, G. R. Whittell, S. Nagiah, C. L. Ho, W. Y. Wong and I. Manners, Chem.–Eur. J., 2010, 16, 3240–3250 CrossRef CAS.
- D. E. Herbert, U. F. J. Mayer, J. B. Gilroy, M. J. Lopez-Gomez, A. J. Lough, J. P. H. Charmant and I. Manners, Chem.–Eur. J., 2009, 15, 12234–12246 CrossRef CAS.
- J. B. Gilroy, S. K. Patra, J. M. Mitchels, M. A. Winnik and I. Manners, Angew. Chem. Int. Ed., 2011, 50, 5851–5855 CrossRef CAS.
- D. E. Herbert, J. B. Gilroy, W. Y. Chan, L. Chabanne, A. Staubitz, A. J. Lough and I. Manners, J. Am. Chem. Soc., 2009, 131, 14958–14968 CrossRef CAS.
- G. Vancso, I. Korczagin, R. Lammertink, M. Hempenius and S. Golze, in Ordered Polymeric Nanostructures at Surfaces, Springer Berlin/Heidelberg, 2006, vol. 200, pp. 91–117 Search PubMed.
- K. Liu, C.-L. Ho, S. Aouba, Y.-Q. Zhao, Z.-H. Lu, S. Petrov, N. Coombs, P. Dube, H. E. Ruda, W.-Y. Wong and I. Manners, Angew. Chem., Int. Ed., 2008, 47, 1255–1259 CrossRef CAS.
- For the preparation of Cp-capped polymers using other methods, see: A. J. Inglis, S. Sinnwell, M. H. Stenzel and C. Barner-Kowollik, Angew. Chem. Int. Ed., 2009, 48, 2411–2414 CAS.
- A. J. Inglis, T. Paulöhrl and C. Barner-Kowollik, Macromolecules, 2010, 43, 33–36 CrossRef CAS.
- L. Gold, J. Chem. Phys., 1958, 28, 91–99 CrossRef CAS.
- W. Y. Chan, S. B. Clendenning, A. Berenbaum, A. J. Lough, S. Aouba, H. E. Ruda and I. Manners, J. Am. Chem. Soc., 2005, 127, 1765–1772 CrossRef CAS.
- W. H. Binder and R. Sachsenhofer, Macromol. Rapid Commun., 2007, 28, 15–54 CrossRef CAS.
- W. H. Binder and R. Sachsenhofer, Macromol. Rapid Commun., 2008, 29, 952–981 CrossRef CAS.
- N. J. Long and C. K. Williams, Angew. Chem., Int. Ed., 2003, 42, 2586–2617 CrossRef CAS.
- J. Vicente, M.-T. Chicote, M.-D. Abrisqueta and P. G. Jones, Organometallics, 1997, 16, 5628–5636 CrossRef CAS.
- M. B. A. Jalil, IEEE Trans. Magn., 2002, 38, 2613–2615 CrossRef CAS.
- M. C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293–346 CrossRef CAS.
- D. Andreeva, Gold Bull., 2002, 35, 82–88 CrossRef CAS.
- M. Mirdamadi-Esfahani, M. Mostafavi, B. Keita, L. Nadjo, P. Kooyman, A. Etcheberry, M. Imperor and H. Remita, Gold Bull., 2008, 41, 98–104 CrossRef CAS.
- S. Bhaviripudi, E. Mile, S. A. Steiner, A. T. Zare, M. S. Dresselhaus, A. M. Belcher and J. Kong, J. Am. Chem. Soc., 2007, 129, 1516–1517 CrossRef CAS.
- A. Berenbaum, A. J. Lough and I. Manners, Organometallics, 2002, 21, 4415–4424 CrossRef CAS.
- A. W. Duff, P. B. Hitchcock, M. F. Lappert, R. G. Taylor and J. A. Segal, J. Organomet. Chem., 1985, 293, 271–283 CrossRef CAS.
- H. E. Gottlieb, V. Kotlyar and A. Nudelman, J. Org. Chem., 1997, 62, 7512–7515 CrossRef CAS.
- B. M. Sutton, E. McGusty, D. T. Walz and M. J. DiMartino, J. Med. Chem., 1972, 15, 1095–1098 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available: 1H NMR and MALDI-TOF spectra of polymer 1d, synthesis and characterization of 3b, GPC traces of 1b, 4a, 5a–c, 6a–c, 6e and 8. EDX analysis of thin films of 7 and 8. 1H NMR spectra of 3b, 4a, 5a, 6a, 7 and 8. See DOI: 10.1039/c1py00298h |
| This journal is © The Royal Society of Chemistry 2011 |