Enhancing the rigidity of a network polymer of intrinsic microporosity by the combined use of phthalocyanine and triptycene components†,‡
Mohammed
Hashem
,
C.
Grazia Bezzu
,
Benson M.
Kariuki
and
Neil B.
McKeown
*
School of Chemistry, Cardiff University, Cardiff, UK. E-mail: mckeownnb@cardiff.ac.uk; Tel: +44 2920 875850
First published on 19th August 2011
Abstract
By combining both triptycene and multifunctional phthalocyanine components within a polymer of intrinsic microporosity (PIM), a polymer network of apparent BET surface of 806 m2 g−1 was obtained that appears to possess a highly rigid structure as determined from the shape of its nitrogen adsorption isotherm, which is similar in appearance to that of a crystalline microporous material such as a zeolite.
The phthalocyanine macrocycle is a useful symmetrical and rigid building unit for making organic materials.1 In addition, metal-containing phthalocyanines are desirable components of nanoporous materials due to their well-established functionality as catalysts and light-harvesting units as illustrated by recent examples of their use within Covalent-Organic-Frameworks (COFs),2,3 nanoporous molecular crystals,4 Metal–Organic-Frameworks (MOFs),5 mesoporous silicas6 and zeolites.7 Over the past decade the concept of Polymers of Intrinsic Microporosity (PIMs) has been developed whereby a microporous material is formed by the inability of the component macromolecules to pack space efficiently due to their rigid and contorted structure.8,9 Indeed, although not labelled as a PIM at the time of publication, a phthalocyanine-based network polymer was the first example of a material designed using this concept.10 Later studies demonstrated the use of these Pc-PIMs as adsorbents to remove organic compounds from water11 and as heterogeneous catalysts for the oxidation of sulfides in aqueous solution12 or cyclohexene by hydrogen peroxide.13 These network polymers were prepared by using the high-temperature, metal cation mediated, phthalocyanine-forming reaction of a spirocyclic bisphthalonitrile precursor. This method is of variable success depending upon the metal cation used as template and provides materials with a broad range of BET surface areas (450–850 m2 g−1) even when employing the same metal cation as template. Since this work, we have found that the formation of dibenzodioxane (p-dibenzodioxin) fused-ring linkages using suitable o-hydroxylated (i.e.catechol) and o-fluorinated monomers is a highly efficient reaction for the preparation of both microporous networks and soluble polymers of high molecular mass.14,15 This synthetic method offers a modular approach due to the large number of potential monomers that can be combined to form the PIM. For example, the low temperature (80 °C) reaction between 2,3,6,7,12,13-hexahydroxytriptycenes, with or without alkyl substitution at the bridgehead positions, and 2,3,5,6-tetrafluoroterephthalonitrile gives highly microporous network polymers (Trip-PIMs) with apparent BET surface areas of up to 1730 m2 g−1.16,17 Inspired by this result, we designed a novel microporous network polymer (Trip-Pc-PIM) that would be prepared from this efficient network-forming reaction using a preformed phthalocyanine monomer that contains appropriate hydroxylated triptycene subunits. It was anticipated that this method would result in the reproducible preparation of PIMs that contain a high concentration of phthalocyanine functionality.
The synthetic route to Trip-Pc-PIM is outlined in Scheme 1.†The Diels–Alder reaction between 2,3,6,7-tetramethoxy-9,10-diethyl anthracene and 4,5-dibromobenzyne, generated in situ from the reaction of n-BuLi with 1,2,4,5-tetrabromobenzene, gives the dibromotriptycene derivative 1. A Rosemund von Braun reaction exchanges the bromine for nitrile substituents to give 2, which is the precursor to the triptycene-substituted phthalocyanine 3. Demethylation of 3 using BBr3 gives monomer 4 that is readily polymerised by the base-mediated reaction with 2,3,5,6-tetrafluoroterephthalonitrile to give a green solid that gave a 13C solid state NMR spectrum (ESI,† Fig. 2) and elemental analysis consistent with the ideal structure of Trip-Pc-PIM.
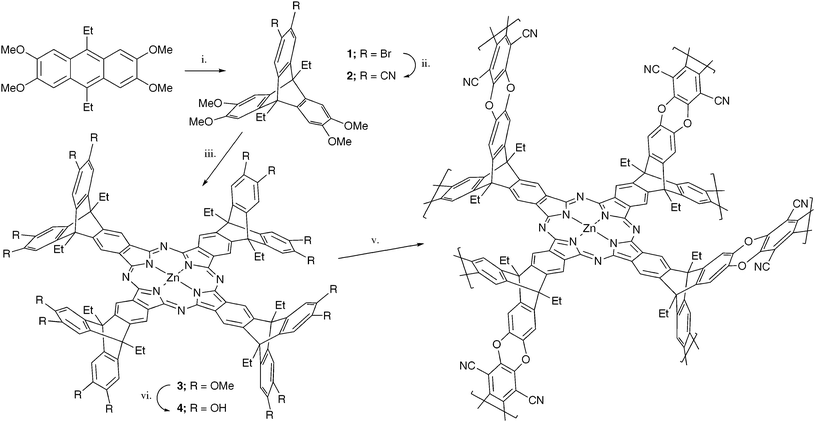 | ||
| Scheme 1 Reagents and conditions: i. 1,2,4,5-tetrabromobenzene, n-BuLi, toluene/hexane, 20 °C, 3h; ii.CuCN, DMF, reflux, 6 h; iii.Zn(OAc)2, DMAC, reflux, 48 h; iv. BBr3, DCM, 20 °C, 24 h; v. 2,3,5,6-tetrafluoroterephthalonitrile, DMF, K2CO3, 80 °C, 48 h. | ||
A single crystal X-ray diffraction (XRD) study of phthalocyanine 3 confirms its highly symmetrical structure (Fig. 1)‡and allows the monomeric unit to be visualised as part of the ideal network structure of Trip-Pc-PIM, within which it would be joined to eight of the fused-ring linking groups (Scheme 1). Triptycene units (in this context previously termed “dibenzobarreleno” substituents) fused to the edge of phthalocyanine have been used previously to prohibit cofacial aggregation of the planar extended macrocycles in solution.18,19 The crystal packing arrangement of 3 demonstrates that cofacial self-association of the phthalocyanine cores is efficiently inhibited by the triptycene units even within the solid state so that the distance between neighbouring zinc cations is over 11 Å (ESI,† Fig. 1). A large number of chloroform molecules (10 per phthalocyanine) fill the cavities of the crystal left by the packing arrangement of 3.
 | ||
| Fig. 1 The molecular structure of phthalocyanine 3 from a single crystal XRD study. The axial ligand (methanol) is disordered and is partially present on each side of the central Zn2+ cation. The carbon of the methanol is further disordered by symmetry. | ||
The microporosity of Trip-Pc-PIM was confirmed by nitrogen adsorption measurements at 77 K. The significant adsorption at low relative pressure (p/po< 0.1) and the classic Type 1 shape of the adsorption isotherm (Fig. 2) are both consistent with a predominantly microporous structure. An apparent BET surface area of 806 m2 g−1 and micropore volume of 0.34 mL g−1 can be calculated from the adsorption data. Although these values are less than that of the equivalent triptycene network PIM (Trip(Et)-PIM) they are significantly greater than those of the best characterised Pc-PIM, designated CoPc20, prepared via the phthalonitrile route (Table 1). Of particular note is the minimal hysteresis between the N2 adsorption and desorption isotherms of Trip-Pc-PIM that is in contrast to the extreme hysteresis and moderate hysteresis demonstrated by the nitrogen adsorption/desorption isotherms of Trip(Et)-PIM and CoPc20, respectively. The hysteresis associated with the N2 adsorption/desorption isotherms of Trip(Et)-PIM and CoPc20 extends to low relative pressures and hence is not due to the presence of mesoporosity but rather is attributed to swelling of the network as it fills with nitrogen. In the extreme case of Trip(Et)-PIM this swelling is facilitated by the layered 2-dimensional structure of this polymer network, as predetermined by the structure of the triptycene monomer within which the three catechol units propagate the network in the same plane. For Trip-Pc-PIM, the lack of hysteresis in the N2 isotherm can be attributed to its rigidity arising both from the rigidity of its phthalocyanine and triptycene components and the fully 3-dimensional connectivity of the network. The reduced hysteresis observed for the isotherm of Trip-Pc-PIM as compared to CoPc20, possibly reflects the greater rigidity of the triptycene unit relative to the 1,1-spirobisindane unit for which it has been established that the spiro-centre provides a site of relative flexibility.20 The minimal hysteresis for the N2 isotherm of Trip-Pc-PIM is unusual for an amorphous microporous polymer and is more characteristic of crystalline nanoporous materials such as zeolites or most Metal–Organic-Frameworks.21 Like these materials it suggests that Trip-Pc-PIM possesses both a rigid non-swelling structure and one that lacks meso- or macroporosity. Hence, the combination of extensive 3-D network connectivity and component rigidity appear to be prerequisites for a Type 1 isotherm - structural features that are satisfied by tetraphenylmethane-based microporous network polymers, which have been demonstrated also to display Type 1 isotherms.22–24
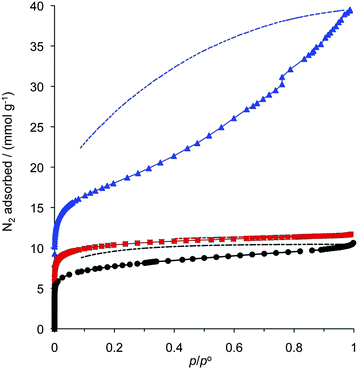 | ||
Fig. 2 The nitrogen adsorption isotherms of Trip-Pc-PIM ( ); CoPc20 (●) and Trip(Et)-PIM ( ); CoPc20 (●) and Trip(Et)-PIM ( ). Partial desorption isotherms are also shown (broken lines) to display the degree of hysteresis attributed to swelling of the polymer network. Note that for Trip-Pc-PIM the desorption curve is almost indivisible from the adsorption curve. ). Partial desorption isotherms are also shown (broken lines) to display the degree of hysteresis attributed to swelling of the polymer network. Note that for Trip-Pc-PIM the desorption curve is almost indivisible from the adsorption curve. | ||
The synthetic approach demonstrated in this work of using a preformed phthalocyanine monomer will allow the reproducible incorporation of a wide variety of metal cations, including those with catalytic activity, within a highly microporous polymer structure. It should be noted that a similar approach using a pre-formed halogenated phthalocyanine monomer gave networks of only modest surface areas, probably due to the aggregation of the phthalocyanines during network formation. The present strategy succeeds due to the use of bulky triptycene units to prohibit aggregation. This synthetic methodology should allow comparative studies using materials of a similar structure and porosity to determine the relative activity of the in-built metal cations rather than the success or otherwise of the network formation. Such studies, similar to those based on related porphyrin-based microporous polymers, are planned.25,26
We acknowledge funding from EPSRC grant EP/H024034/1 (CGB) and thank the EPSRC National Solid State NMR Research Service (Durham University) for the analysis ofTrip-Pc-PIM.
Notes and references
- N. B. McKeown, Phthalocyanine Materials: Synthesis, Structure and Function, CUP, Cambridge, 1998 Search PubMed.
- E. L. Spitler and W. R. Dichtel, Nat. Chem., 2010, 2, 672–677 CrossRef CAS.
- X. S. Ding, J. Guo, X. A. Feng, Y. Honsho, J. D. Guo, S. Seki, P. Maitarad, A. Saeki, S. Nagase and D. L. Jiang, Angew. Chem., Int. Ed., 2011, 50, 1289–1293 CrossRef CAS.
- C. G. Bezzu, J. E. Warren, M. Helliwell, D. R. Allan and N. B. McKeown, Science, 2010, 327, 1627–1630 CrossRef CAS.
- O. V. Zalomaeva, K. A. Kovalenko, Y. A. Chesalov, M. S. Mel'gunov, V. I. Zaikovskii, V. V. Kaichev, A. B. Sorokin, O. A. Kholdeeva and V. P. Fedin, Dalton Trans., 2011, 40, 1441–1444 RSC.
- J. T. Li, L. M. Guo and J. L. Shi, Phys. Chem. Chem. Phys., 2010, 12, 5109–5114 RSC.
- I. Lopez-Duarte, L. Q. Dieu, I. Dolamic, M. V. Martinez-Diaz, T. Torres, G. Calzaferri and D. Bruhwiler, Chem.–Eur. J., 2011, 17, 1855–1862 CrossRef CAS.
- N. B. McKeown and P. M. Budd, Macromolecules, 2010, 43, 5163–5176 CrossRef CAS.
- N. B. McKeown and P. M. Budd, Chem. Soc. Rev., 2006, 35, 675–683 RSC.
- N. B. McKeown, S. Makhseed and P. M. Budd, Chem. Commun., 2002, 2780–2781 RSC.
- A. V. Maffei, P. M. Budd and N. B. McKeown, Langmuir, 2006, 22, 4225–4229 CrossRef CAS.
- S. Makhseed, F. Al-Kharafi, J. Samuel and B. Ateya, Catal. Commun., 2009, 10, 1284–1287 CrossRef CAS.
- H. J. Mackintosh, P. M. Budd and N. B. McKeown, J. Mater. Chem., 2008, 18, 573–578 RSC.
- N. B. McKeown, S. Hanif, K. Msayib, C. E. Tattershall and P. M. Budd, Chem. Commun., 2002, 2782–2783 RSC.
- P. M. Budd, B. S. Ghanem, S. Makhseed, N. B. McKeown, K. J. Msayib and C. E. Tattershall, Chem. Commun., 2004, 230–231 RSC.
- B. S. Ghanem, M. Hashem, K. D. M. Harris, K. J. Msayib, M. C. Xu, P. M. Budd, N. Chaukura, D. Book, S. Tedds, A. Walton and N. B. McKeown, Macromolecules, 2010, 43, 5287–5294 CrossRef CAS.
- B. Ghanem, N. B. McKeown, K. D. M. Harris, Z. Pan, P. M. Budd, A. Butler, J. Selbie, D. Book and A. Walton, Chem. Commun., 2007, 67–69 RSC.
- Vn Kopranen, Gi Rumyants and Ea Lukyanet, Zh. Obshch. Khim., 1972, 42, 2586–2586 Search PubMed.
- M. G. Galpern, V. F. Donyagina, V. K. Shalaev, V. R. Skvarchenko, N. I. Bundina, N. G. Mekhryakova, O. L. Kaliya and E. A. Lukyanets, Zh. Obshch. Khim., 1982, 52, 715–716 CAS.
- M. Heuchel, D. Fritsch, P. M. Budd, N. B. McKeown and D. Hofmann, J. Membr. Sci., 2008, 318, 84–99 CrossRef CAS.
- P. A. Wright, Microporous Framework Solids, RSC, Cambridge, 2007 Search PubMed.
- T. Ben, H. Ren, S. Q. Ma, D. P. Cao, J. H. Lan, X. F. Jing, W. C. Wang, J. Xu, F. Deng, J. M. Simmons, S. L. Qiu and G. S. Zhu, Angew. Chem., Int. Ed., 2009, 48, 9457–9460 CrossRef CAS.
- E. Stockel, X. F. Wu, A. Trewin, C. D. Wood, R. Clowes, N. L. Campbell, J. T. A. Jones, Y. Z. Khimyak, D. J. Adams and A. I. Cooper, Chem. Commun., 2009, 212–214 RSC.
- J. R. Holst, E. Stockel, D. J. Adams and A. I. Cooper, Macromolecules, 2010, 43, 8531–8538 CrossRef CAS.
- L. Chen, Y. Yang and D. L. Jiang, J. Am. Chem. Soc., 2010, 132, 9138–9143 CrossRef CAS.
- A. M. Shultz, O. K. Farha, J. T. Hupp and S. T. Nguyen, Chem. Sci., 2011, 2, 686–689 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic procedures and physical methodology. CCDC 831067. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1py00288k/ |
‡ Crystals of 3 were prepared by the slow diffusion of MeOH into CHCl3 solution. Crystal size 0.3 × 0.2 × 0.2 mm, triclinic, space groupP![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 13.0853, b = 18.0594, c = 18.6508 Å, α = 79.027, β = 75.275, γ = 72.973, V = 4043.7 Å3, Z = 1, μ = 0.740 μm−1, 22912 reflections measured, 15 , a = 13.0853, b = 18.0594, c = 18.6508 Å, α = 79.027, β = 75.275, γ = 72.973, V = 4043.7 Å3, Z = 1, μ = 0.740 μm−1, 22912 reflections measured, 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 100 unique reflections (Rint = 0.0553), 9528 reflections with I >2σ(I), R = 0.1629 and ωR2 = 0.4436 (observed data), R = 0.2130 and ωR2 = 0.4765. 100 unique reflections (Rint = 0.0553), 9528 reflections with I >2σ(I), R = 0.1629 and ωR2 = 0.4436 (observed data), R = 0.2130 and ωR2 = 0.4765. |
| This journal is © The Royal Society of Chemistry 2011 |