DOI:
10.1039/C1PY00283J
(Review Article)
Polym. Chem., 2011,
2, 2741-2757
Recent advances in block copolymer-assisted synthesis of supramolecular inorganic/organic hybrid colloids
Received
22nd June 2011
, Accepted 28th July 2011
First published on 30th August 2011
Abstract
Synthesis of supramolecular inorganic/organic hybrid colloids with the assistance of block copolymer self-assembly motifs has attracted increasing research interest. Two strategies of multicomponent-assemblies, concurrent self-assembly and sequential self-assembly, are usually used for the investigation. Such co-assembly concept broadens the scope of block copolymer self-assembly studies and provides new routes for hybrid colloid design. Moreover, this kind of self-assembly opens unlimited possibilities for the development of inorganic/organic hybrid functional materials. In this article, we briefly review the recent progresses of this area through highlighting a number of representative literature reports.
 Yibo Liu | Yibo Liu received her Bachelor's degree from Heilongjiang University in China. She is currently a graduate student in Dr Xiaosong Wang's group at the University of Waterloo. She works on metallopolymer synthesis and self-assembly of functional nanomaterials. |
 Xiaosong Wang | Dr Xiaosong Wang obtained his PhD degree from the East China University of Science and Technology, China. After postdoctoral research in the University of Sussex and the University of Toronto, he started to work at the University of Leeds in 2006. He is currently an Associate Professor at the University of Waterloo working in the department of chemistry and the institute of nanotechnology. He has developed a number of new concepts for the creation, modification and manipulation of organometallic, inorganic and organic polymer nanoparticles with numerous potential applications. Recent examples include living self-assembly and miniemulsion periphery polymerization. His group currently focuses on the development of functional nanomaterials using block copolymer supramolecular chemistry. |
1. Introduction
Amphiphilic block copolymers have been extensively studied and used for the preparation of polymer colloids with various morphologies and functionalities.1,2 For example, in a selective solvent, the polymers are able to aggregate into micelles with solvent insoluble blocks as cores and solvent soluble blocks acting as coronas;3–5 In oil/water heterogeneous solution, the polymers function as stabilizers for the formation of oil droplets, known as emulsions.6–8 Depending on the functional groups presented in the backbones, the polymers are able to associate with a second component via noncovalent interactions, such as hydrogen bonding, electrostatic and hydrophobic interactions, leading to complex self-assembly behaviour and the creation of a number of interesting novel nanostructures.9–12 Taking advantages of these studies, block copolymer self-assembly motifs have been explored further and adapted for the preparation of inorganic/organic supramolecular hybrid colloids in an attempt to develop novel functional nanomaterials.
Since inorganic/organic hybrid colloids with synergetic functions of the two components have been a long term research interest,13–22 supramolecular chemistry of block copolymers has been involved in the synthesis of hybrid colloids many years ago.23,24 The major approach involves in situ synthesis of inorganic nanoparticles, such as quantum dots,25 metal nanoparticles,26,27silica particles,28,29etc., using self-assembled block copolymers as templates or nanoreactors. Self-assembly of inorganic block copolymers is another growing technique to access inorganic/organic colloids with novel morphologies and properties.30–32
In addition to the above approaches, supramolecular chemistry of multicomponent systems based on well-studied block copolymer self-assembly motifs has been explored in an attempt to incorporate inorganic components into the colloids in a designed fashion. The co-assembly approach allows a broad range of inorganic species to be involved, offering a great deal of flexibility for potential material development. Two strategies are usually used for the investigation: (1) concurrent self-assembly of block copolymers with a second component;33–40 (2) sequential self-assembly of inorganic species with assembled block copolymer colloids.41–51 The early stage investigation of these strategies has resulted in interesting, sometime unexpected, self-assembly behaviour, which has enriched our knowledge of supramolecular chemistry on multicomponent complex systems. Consequently, a range of novel hybrid nanostructures have been prepared with varied sizes, morphologies, and chemical compositions depending on the choice of polymers, self-assembly conditions, and details of processing. A particularly notable example is the synthesis of metal coordination/organic polymer nanoparticles. Several researches including our contributions have demonstrated that, with the assistance of supramolecular chemistry of block copolymers, it has become possible to prepare metal coordination polymer nanoparticles in a designed fashion in terms of their size, morphology, solubility and functionality.45–47,49,50,52 This achievement has addressed a long term challenge in the area of metal coordination polymer synthesis.32 Moreover, a number of research examples have demonstrated that supramolecular hybrid colloids produced from block copolymers are particularly useful as functional materials that cannot be achieved by using other synthetic techniques.53–56
In this article, we aim to briefly review the recent progress in the synthesis of inorganic/organic hybrid colloids self-assembled from multicomponent systems using block copolymers. It is not intended to be comprehensive, but instead illustrates the general synthetic concepts via highlighting some recent representative reports. Research examples demonstrating novel properties and potential applications of such ensembles are also highlighted.
Concurrent self-assembly is a process whereby the block copolymer self-assembly occurred in the presence of a second component such as metal nanoparticles (NPs). A typical experiment involves two steps: (1) amphiphilic block copolymers are mixed with a second component in a common solvent; (2) upon the addition of a selective solvent, self-assembly of the two components occurred simultaneously, leading to the formation of hybrid colloids. This facile self-assembly technique has been proved to be a very effective approach to incorporate different types of inorganic NPs, such as gold, semiconductors and magnets, into block copolymer colloids.33–40
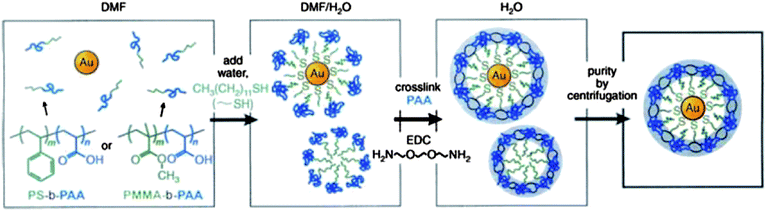
A representative example of concurrent self-assembly was reported by Taton and co-workers.33–35,57–59 They constructed hybrid colloids with gold NPs (Au NPs) as cores and block copolymers as shells. The preparation procedure is shown in Scheme 1, polystyrene-b-poly(acrylic acid) (PS-b-PAA) (or poly(methylmethacrylate)-b-poly(acrylic acid) (PMMA-b-PAA)) amphiphilic block copolymers were initially dissolved in N,N-dimethylformamide (DMF), which is a good solvent for all polymer blocks.33Citrate-capped Au NPs, prepared in water, were centrifuged and transferred to DMF.60 The DMF solution of Au NPs was mixed with the above block copolymer solution in the presence of a small amount of 1-dodecanethiol that is able to hydrophobically functionalize the surfaces of Au NPs and direct the Au NPs to the hydrophobic interior of the micelle. To induce the self-assembly, water, a poor solvent for dodecanethiol functionalized Au NPs, PS (or PMMA) blocks, was added gradually to the mixture. As a result, assembled hybrid colloids were produced. The colloids possess Au NP cores covered by shells of hydrophobic PS (or PMMA) blocks, while hydrophilic PAA chains remain soluble and stabilize the particles. The assembled hybrid micelles could be permanently fixed by cross-linking the soluble PAA blocks using 2,2′-(ethylenedioxy)bis(ethylamine) as the crosslinking agent and 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide methiodide as the coupling reagent in water. After cross-linking, excess crosslinking agents and DMF were removed via a dialysis process. Fixed core/shell NPs were separated from empty micelles by centrifugation and could be resuspended in H2O.
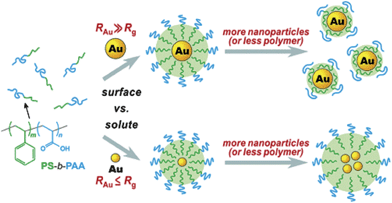
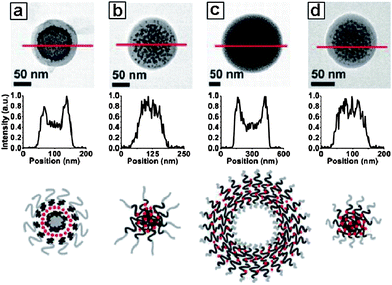
It was found that the size of Au NPs strongly determined the self-assembly behaviour (Scheme 2).34 By using smaller Au NPs (ca. 4 nm), hybrid colloids containing multiple NPs were produced as evidenced by the TEM image (Fig. 1a), because the smaller Au NPs behaved like solutes swelling the micelle cores. This random encapsulation of nanoparticles led to hybrid colloids having very broad size distribution. In contrast, when larger Au NPs (>10 nm) were used, single particle was encapsulated within each micelle (Fig. 1b–i), suggesting these larger NPs behaved like a surface, rather than solutes, to direct the assembly of block copolymers. Further research has demonstrated that the thickness of the polymer shell can be varied by controlling several factors34 including: (1) the size of NPs. As shown in Fig. 1b and c, shell thickness gradually increased with increasing the diameter of NPs when the ratio of polymer to particle surface area was kept constant; (2) the available surface area of the NPs. As shown in Fig. 1d–f, shell thickness of the encapsulated NPs gradually increased with decreasing the amount of NPs while keeping the concentration of the polymer constant; (3) the block ratio of the polymers. As shown in Fig. 1g–i, the thickness of the shell increased with increasing the chain length of PS with respect to the PAA block.
 |
| | Scheme 2 Schematic illustration of the preparation of core/shell Au NPs containing hybrids using Au NPs with different NP diameters. | |
 |
| | Fig. 1
TEM images for Au@PS100-b-PAA13 hybrid colloids. The influence of (a–c) the NPs size, (d–f) the relative available surface area of the NPs and (g–h) the block copolymer composition on the polymer shell thickness. | |
Chen and co-workers reported a facile approach for the synthesis of Janus-shaped hybrid colloids via the assembly of block copolymers and Au NPs in the presence of hydrophobic (LA) and hydrophilic ligands (LB) (Fig. 2).36 The binding competition between LA and LB on the surface of Au NPs broke the symmetry of surface attachments of the block copolymers of PS154-b-PAA60 on the Au NPs. As shown in Fig. 2c and d, both LA and LB are able to coordinate with the NPs, whereas comparing with LB, LA has a higher tendency to be embedded in the PS layer due to stronger hydrophobic interactions. Consequently, during the self-assembly, the block copolymers preferred to associate with the surfaces of the NPs covered by LA, leaving the LB-attached surface exposed to water. The relative concentrations of LA and LB used for the self-assembly determined the morphology of the resulting Au NPs hybrid colloids (Fig. 2b–d). As shown in TEM images (Fig. 2e–g), when [LA]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[LB] varied from 1:0, 1:22, to 1:132, the core/shell morphology evolved from homocentric (Fig. 2e) to slightly eccentric (Fig. 2f) and then to highly eccentric (Fig. 2g). The observed anisotropic polymer coverage of Au NPs opens up opportunities for selective functionalization and controlled assembly of NPs in the future.
:[LB] varied from 1:0, 1:22, to 1:132, the core/shell morphology evolved from homocentric (Fig. 2e) to slightly eccentric (Fig. 2f) and then to highly eccentric (Fig. 2g). The observed anisotropic polymer coverage of Au NPs opens up opportunities for selective functionalization and controlled assembly of NPs in the future.
![Structures of LA and LB, and schematic diagram showing the formation of (a and b) homocentric and (a, c, and d) eccentric AuNP@polymer. TEM images of purified AuNP@polymer when [LA] : [LB] = (e)1 : 0, (f) 1 : 22 and (g) 1 : 132, corresponding to (b–d), respectively. All scale bars are 50 nm.](/image/article/2011/PY/c1py00283j/c1py00283j-f2.gif) |
| | Fig. 2 Structures of LA and LB, and schematic diagram showing the formation of (a and b) homocentric and (a, c, and d) eccentric AuNP@polymer. TEM images of purified AuNP@polymer when [LA]:[LB] = (e)1:0, (f) 1:22 and (g) 1:132, corresponding to (b–d), respectively. All scale bars are 50 nm. | |
Incorporating NPs into the walls of polymer vesiclesvia concurrent self-assembly was reported by Eisenberg and co-workers.39 Two types of metal NPs stabilized by diblock copolymers were used for the investigation. As shown in Fig. 3, one type of NPs is PEO45-b-PS155-b-P(APb)25 micelles with cross-linked Pb acrylate cores, P(APb)25, surrounded by PS155-b-PEO45 coronas. The other type of NPs is Au NPs stabilized by PS270-b-PAA15 copolymers with a thioctate ester (TE) end group (TE-PS270-b-PAA15Au NPs). These metal NPs were used respectively to assemble with PS235-b-PEO45 by adding water to a dioxane solution containing both the polymers and the NPs. It is well-known that PS235-b-PEO45 are able to aggregate into vesicles with PS chains acting as walls under this condition.61 The metal NPs also contain PS chains that provided a chance to integrate the NPs into the PS walls of the vesiclesvia hydrophobic interactions. Fig. 4 shows TEM photographs of the vesicles prepared from the self-assembly of PS235-b-PEO45 and PEO45-b-PS155-b-P(APb)25 micelles. The average diameter and wall thickness of the vesicles are given in the upper right corner of Fig. 4a. As shown in Fig. 4, a large number of Pb acrylate cores can be seen as black dots in the vesicles. It appeared likely that the NPs localized toward the centre of the walls with the attached diblock copolymers partitioning to both interfaces. This proposed structural feature of the hybrid vesicles was supported by TEM analysis: an empty space on the outside of the vesicle walls is observed in Fig. 4a and a similar empty distance also exists on the inside of the wall as shown in the TEM image of indented vesicles (Fig. 4c). Another argument in favour of the existence of the empty distance on both interfaces of the vesicle walls is based on consideration of symmetry. Two possible types of wall incorporation are illustrated in Fig. 5. The upper half on the right of Fig. 5 shows the situation of each NP located in the central portion of the wall, with the corona chains extended more or less equally toward each interface. The lower half on the right of Fig. 5 illustrates the situation for the asymmetric placement of the NPs, i.e. a structure in which all of the corona chains are pointing only to one side of the NP, drawing the particles to either the inside or outside interface, but not to both. Such a unilateral segregation is highly unlikely for both entropic and steric reasons, so that the placement of the NPs in the central portion of the wall appears most reasonable.
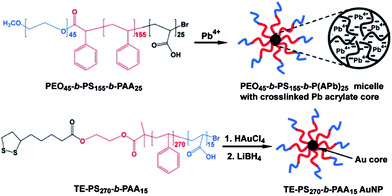 |
| | Fig. 3 Schematic illustration of the preparation of the two types of diblock copolymer coated NPs. | |
 |
| | Fig. 4
TEM micrographs of the vesicles with the incorporated NPs prepared from the combined solution of the PS235-b-PEO45 copolymer (0.5 wt%) and the PEO45-b-PS155-b-P(APb)25 micelles (0.5 wt%). | |
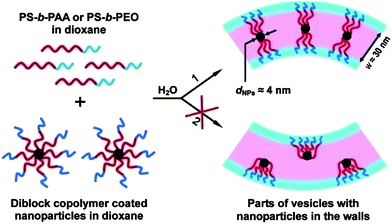 |
| | Fig. 5 Schematic illustration of two possible types of incorporation of the diblock copolymer coated NPs into vesicle walls. The red color represents the hydrophobic composition, and the blue color represents the hydrophilic composition. Possibility 2 is believed to be highly unlikely. | |
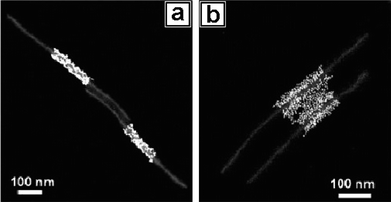
In addition to spherical metal NPs, Au nanowires (Au NWs) were also used to self-assemble with block copolymers (Fig. 6).38 The oleylamine-stabilized Au NWs as shown in Fig. 6b were synthesized and purified as reported,62,63 which were mixed with PS154-b-PAA49 dissolved in a ternary solvent mixture of DMF/THF/H2O (4:4:1, v/v/v). THF was used to improve the solubility of the Au NWs, and the large percentage of organic solvents was able to swell the polymer. After the solution was incubated for 24 h, the sample was then diluted by water to deswell the polymer shells and centrifuged to remove the excess solvent and free PS-b-PAA micelles. The resulting Au NWs hybrid colloids were characterized by TEM. As shown in Fig. 6c, the originally straight Au NWs were unexpectedly transformed into orderly structures with each polymer micelle containing one circular spring-like coil of 5–10 loops. The side views of the hybrid colloids show that the overall PS-b-PAA shells have shapes of circular discs and that the metal coils consist of closely packed coplanar rings (Fig. 6d). Calculation based on the radii of the nanosprings (52.1 ± 6.7 nm) (Fig. 6i) and the overall circumferences of the loops are in general agreement with the original length of the Au NWs (ca. 1–2 μm). Close inspection shows that the nanosprings often reveal two ends (Fig. 6e and f) with arguably chiral structures, confirming that they each originated from a single wire. The transformation of straight wires to circular rings was induced by the contraction of encapsulating shells as a result of PS-b-PAA block copolymer micellization. The mechanical energy stored in this process could be released upon removal or swelling of the polymer shells.
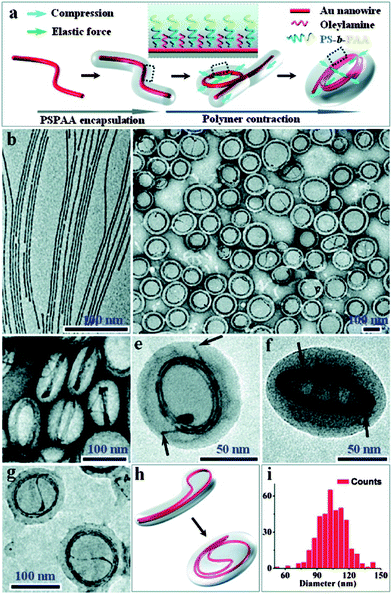 |
| | Fig. 6 (a) Schematics showing the polymer-induced coiling of Au NWs. TEM images of (b) as-synthesized oleylamine-stabilized Au NWs; (c and d) coiled nanosprings embedded in micelles; (e and f) two typical Au nanosprings from different perspectives, with arrows indicating their ends; (g and h) TEM image and schematic illustration of a typical Au nanospring that did not coil along its first loop but via a new loop formed by the folded double-wire; (i) diameter distribution of the Au nanosprings. | |
Concurrent self-assembly technique was also used to prepare quantum dots (QDs) containing hybrid colloids.64,65 Winnik et al. reported a simple and versatile approach for spatially defined organization of colloidal CdSe QDs and poly(3-hexylthiophenes) (P3HTs) conjugated polymer using micelles of polystyrene-b-poly(4-vinylpyridine) (PS-b-P4VP) as the main structural motif (Fig. 7).37PS-b-P4VP spherical micelles with PS cores and P4VP coronas could solubilize P3HT in the PS cores due to their common insolubility. Meanwhile, the micelles could bind a variety of different QDs within the coronas because of their favourable interaction with the P4VP blocks. This created a spatial organization of the two components. Steady-state fluorescence measurements demonstrated excited-state interactions between the QDs and the conducting polymer.
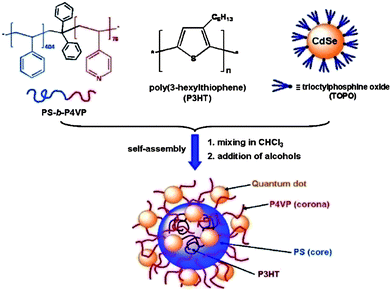 |
| | Fig. 7 Schematic presentation of the self-assembly of PS404-b-P4VP76 diblock copolymer, P3HT, and CdSe/TOPO QDs into composite micelles in which P3HT chains are in the core and QD particles are in the corona. | |
Moffitt and co-workers performed experiments to assemble CdS NPs, prepared from polystyrene-b-poly(cadmium acrylate) (PS-b-PACd) block copolymers, with polystyrene-b-poly(ethylene oxide) (PS-b-PEO) at an air–water interface.66 Preferential interactions between PS-functionalized NPs and the PS blocks of an amphiphilic PS-b-PEO block copolymer resulted in synergistic self-assembly at the air–water interface, forming a range of highly stable one-dimensional NP/polymer surface features, including branched nanowires, nanocables up to 100 μm in length, and nanowires with nanoring connectors. This strategy offers new routes to hierarchical hybrid assemblies with potential photonics applications.
Park and co-workers reported the synthesis of well-defined polymersomes densely packed with iron oxide NPs for the first time via the concurrent self-assembly of magnetic NPs and PAA-b-PS block copolymers. Importantly, they were able to vary the nanostructures of the hybrid colloids by using different common solvents for the assembly.40 These structures include core–shell type assemblies where magnetic NPs are radially localized at the interface between the polymer core and the shell (magneto-core–shell) (Fig. 8a), micelles solubilizing magnetic NPs (magneto-micelles) (Fig. 8b) and polymersomes packed with magnetic NPs (magneto-polymersomes) (Fig. 8c). A number of organic solvents were used as common solvents to dissolve both the block copolymers and NPs. Upon the addition of water, the self-assembly of the components starting from the solutions in DMF, THF or dioxane led to the generation of magneto-core–shell, magneto-micelle and the mixture of magneto-micelle and magneto-polymersomes, respectively. These structures were proved by energy-dispersive X-ray spectroscopy (EDS) (Fig. 8). The different morphologies produced from different solvents could be attributed to solvent–polymer interaction. Depending on the degree of stretching for PS and PAA chains, it is predicated that the relative volume taken by PAA is smaller in dioxane than in DMF and THF leading to the formation of vesicles in dioxane and micelles in DMF and THF. In addition, NP–PS interaction in system containing different common solvents strongly affected the NPs arrangement in the polymer matrix. PS has a relatively lower solubility in DMF than in THF and dioxane, leading to a compact PS core of the micelles prepared from DMF solution. As a result, NPs are pushed out of the compact PS cores and sit at the interface between the core and the shell during the formation of the assemblies from DMF/THF (Fig. 8a). In contrast, THF or dioxane common solvents can swell the PS cores of the micelles, allowing the NPs distribute throughout the polymer micelles or vesicle walls (Fig. 8b and c). Furthermore, they have also demonstrated that these morphologies significantly affected their magnetic relaxation properties, emphasizing the importance of the self-assembly structure for the functional nanomaterials design.67,68
 |
| | Fig. 8 Three different self-assembly structures of (a) magneto-core–shell, (b) magneto-micelles and (c and d) magneto-polymersomes and magneto-micelles hybrid colloids prepared from the solution in DMF/THF (96.8% DMF), in THF and dioxane/THF (96.8% dioxane) respectively. EDS data of these different hybrid colloids are describe below the corresponding TEM images. | |
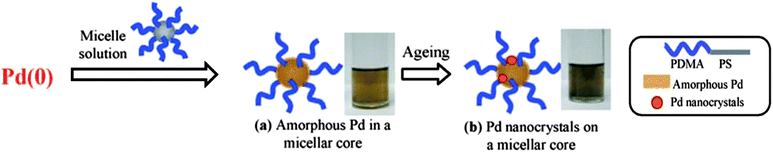 |
| | Scheme 3 Micelle-mediated nucleation. (a) Pd-micelle assembly; (b) slow nucleation of Pd in the cores of polymer micelles. | |
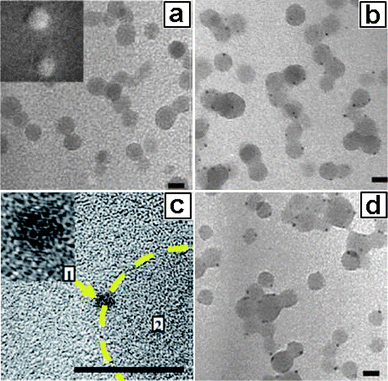 |
| | Fig. 10
TEM images for (a) a fresh sample with amorphous Pd in micellar cores (inset: Pd element mapping); (b) an aged sample with Pd nanocrystals in micellar cores; (c) HR-TEM for a Pd nanocrystal attached on a micellar core; and (d) a heated sample. The scale bars represent 20 nm. | |
The sequential self-assembly of semicrystalline polyferrocenylsilane (PFS) block copolymers has lead to the discovery of living self-assembly,30,69 driven by PFS crystallization forces in conjugation with its hydrophobic association. As a result, designed synthesis of block comicelles with linear and scarf architectures becomes possible.69–71 Further assembly of PFS block comicelles with metal NPs yielded new types of nanomaterials with selective functionalization.42,72 For example, selective patterning of the PFS block co-micelles with Au NPs and PbS QDs via electrostatic interactions has been demonstrated.42 In this work, PFS17-b-P2VP170 was used to create cylindrical micelles in 2-propanol with PFS as cores and P2VP as coronas.73 Quaternization of the P2VP coronas with MeI rendered the cylinder with positive charges. Afterwards, PFS17-b-P2VP170 was assembled to the two ends of the positively charged micelles driven by epitaxial crystallization of PFS blocks, leading to the generation of tri-block co-micelles with positively charged central block and non-charged end blocks. Subsequently, negatively charged Au NPs74,75 or PbS QDs76,77 were added to the tri-block co-micelle solution, allowing the negatively charged NPs to selectively bind to the positively charged central blocks of the co-micelles by the electrostatic interactions (Scheme 4). TEM images of the final composites are shown in Fig. 11. It is clear that Au NPs or PbS QDs were exclusively located on the central blocks of the co-micelles. No unreacted NPs were observed on the carbon film. This method can be extended to related architectures for possible design of functional nanomaterials.
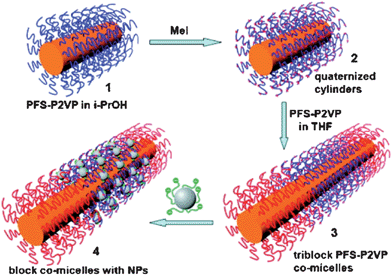 |
| | Scheme 4 Schematic illustration of the formation of block co-micelles with NPs. | |
 |
| | Fig. 11 Dark-field TEM mages show block co-micelles with the central block functionalized with (a) Au NPs and (b) PbS QDs. | |
In addition to metal NPs42 or atoms,41 sequential introduction of metal cations to block copolymer colloids was also tried to produce novel supramolecular structures.43,44,78 Discher and co-workers produced spotted vesicles and striped micelles via the assembly of polymer micelles with metal ions which can induce phase separation.43,44 Two diblock copolymers, poly(acrylic acid)-b-poly(butadiene) (PAA-b-PBD) and poly(ethylene oxide)-b-poly(butadiene) (PEO-b-PBD), were blended to produce polymer vesicles. The different block copolymers remained miscible until the addition of divalent cations Ca2+ which essentially served as metal centers and strongly bound to the PAA side chains. Critically, this binding induced PAA-b-PBD polymers to phase separate from PEO-b-PBD blocks, producing patterns on the surface layer of the particles (Fig. 12). The types of the pattern formed on the surface of the vesicles could be controlled by simply varying the blending ratios of the respective block copolymers. It was found that the deformability of the two different domains was different. Aspirated projections of PEO-b-PBD continuous phase were seen at a pressure of 70 Pa (Fig. 12a), whereas no significant projection could be obtained in the Ca2+-bound PAA-b-PB continuous phase in micropipette aspiration (Fig. 12b) due to an increase in the membrane stiffness caused by calcium-mediated bridging between polyanionic chains. They indicated that the effective rigidity for the PAA-b-PBD membranes was in correlation with the pH and the calcium concentration, but is more sensitive to calcium concentration than to pH. On the other hand, when Cu2+ was added, the domain formation was also observed, but the domains appear more fractured (Fig. 12c), probably as a result of the stronger interactions between Cu2+ and PAA.
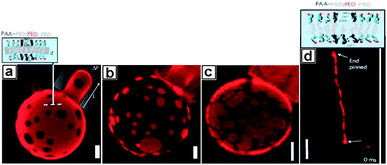 |
| | Fig. 12 Z-Sectioning confocal microscopy images of (a) Ca2+-induced patchy particles during the micropipette aspiration of continuous PEO-b-PBD phase; (b) Ca2+-induced patchy particles during the micropipette aspiration of continuous Ca2+-bond PAA-PB phase. (c) Cu2+-induced patchy particles and (d) Ca2+-induced striped worm-like cylinder micelle. Scale bar: (a–c) 2 μm (d) 4 μm. | |
By using the same polymer system but at high pH, PAA becomes highly charged and the electrostatic repulsion of PAA blocks is increased, leading to an increase in the curvature of core/brush interface and the generation of cylindrical micelles. Upon the addition of Ca2+, the phase-separation was also observed in the cylinders (Fig. 12d). The complexity of this kind of particles will push the particles towards multifunctional uses.
Sequential self-assembly using block copolymers has been recently extended to the area of metal coordination polymers in an attempt to develop controlled synthesis. Metal coordination polymers are inorganic/organic hybrid compounds of ordered structures constructed from metal–organic ligand coordination interactions.32 Depending on the nature of their chemical compositions and network structures, the polymers have many unique properties centered on the applications in gas storage, ion-exchange, catalysis, conductivity, luminescence and magnetism etc.79–81 However, synthesis of the polymers with designed morphology, solubility and chemical composition is a challenge task. Most of coordination polymers only form bulky powder upon precipitation and insoluble in most of solvents,80–82 which limited their practical applications. Using block copolymer, two approaches of sequential assembly techniques have been explored to address the challenge: (1) the first approach involves the self-assembly of block copolymers followed by the addition of the second components to induce a coordination polymerization; (2) the second approach is in a reverse order, in which metal coordination polymers were created first, then block copolymers were added to provide driving forces for desired nanostructure variation.
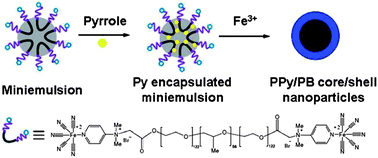
Taking advantages of well-developed miniemulsion technique, miniemulsion periphery polymerization (MEPP) was developed by our group. Generally, MEPP involves the creation of oil phase in water miniemulsion stabilized by block copolymer surfactants with polymerizable end groups, followed by a polymerization confined on the periphery of the oil droplets.45,83 In one of our work, we prepared miniemulsion using a metallo-surfactant of poly(ethylene glycol)-b-poly(propylene glycol)-b-poly(ethylene glycol) terminated with pentacyano(4-(dimethyl amino)pyridine)ferrate (EPE-Fe). Due to the presence of pentacyanoferrate end groups, the addition of Fe3+ can induce a metal coordination polymerization on the surface of miniemulsion oil droplets, led to the generation of the first metal coordination nanoshells (Scheme 5). Fig. 13a shows a typical TEM image of the coordination polymer nanoshells with a diameter of ca. 65 nm. Particles in the TEM image at higher magnification (Fig. 13b) exhibit a ring-like morphology suggesting shell structures of the particles. Element mapping of the particles reveals that both carbon and iron elements are uniformly distributed over the surface of the shells, confirming the formation of shells of organometallic networks. From a tapping mode AFM image, the dried shells appeared as short rods with hemisphere caps, suggesting the shells are rigid (Fig. 13c). This rigidity is also observed from the SEM image (Fig. 14). The observed rigidity of the shells could be attributed to directional coordination interactions. WAXD indicated that the resulted particles are amorphous, probably because the curved structures prevent the PB from crystallization.
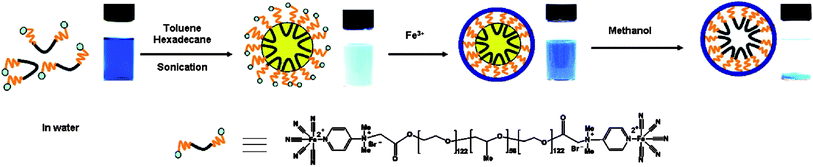 |
| | Scheme 5 Schematic illumination of Prussian blue (PB) nanoshells produced through miniemulsion periphery polymerization (MEPP). | |
 |
| | Fig. 13
TEM micrographs of the PB nanoshells (a) at lower magnification, (b) at higher magnification, (c) size of AFM micrograph is 1 × 1 μm2. Particle size is determined to be ca. 65 nm. | |
 |
| | Fig. 14
SEM micrographs of the nanoshells. The inset is the SEM micrograph at higher magnification. | |
The advent of MEPP has made it possible to prepare metal coordination particles with designed nanostructures in terms of their size, shape and chemical composition. It is well-known that the diameters of miniemulsion droplets can be readily varied in a wide range from 50 nm to 500 nm. It is therefore possible to manipulate the size of the nanoshells. To demonstrate this possibility, miniemulsions were prepared by using the same amount of EPE-Fe (0.5 wt%), but with a different toluene content of 5 wt% and 20 wt%, respectively. The shells prepared from the two systems were characterized by TEM. As shown in Fig. 15, the particles prepared from the solution containing 20 wt% toluene are ca. 198 nm, which is about twice as large as those in the system with 5 wt% toluene.
 |
| | Fig. 15
TEM micrographs of the nano-shells produced using the same amount of EPE-Fe (0.5% by weight) but different amount of toluene. (a) 20% by weight of toluene and (b) 5% by weight of toluene. Particle sizes are determined to be ca. 198 nm and ca. 100 nm, respectively. | |
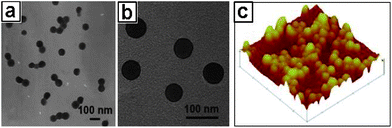
We have also demonstrated that the crystalline nature of Prussian blue (PB) could be harnessed in conjunction with the MEPP process, which led to the synthesis of the first example of metal coordination polymer nanoboxes.46 To induce PB crystallization, it was found that reducing the overall end functionality of the EPE-Fe surfactant from ∼100% to 60% was crucial. Further reducing EPE-Fe to a level below 60% failed to attain fully crosslinked shell structures.47 The nanobox structure was confirmed by electronic microscopy characterization. As shown in Fig. 16, it is evident from the SEM images that these NPs are cubic in nature. The strong contrast between the dark edges and brighter centers is clearly indicative of the hollow nature (Fig. 16a and b). The TEM image at low magnification reveals the uniform size of the PB nanoboxes (Fig. 16d).
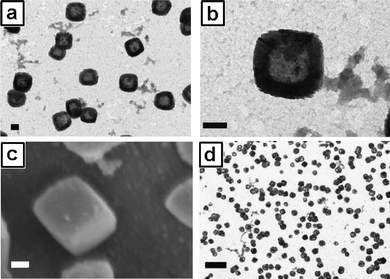 |
| | Fig. 16
TEM images (a) scale bar = 100 nm and (b) scale bar = 100 nm, SEM image (c) scale bar = 100 nm and TEM image (d) scale bar = 500 nm of the PB nanoboxes obtained using a 4 wt% loading of EPE (60% EPE-Fe:40% EPE-Br), 5 wt% oil phase and a 5 molar excess of FeCl3 relative to total surfactant end-groups. | |
The formation of the non-spherical structures was attributed to the crystallization of PB domain, which was supported by WADX analysis of the particles and confirmed by the observation of the crystalline lattice in the high resolution TEM image (Fig. 17). Fast Fourier transform (FFT) power spectra (Fig. 17) taken from a number of regions either at the edge or in the central face of the particle reveal diffraction patterns, suggesting that nanoboxes are crystalline across their entire surface. This discovery underpins a fundamental property of coordination polymers in illustrating how their directional interactions can be utilized to overcome solvent–particle interfacial tension and produce non-spherical structure at ambient conditions.
 |
| | Fig. 17 The TEM image (left) depicting the edge of a single nanobox. Note parallel lines depicting crystalline order (inset). Numbered FFT regions (right) denote areas from which FFT data were taken (scale bar = 10 nm). | |
Due to the nature of the technique, PB NPs produced from MEPP comprised more than 90 wt% organic components. Recently, we have prepared surfactant free pure metal coordination shells of PB analogue (PBA).84 In this case, lanthanide ions (Gd3+ or Er3+) were used as the crosslinking metals instead of the d-block metal ions (Fe3+), because lanthanide is not only able to crosslink metallo-surfactant miniemulsion droplets but also able to promote hydrolysis of ester linkages to assistant the removal of organic surfactant. The successes of this strategy also rely on a rational design of a second generation MEPP metallo-surfactant, namely EPE-Py-Fe (Fig. 18). As shown in the figure, the carbonyl group in the EPE-Py-Fe directly links to an aromatic pyridyl group, the pentacyanoferrate would donate the electrons through the π-conjugated system and increase electron density on the carbonyl, thus decreasing its electrophilic character and reducing the hydrolysis rate.85 The reduction of ester linkage hydrolysis rate ensures that the nanoshells formation can be completed prior to surfactant detachment. The complete removal of the surfactants was evidenced by FT-IR, NMR and TGA analysis, and the hollow structures were clearly confirmed by TEM analysis. Nitrogen adsorption experiments of the nanocages indicated the cage wall is a mesoporous structure. The internal hollow and porosity of the nanocage, coupled with the various properties of the coordination polymer building blocks, promise a wide variety of potential applications for such kind of nanostructure.
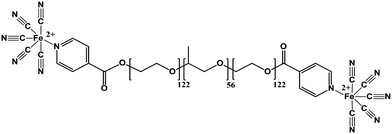 |
| | Fig. 18 Structure of EPE-Py-Fe. | |
MEPP is not only amenable to structure variation, but also provides the possibility of integrating other chemical components into the shells for designed synthesis of Prussian blue (PB) nanocomposites. In this regard, we have prepared polypyrrole (PPy)/PB core/shell NPs.48 In the process of the synthesis, pyrrole (Py) monomers were introduced into the miniemulsion system before the addition of Fe3+ ions. The cyclic voltammetric (CV) profile indicated that Py is mainly located in the interior of the miniemulsion droplets, because no signal for electro-polymerization of Py was observed. The formation of PPy/PB core/shell NPs was accomplished by the addition of Fe3+ which can induce the inner oxidation polymerization of Py for the formation of PPy cores and the periphery metal coordination polymerization for the formation of PB nanoshells simultaneously (Scheme 6). The core/shell structure of the PPy/PB NPs was confirmed by TEM characterization. As shown in Fig. 19b, the NPs have a diameter of 200 nm with a darker periphery attributed to the PB shells. Element mapping (Fig. 19b, inset) of the particles revealed that nitrogen and iron elements were uniformly distributed over the surface of the particles supporting the formation of PB shells. Interestingly, the resulting composites show strong fluorescence that is not a property usually associated with PB polymers. IR and Raman spectra have provided preliminary evidence for the occurrence of charge transfer between the two components accounting for the observed optical function.48,86–88Cell studies (Fig. 19c) indicated that the particles were able to enter the cells and remained photoluminescent, suggesting their possible application as biomaterials.
 |
| | Fig. 19
TEM micrographs of the PPy/PB core/shell NPs (a) at lower magnification, (b) at higher magnification (inset: EDX mapping images of PPy/PB core/shell NPs, upside: nitrogen element: downside: iron), (c) confocal laser scanning microscopy (CLSM) image of Bel-7402 cells incubated with PPy/PB core/shell NPs. | |
PBA hollow structures have been also prepared in organic solvent using block copolymer stabilized reverse emulsion.50 As shown in Scheme 7, MacLachlan and co-workers prepared pentacyanoferrate coordinated block copolymers, named Fe-BI-75 (Scheme 7), via a ligand replacement reaction of NH3 ligand in Na3[Fe(CN)5NH3] with bipyridium moieties in the block copolymer of BI-75 (Scheme 7). The reaction was carried out in the mixed solvents of THF and H2O. To this solution, chlorobenzene was added to induce a water-in-oil reverse emulsion stabilized by Fe-BI-75. Followed by the addition of Zn(NO3)2, [FeII(CN)5]3− moieties in the Fe-BI-75 were crosslinked via metal coordination reactions, leading to the generation of PBA nanoshells. The final crosslinked spheres contain PS brushes and hence have good solubility in many organic solvents, including chlorobenzene, benzene, toluene, THF and chlorinated hydrocarbons. As characterized by TEM and SEM, the nanoobjects dried from unlinked reverse emulsion are deformed hollow capsules (Fig. 20a). After coordination crosslinking, the hollow structure remained and looks less deformative (Fig. 20b) due to the formation of metal coordination structures. SEM image (Fig. 20c) of the particles reveals a ring-like structure probably due to the collapse of PBA vesicles. Compared with the hollow structure prepared using MEPP (Fig. 14), the PBA vesicles are less rigid probably because the metal coordination structures were built on the scaffold of flexible polymer chains. The encapsulation behaviour of the PBA was studied by encapsulating a water-soluble organic dye of methylene blue (MB). It was found that the dye could be released from the particles in methanol, but remained encapsulated when the particles dispersed in water. This experiment demonstrates a possibility to construct hollow NPs with selective permeability.
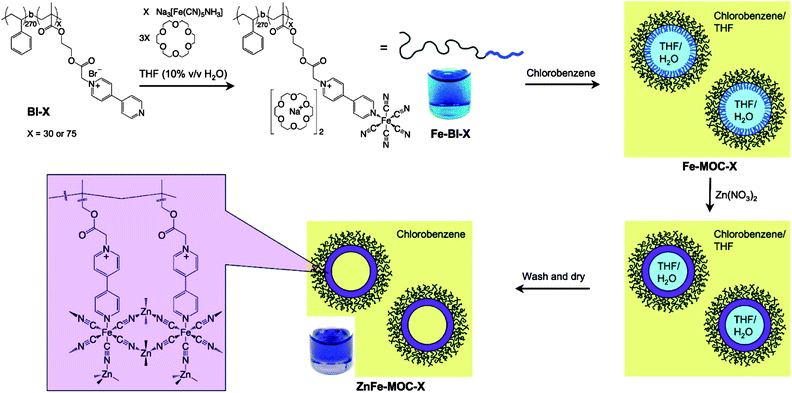 |
| | Scheme 7 Synthetic approach to soluble block ionomer–PBA capsules through emulsion-induced assembly. | |
 |
| | Fig. 20
TEM images of (a) nanocapsules before crosslinking with metal ions and (b) PBA-type nanocapsules. (c) SEM image of PBA-type nanocapsules. | |
Using the similar polymers, they prepared worm-like metal coordination nanostrucutres.49 The BI polymers form micelles in THF, to which aqueous solution of Na3[Fe(CN)5NH3] and [18]crown-6 was added to introduce [FeII(CN)5]3− moieties via ligand exchange reaction inside the core of the micelles. The resulting structure was denoted as [FeII(CN)5]BI. Subsequently, Mn+ (Fe3+, Zn2+, and Co2+) was added to crosslink the [FeII(CN)5]3−groups into PB-type frameworks (M[Fe(CN)5]BI). The worm-like nanostructures were observed for both [FeII(CN)5]BI (Fig. 21a) and metal coordinated M[FeII(CN)5]BI (Fig. 21b), but M[FeII(CN)5]BI has better contrast than [FeII(CN)5]BI due to higher density of metal elements. This experiment seems to suggest that metal coordination polymerization is not a decisive reason for the formation of one-dimensional structures. Interestingly, when K4[Fe(CN)6], which is not able to bind to the polymer (BI), was used, wormlike structures were still observed (Fig. 21d). In a control experiment of using triethylammonium ion functionalized block copolymer (denoted as BI*) (Fig. 21e) that cannot coordinate to either K4[Fe(CN)6] or Mn+, wormlike micelles were also formed. However, the cores of the nanoworms do not contain PB that is observed as crystal outside the micelles (Fig. 21f). These experiments demonstrated that the ability of polymer to bind to either metal coordination species or metal ions seems to be an important factor for the formation of the integrated hybrid structures.
![TEM images of (a) [FeII(CN)5]BI and (b) M[FeII(CN)5]BI. (c) High magnification image of FeIII[FeII(CN)5BI] nanoworm. (d) TEM image of FeIII[FeII(CN)6]BI. (e) The chemical structure of the triethylammonium ion functionalized block ionomer (BI*). (f) TEM image when BI* is used instead of BI in the synthesis of PB nanoworms.](/image/article/2011/PY/c1py00283j/c1py00283j-f21.gif) |
| | Fig. 21
TEM images of (a) [FeII(CN)5]BI and (b) M[FeII(CN)5]BI. (c) High magnification image of FeIII[FeII(CN)5BI] nanoworm. (d) TEM image of FeIII[FeII(CN)6]BI. (e) The chemical structure of the triethylammonium ion functionalized block ionomer (BI*). (f) TEM image when BI* is used instead of BI in the synthesis of PB nanoworms. | |
Stuart and co-workers prepared metal coordination nanostructures by mixing aqueous solutions of polyion-neutral diblock copolymers and oppositely charged metal coordination polyelectrolytes.52 The structures were named as complex coacervate core micelles (C3Ms). In their experiments, a solution of zinc coordinated pyridine-2,6-dicarboxylic acid groups based bisligand (Zn-L2EO4) (Scheme 8) was prepared and used as the coordination polyelectrolyte. Upon mixing with poly(2-vinyl-N-methylpyridinium iodide)-b-poly(ethylene oxide) (P2MVP41-b-PEO205), the positively charged P2MVP block interacted with the negatively charged Zn-L2EO4 electrostatically, which provides the driving force for the formation of the self-assembled nano-objects (Scheme 8). No self-assembled structures were formed from the individual constituent components under the same conditions, which further supported that the electrostatic interactions between the two components act as a driving force.
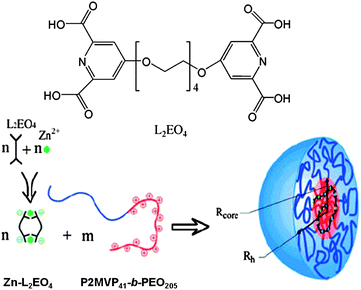 |
| | Scheme 8 The structure of pyridine-2,6-dicarboxylic acid based bisligand (L2EO4) and illustration of the formation of complex coacervate core micelles in the Zn-L2EO4 and P2MVP41-b-PEO205 mixed system. | |
It was found that, at an optimal condition with an equal amount of PMVP41-b-PEO205 and Zn-L2EO4, i.e. f− = 0.5 (f− = [−]/([−] + [+]), [−] and [+] are the molar concentrations of charges for negatively charged Zn-L2EO4 and positively charged P2MVP41-b-PEO205 respectively), spherical objects with 10 nm core and 25–27 nm corona were observed by using cryo-TEM (Fig. 22a) and DLS. By varying the ratio of negatively and positively charged species, they successfully transformed the spherical objects to wormlike structures (Fig. 22b).51 When f− = 0.33, it was found that some wormlike micelles coexisted with spherical micelles. Since their cross-sectional radius is almost the same as that of spherical, it was assumed that the wormlike micelles were aggregates of spherical micelles. A possible mechanism, centering on a ‘bridging’ effect of Zn-L2EO4 complexes between the positively charged pre-C3Ms, has been put forward by the authors. As shown in Scheme 9, in a system (f− = 0.33) containing less amount of negatively charged coordination polymers, positively charged pre-micelle in the solution have to share few [Zn2(L2EO4)]4−, leading to the formation of one-dimensional structures as a result of inter-micelle association.
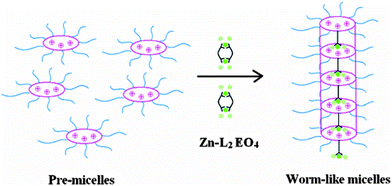 |
| | Scheme 9 Illustration of the bridging effect of Zn-L2EO4 coordination oligomers (small rings) between positively charged pre-micelles. | |
![TEM images for the complex coacervate core micelles assembled from Zn-L2EO4 and P2MVP41-b-PEO205. (a) 0.02% P2MVP41-b-PEO205 with [+] = 0.44 mM, f− = 0.5; (b) 0.04% P2MVP41-b-PEO205 with [+] = 0.87 mM, f− = 0.33. The scaling bar represents 200 nm.](/image/article/2011/PY/c1py00283j/c1py00283j-f22.gif) |
| | Fig. 22
TEM images for the complex coacervate core micelles assembled from Zn-L2EO4 and P2MVP41-b-PEO205. (a) 0.02% P2MVP41-b-PEO205 with [+] = 0.44 mM, f− = 0.5; (b) 0.04% P2MVP41-b-PEO205 with [+] = 0.87 mM, f− = 0.33. The scaling bar represents 200 nm. | |
4. Properties and applications
Developing new concepts for functional nanomaterial design is a major impetus for the researches of block copolymer-assisted hybrid colloid synthesis. In addition to searching synergetic effects arisen from intimate contact of both inorganic/organic components for new properties, flexibility in incorporating various inorganic species such as metal compounds, ions and NPs into block copolymers nanostructures39–43via supramolecular chemistry provides unlimited opportunities for structure variation, including the size and morphology of resulting hybrid colloids. The combination of these structure parameters with the intrinsic properties of each component in the colloids opens unlimited possibilities for new functions applicable for modern technologies, such as biomedicine, electronic, magnetic nanodevices, sensors and stimulated catalysts.53–56,89 Several recent examples of this type of materials prepared via mainly supramolecular routes are highlighted to demonstrate this idea of material development.
4.1
Drug delivery
Carbon monoxide (CO) plays versatile roles in tissue protection via anti-inflammatory, antiproliferative, and antiapoptotic effects,90,91 but difficult to be delivered. By incorporating CO into block copolymer micelles, Hubbell and co-workers recently developed a CO-delivery system. PEG-b-OrnNa-b-nBu triblock copolymers composed of hydrophilic poly(ethylene glycol) (PEG), poly(ornithine acrylamide) (OrnNa) with metal coordination sites, and hydrophobic poly(n-butylacrylamide) (nBu) block was prepared and complexed with [Ru(CO)3Cl2]2, leading to triblocks bearing Ru(CO)3Cl(ornithinate) moieties. The metal coordinated triblock copolymers were able to associate into micelles in water, resulting in hybrid colloids with nBu as cores, middle block containing Ru(CO)3Cl(ornithinate) moieties forming shells and PEO acting as corona (Scheme 10).53 Their sizes varied in the range of 30–40 nm in hydrodynamic diameters. Structure characterization revealed a high CO-loading capacity of the micelles. CO-release studies showed that the micelles were stable in the physiological buffer and serum and released CO in response to thiol-containing compounds such as cysteine, which may be potential candidates to induce CO release in the body. The CO-release of the micelles was slower than that of several transition-metal carbonyl complexes such as Ru(CO)3Cl(glycinate) that have been used as CO-releasing molecules both in vitro and in vivo. Cell viability assays revealed that the micelles also significantly reduced the cytotoxicity of the Ru(CO)3Cl(amino acidate) moiety.
 |
| | Scheme 10 Schematic illustration of micelle formation of PEG-b-OrnRu-b-nBu triblock copolymer and CO release after cellular uptake. | |
4.2 Nanoelectronics
Park and coworkers reported a new practical and robust method for fabricating transparent SWNT/polymer nanocomposites with a very low electric resistance.54 The film was prepared by spin-casting a well-dispersed SWNT solution with self-assembled polystyrene-b-poly(4-vinylpyridine) (PS-b-P4VP) copolymers as a dispersant. Selective incorporation of HAuCl4·3H2O doping agent into P4VP domains did not harm the film transparency, but improved electron conductance significantly. TEM analysis revealed that SWNTs were well dispersed with PS-b-P4VP micelles (Fig. 23a) and Au NPs (reduced from HAuCl4·3H2O) with a diameter of approximately 5 nm were selectively located in the core regions of the micelles (Fig. 23b). The conductance of the thin transparent films could be tuned ranging from 0.01 to approximately order of 1 S cm−1 by controlling the amount of the Au NPs. An OTFT with a pentacene channel and transparent source/drain SWNT/PS-b-P4VP composite films exhibited a good mobility of approximate 0.05 cm2 V−1s−1 with an on/off ratio of approximately 105, suggesting the nanocomposite films are potential useful as electrodes with either folding or rolling capability.92
 |
| | Fig. 23
Bright-field TEM images of SWNTs dispersed and stabilized by (a) PS-b-P4VP copolymer, and (b) PS-b-P4VP with the maximum doping of HAuCl4·3H2O with respect to P4VP domains. The insets of (a) and (b) show a schematic illustration of SWNT stabilization by PS-b-P4VP and a high-resolution (HR) TEM image of a micelle adhering to the surface of nanotubes with the reduced Au NPs, respectively. | |
4.3 Magnetic recording materials
Dai and coworkers synthesized monodisperse PAA-b-PS block copolymer stabilized cobalt ferrite NPs (CoFe2O4) using solution-based methods and then stabilized in THF solution using PAA-b-PS (Scheme 11).55 The acid groups of the acrylate block bound the polymer to the surface of NP via multivalent interactions, while the styrene block afforded the magnetic NP–polymer complex solubility in organic solvents such as cyclohexane, THF and toluene. The incorporation of block copolymers also prevents the ferrimagnetic NPs (FMNP) from irreversible aggregation by reducing the strong interparticle magnetic interactions. Thin film, assembled from the hybrid magnetic colloids, was prepared by spin coating from a toluene solution onto a silicon wafer.
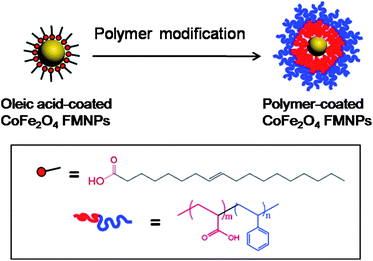 |
| | Scheme 11 Schematic representation of PAA-b-PS diblock copolymer modification of 18 nm oleic acid-coated CoFe2O4 FMNPs. | |
High glass transition temperature (Tg) value of PS chain allows the particle being fixed at room temperature and mobile at a temperature above the Tg. By taking advantage of this property, they developed a thermal approach for the realignment of the magnetic axis of the individual FMNP. When the sample was heated to 378 K (above the Tg of polystyrene) and simultaneously subjected to a magnetic field, the FMNPs physically reoriented to align themselves in the direction of the applied magnetic field (Fig. 24a). As indicated from magnetic measurement, the squareness of the in-plane hysteresis loops of the self-assembled composite films increased following the magnetic alignment process (Fig. 24b). The normalized magnetic remanence increased from 0.5 to 0.85 after the as-cast composite film was heated at 378 K in a magnetic field (2800 G), suggesting that the FMNPs were physically reoriented to align themselves in the direction of the magnetic field. Control experiments using polymer coated NPs at an alignment temperature lower than the Tg of PS or using oleic acid-coated CoFe2O4 FMNP films (without block copolymers) but at an alignment temperature higher than the Tg of PS were performed. In both cases, the magnetic remanence and squareness of the samples did not change (Fig. 24c), which confirms that the polymer shell is necessary to provide the particles with mobility when the sample is simultaneously heated and exposed to an external magnetic field. Because the relative orientation of the magnetic easy axis of the individual magnetic NPs is essential for magnetic recording properties, the discovered magnetic thermal responsive properties of the film are particularly useful for functional magnetic storage. Demonstrations with a contact magnetic tester show that the FMNP–polymer composite was suitable as a self-assembled magnetic recording media.
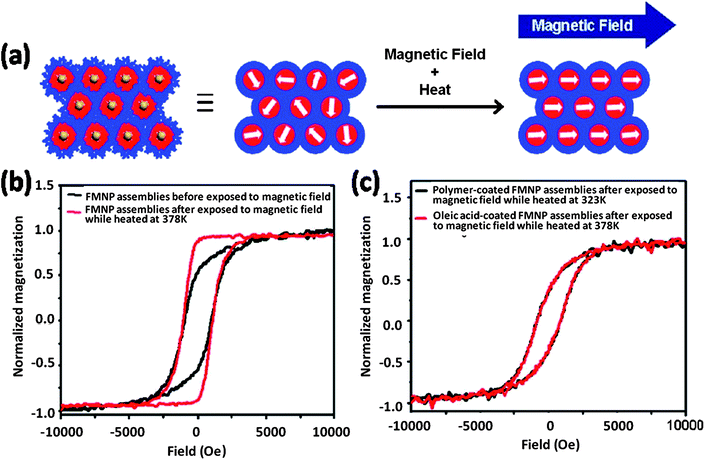 |
| | Fig. 24 (a) A schematic representation of the magnetic alignment process for the assembly of CoFe2O4 FMNPs modified with PAA33-b-PS340. (b) The hysteresis loops (measured by VSM, 298 K) of the FMNP–polymer composite before (black line) and after (red line) magnetic alignment at 378 K. (c) Control experiments showing the hysteresis curves (measured by VSM, 298 K) of the FMNP–polymer composite heated to 323 K while exposed to a magnetic field (black line), and the oleic acid-coated CoFe2O4 FMNPs exposed to a magnetic field and heated at 378 K. A 2800 G magnetic field was applied during alignment. | |
4.4 Responsive nanocatalysts
Another promising application for inorganic/organic hybrid colloids is to develop catalysts with special catalyzing behaviour in reactivity, stability, and selectivity.93–97 Zhang and co-workers synthesized Au NPs inside the core of thermoresponsive micelles of poly(N-isopropylacrylamide)-b-poly(4-vinyl pyridine) (PNIPAM-b-P4VP).98 Temperature responsive solubility of PNIPAM corona chains was used to adjust the catalytic activity. Below LCST, the PNIPAM chains were hydrophilic and extended which exposed the Au NPs to the hydrophilic reactants and accelerated the reaction. On the other hand, above LCST, the solubility of PNIPAM chains decreased and the Au NPs were covered by the collapsed PNIPAM chains, which decelerate the reaction.
Taking advantages of recent progress in the synthesis of well-defined boron-containing polymer,99–102 Hest and co-workers prepared polymer vesicles (polymersomes) using PEG-b-PS in the presence of small amount of boronic acid containing block copolymers of poly(ethylene glycol)-b-poly(styrene boronic acid) (PEG-b-PSBA).56 The nonpolar PS blocks from PEO-b-PS and the polar PSBA from PEO-b-PSBA hydrophobically associated together, forming the membrane of the polymersomes. At high pH or in the presence of sugar molecules, the solubility of the PSBA block in water increased. Subsequently, PEO-b-PSBA was extracted from the membrane of the polymersomes, generating pores on the polymersome walls (Scheme 12). Using this co-assembly approach, polymersomes with stimulated permeability has been successfully achieved. This type of polymersomes was used to encapsulate enzymes of Candida Antarctica Lipase B (CALB). As a result, the catalytic behavior of the enzymes can be manipulated. For example, the encapsulated enzymes did not show catalytic activity. After removal of PEO-b-PSBA, the membrane is permeable and the enzyme become available for catalytic reactions. The permeability, determined by the amount of PEO-b-PSBA in the hybrid colloids, could be used to adjust the activity of the enzymes.
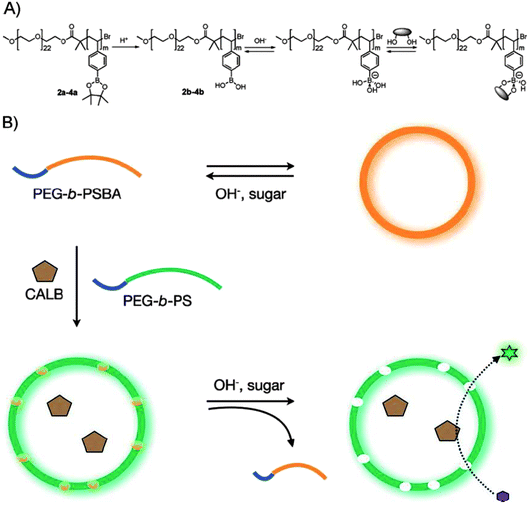 |
| | Scheme 12 (a) Molecular structure of block copolymers, PEG-b-PSBA, and their equilibrium with sugar molecules in a basic aqueous phase. Complete ionization of boronic acid groups was assumed for simplicity. (b) Schematic representation of the formation of bioreactors with a permeable membrane utilizing the sugar responsiveness of the block copolymers. | |
5. Conclusions and outlook
Building on well-studied block copolymer self-assembly behavior, block copolymer-assisted synthesis of inorganic/organic supramolecular hybrid colloids has been initiated. Through either concurrent or sequential assembling, second components could be readily incorporated into the resulting hybrid colloids, leading to a number of new types of hybrid colloids with spatial control of each components, novel morphologies and unique properties. However, although many interesting supramolecular behaviour has been identified through experimental exploration, explicitly understanding these experiments usually is difficult as the systems involve multiple non-covalent interactions with varied strengths. To achieve designed synthesis, it is essential to manipulate these forces working co-operatively, sequentially or orthogonally. Therefore, how to thermodynamically or kinetically control inter- or intra-molecular forces in the systems is no doubt a central concern of future research for a desired breakthrough of this area. In the process of this research, it is important to take advantages of current progress in block copolymer synthesis and self-assembly. As a matter of fact, the developed knowledge of block copolymer supramolecular chemistry has been commonly used to help us explain experimental results or to design multicomponent systems for investigation. In this context, it is worth to note that block copolymer containing inorganic elements often possess unusual supramolecular behaviour.30,31,70,101,102 Involving this type of block copolymers as building blocks in the study will not only extend the scope of research, but also bring the functions associated with inorganic elements into hybrid colloids. As for the choice of the second components, there is virtually no limitation. Metal NPs are commonly used due to well-developed particle synthetic chemistry. Other species including metal atoms,41 ions43 and organometallics45,46,49–52 are highly encouraged in an attempt to build up supramolecular knowledge and develop novel materials. Current efforts from our group and others to introduce block copolymer self-assembly into metal coordination polymer synthesis is a good example, which, in fact, has opened up possibilities for designed synthesis of metal coordination NPs with desired functions.53–56 Finally, although it is still a long way to develop effective strategies for rational synthesis of hybrid colloids using block copolymers, several exciting novel materials have been realized and highlighted in the article. These discoveries will foster application-orientated researches which are also important and essential to bridge fundamental supramolecular studies and the exploration of highly demanded material for modern technologies.
Acknowledgements
The authors would like to thank The Engineering and Physical Sciences Research Council, UK (EPSRC), Natural Sciences and Engineering Research Council of Canada (NSERC) and the University of Waterloo for financial support. X.S.W. thanks the EPSRC for the award of a Roberts Fellowship.
References
-
P. Alexandridis and B. Lindman, Amphiphilic Block Copolymers: Self-Assembly and Applications, Elsevier, Amsterdam, 2000 Search PubMed.
-
I. W. Hamley, The Physics of Block Copolymers, Oxford University Press, 1998 Search PubMed.
- J. Rodríguez-Hernández, F. Chécot, Y. Gnanou and S. Lecommandoux, Prog. Polym. Sci., 2005, 30, 691–724 CrossRef.
- A. Blanazs, S. P. Armes and A. J. Ryan, Macromol. Rapid Commun., 2009, 30, 267–277 CrossRef CAS.
- G. Riess, Prog. Polym. Sci., 2003, 28, 1107–1170 CrossRef CAS.
- J. A. Hanson, C. B. Chang, S. M. Graves, Z. Li, T. G. Mason and T. J. Deming, Nature, 2008, 455, 85–88 CrossRef CAS.
- A. Muñoz-Bonilla, A. M. v. Herk and J. P. A. Heuts, Macromolecules, 2010, 43, 2721–2731 CrossRef.
- W. Li, K. Min, K. Matyjaszewski, F. Stoffelbach and B. Charleux, Macromolecules, 2008, 41, 6387–6392 CrossRef CAS.
- Z. Sun, F. Bai, H. Wu, S. K. Schmitt, D. M. Boye and H. Fan, J. Am. Chem. Soc., 2009, 131, 13594–13595 CrossRef CAS.
- H. Cui, Z. Chen, S. Zhong, K. L. Wooley and D. J. Pochan, Science, 2007, 317, 647–650 CrossRef CAS.
- D. J. Pochan, Z. Chen, H. Cui, K. Hales, K. Qi and K. L. Wooley, Science, 2004, 306, 94–97 CrossRef CAS.
- C. Cai, J. Lin, T. Chen, X.-S. Wang and S. Lin, Chem. Commun., 2009, 2709–2711 RSC.
- H. Cölfen and S. Mann, Angew. Chem., Int. Ed., 2003, 42, 2350–2365 CrossRef.
- M. Lazzari and M. A. López-Quintela, Adv. Mater., 2003, 15, 1583–1594 CrossRef CAS.
- D. Astruc, F. Lu and J. R. Aranzaes, Angew. Chem., Int. Ed., 2005, 44, 7852–7872 CrossRef CAS.
- A. Böker, J. He, T. Emrick and T. P. Russell, Soft Matter, 2007, 3, 1231–1248 RSC.
- T. Jamieson, R. Bakhshi, D. Petrova, R. Pocock, M. Imani and A. M. Seifalian, Biomaterials, 2007, 28, 4717–4732 CrossRef CAS.
- J. H. Park, S. Lee, J.-H. Kim, K. Park, K. Kim and I. C. Kwon, Prog. Polym. Sci., 2008, 33, 113–137 CrossRef.
- Y. H. Kim, D. K. Yoon and H.-T. Jung, J. Mater. Chem., 2009, 19, 9091–9102 RSC.
- J. Liu, J. Liu, L. Yang, X. Chen, M. Zhang, F. Meng, T. Luo and M. Li, Sensors, 2009, 9, 7343–7364 CrossRef CAS.
- S. Mann, Nat. Mater., 2009, 8, 781–792 CrossRef CAS.
- W. Schärtl, Nanoscale, 2010, 2, 829–843 RSC.
- M. Möller and J. P. Spatz, Curr. Opin. Colloid Interface Sci., 1997, 2, 177–187 CrossRef.
- M. Antonietti, E. Wenz, L. Bronstein and M. Seregina, Adv. Mater., 1995, 7, 1000–1005 CrossRef CAS.
- E. D. Sone and S. I. Stupp, J. Am. Chem. Soc., 2004, 126, 12756–12757 CrossRef CAS.
- H.-D. Koh, S. Park and T. P. Russell, ACS Nano, 2010, 4, 1124–1130 CrossRef CAS.
- H. Wang, X. Wang, M. A. Winnik and I. Manners, J. Am. Chem. Soc., 2008, 130, 12921–12930 CrossRef CAS.
- J.-J. Yuan, O. O. Mykhaylyk, A. J. Ryan and S. P. Armes, J. Am. Chem. Soc., 2007, 129, 1717–1723 CrossRef CAS.
- A. Khanal, Y. Inoue, M. Yada and K. Nakashima, J. Am. Chem. Soc., 2007, 129, 1534–1535 CrossRef CAS.
- J. B. Gilroy, T. Gädt, G. R. Whittell, L. Chabanne, J. M. Mitchels, R. M. Richardson, M. A. Winnik and I. Manners, Nat. Chem., 2010, 2, 566–570 CrossRef CAS.
- G. R. Whittell and I. Manners, Adv. Mater., 2007, 19, 3439–3468 CrossRef CAS.
- X. Wang and R. McHale, Macromol. Rapid Commun., 2010, 31, 331–350 CrossRef CAS.
- Y. Kang and T. A. Taton, Angew. Chem., Int. Ed., 2005, 44, 409–412 CrossRef CAS.
- Y. Kang and T. A. Taton, Macromolecules, 2005, 38, 6115–6121 CrossRef CAS.
- B.-S. Kim and T. A. Taton, Langmuir, 2007, 23, 2198–2202 CrossRef CAS.
- T. Chen, M. Yang, X. Wang, L. H. Tan and H. Chen, J. Am. Chem. Soc., 2008, 130, 11858–11859 CrossRef CAS.
- M. Wang, S. Kumar, A. Lee, N. Felorzabihi, L. Shen, F. Zhao, P. Froimowicz, G. D. Scholes and M. A. Winnik, J. Am. Chem. Soc., 2008, 130, 9481–9491 CrossRef CAS.
- J. Xu, H. Wang, C. Liu, Y. Yang, T. Chen, Y. Wang, F. Wang, X. Liu, B. Xing and H. Chen, J. Am. Chem. Soc., 2010, 132, 11920–11922 CrossRef CAS.
- Y. Mai and A. Eisenberg, J. Am. Chem. Soc., 2010, 132, 10078–10084 CrossRef CAS.
- R. J. Hickey, A. S. Haynes, J. M. Kikkawa and S.-J. Park, J. Am. Chem. Soc., 2011, 133, 1517–1525 CrossRef CAS.
- T. Tang, T. Metanawin, A. Hebden, P. McGowan and X.-S. Wang, Chem. Commun., 2010, 46, 6663–6665 RSC.
- H. Wang, W. Lin, K. P. Fritz, G. D. Scholes, M. A. Winnik and I. Manners, J. Am. Chem. Soc., 2007, 129, 12924–12925 CrossRef CAS.
- D. A. Christian, A. Tian, W. G. Ellenbroek, I. Levental, K. Rajagopal, P. A. Janmey, A. J. Liu, T. Baumgart and D. E. Discher, Nat. Mater., 2009, 8, 843–849 CrossRef CAS.
- D. J. Pochan, Nat. Mater., 2009, 8, 773–774 CrossRef CAS.
- G. Liang, J. Xu and X. Wang, J. Am. Chem. Soc., 2009, 131, 5378–5379 CrossRef CAS.
- R. McHale, N. Ghasdian, Y. Liu, M. B. Ward, N. S. Hondow, H. Wang, Y. Miao, R. Brydson and X. Wang, Chem. Commun., 2010, 46, 4574–4576 RSC.
- R. McHale, N. Ghasdian, Y. Liu, H. Wang, Y. Miao and X. Wang, Macromol. Rapid Commun., 2010, 31, 856–860 CrossRef CAS.
- S. Ye, Y. Liu, S. Chen, S. Liang, R. McHale, N. Ghasdian, Y. Lu and X. Wang, Chem. Commun., 2011, 47, 6831–6833 RSC.
- X. Roy, J. K.-H. Hui, M. Rabnawaz, G. Liu and M. J. MacLachlan, Angew. Chem., Int. Ed., 2011, 50, 1597–1602 CrossRef CAS.
- X. Roy, J. K.-H. Hui, M. Rabnawaz, G. Liu and M. J. MacLachlan, J. Am. Chem. Soc., 2011, 133, 8420–8423 CrossRef CAS.
- Y. Yan, N. A. M. Besseling, A. D. Keizer, M. Drechsler, R. Fokkink and M. A. C. Stuart, J. Phys. Chem. B, 2007, 111, 11662–11669 CrossRef CAS.
- Y. Yan, N. A. M. Besseling, A. D. Keizer, A. T. M. Marcelis, M. Drechsler and M. A. C. Stuart, Angew. Chem., Int. Ed., 2007, 46, 1807–1809 CrossRef CAS.
- U. Hasegawa, A. J.v. d. Vlies, E. Simeoni, C. Wandrey and J. A. Hubbell, J. Am. Chem. Soc., 2010, 132, 18273–18280 CrossRef CAS.
- J. Sung, P. S. Jo, H. Shin, J. Huh, B. G. Min, D. H. Kim and C. Park, Adv. Mater., 2008, 20, 1505–1510 CrossRef CAS.
- Q. Dai, D. Berman, K. Virwani, J. Frommer, P.-O. Jubert, M. Lam, T. Topuria, W. Imaino and A. Nelson, Nano Lett., 2010, 10, 3216–3221 CrossRef CAS.
- K. T. Kim, J. J. L. M. Cornelissen, R. J. M. Nolte and J. C. M.v. Hest, Adv. Mater., 2009, 21, 2787–2791 CrossRef CAS.
- Y. Kang and T. A. Taton, J. Am. Chem. Soc., 2003, 125, 5650–5651 CrossRef CAS.
- C. S. T. Laicera, R. A. Mrozeka and T. A. Taton, Polymer, 2007, 48, 1316–1328 CrossRef.
- Y. Shibasaki, B.-S. Kim, A. J. Young, A. L. McLoon, S. C. Ekker and T. A. Taton, J. Mater. Chem., 2009, 19, 6324–6327 RSC.
- K. R. Brown, D. G. Walter and M. J. Natan, Chem. Mater., 2000, 12, 306–313 CrossRef CAS.
- Y. Yu, L. Zhang and A. Eisenberg, Macromolecules, 1998, 31, 1144–1154 CrossRef CAS.
- H. Feng, Y. Yang, Y. You, G. Li, J. Guo, T. Yu, Z. Shen, T. Wu and B. Xing, Chem. Commun., 2009, 1984–1986 RSC.
- L. Bai, X. Ma, J. Liu, X. Sun, D. Zhao and D. G. Evans, J. Am. Chem. Soc., 2010, 132, 2333–2337 CrossRef CAS.
- M. Wang, N. Felorzabihi, G. Guerin, J. C. Haley, G. D. Scholes and M. A. Winnik, Macromolecules, 2007, 40, 6377–6384 CrossRef CAS.
- M. Zhang, M. Wang, S. He, J. Qian, A. Saffari, A. Lee, S. Kumar, Y. Hassan, A. Guenther, G. Scholes and M. A. Winnik, Macromolecules, 2010, 43, 5066–5074 CrossRef CAS.
- R. B. Cheyne and M. G. Moffitt, Langmuir, 2005, 21, 10297–10300 CrossRef CAS.
- X.-S. Wang, T. Metanawin, X.-Y. Zheng, P.-Y. Wang, M. Ali and D. Vernon, Langmuir, 2008, 24, 9230–9232 CrossRef CAS.
- T. Metanawin, T. Tang, R. Chen, D. Vernon and X. Wang, Nanotechnology, 2011, 22, 235604 CrossRef.
- X. Wang, G. Guerin, H. Wang, Y. Wang, I. Manners and M. A. Winnik, Science, 2007, 317, 644–647 CrossRef CAS.
- F. He, T. Gädt, I. Manners and M. A. Winnik, J. Am. Chem. Soc., 2011, 133, 9095–9103 CrossRef CAS.
- T. Gädt, N. S. Ieong, G. Cambridge, M. A. Winnik and I. Manners, Nat. Mater., 2009, 8, 144–150 CrossRef.
- H. Wang, A. J. Patil, K. Liu, S. Petrov, S. Mann, M. A. Winnik and I. Manners, Adv. Mater., 2009, 21, 1805–1808 CrossRef CAS.
- H. Wang, M. A. Winnik and I. Manners, Macromolecules, 2007, 40, 3784–3789 CrossRef CAS.
- N. R. Jana and X. Peng, J. Am. Chem. Soc., 2003, 125, 14280–14281 CrossRef CAS.
- A. M. Kalsin, M. Fialkowski, M. Paszewski, S. K. Smoukov, K. J. M. Bishop and B. A. Grzybowski, Science, 2006, 312, 420–424 CrossRef CAS.
- M. A. Hines and G. D. Scholes, Adv. Mater., 2003, 15, 1844–1849 CrossRef CAS.
-
W. J. Lin, MSc thesis, University of Toronto, 2007.
- E. Lee, B. Hammer, J.-K. Kim, Z. Page, T. Emrick and R. C. Hayward, J. Am. Chem. Soc., 2011, 133, 10390–10393 CrossRef CAS.
- W. Lin, W. J. Rieter and K. M. L. Taylor, Angew. Chem., Int. Ed., 2009, 48, 650–658 CrossRef CAS.
- A. Y. Robin and K. M. Fromm, Coord. Chem. Rev., 2006, 250, 2127–2157 CrossRef CAS.
- W. L. Leong and J. J. Vittal, Chem. Rev., 2011, 111, 688–764 CrossRef CAS.
-
S. R. Batten, S. M. Neville and D. R. Turner, Coordination polymers: Design, Analysis and Application, Royal Society of Chemistry, Cambridge, 2008 Search PubMed.
- R. McHale, N. Ghasdian, N. S. Hondow, P. M. Richardson, A. M. Voice, R. Brydson and X. Wang, Macromolecules, 2010, 43, 6343–6347 CrossRef CAS.
- R. McHale, Y. Liu, N. Ghasdian, N. S. Hondow, S. Ye, Y. Lu, R. Brydson and X. Wang, Nanoscale, 2011 Search PubMed , in press.
- N. D. L.d. Katz and N. E. Katz, Monatsh. Chem., 1982, 113, 745–750 CrossRef.
-
J. Z. Zhang, Optical Properties and Spectroscopy of Nanomaterials, World Scientific Publishing Co. Pte. Ltd, 2009 Search PubMed.
- M. Yamada, N. Ohnishi, M. Watanabe and Y. Hino, Chem. Commun., 2009, 7203–7205 RSC.
- Y. Zhang, Y. Wen, Y. Liu, D. Li and J. Li, Electrochem. Commun., 2004, 6, 1180–1184 CrossRef CAS.
- M. Prabaharan, J. J. Grailer, S. Pilla, D. A. Steeber and S. Gong, Biomaterials, 2009, 30, 6065–6075 CrossRef CAS.
- S. W. Ryter and L. E. Otterbein, BioEssays, 2004, 26, 270–280 CrossRef CAS.
- S. W. Ryter, J. Alam and A. M. K. Choi, Physiol. Rev., 2006, 86, 583–650 CrossRef CAS.
-
G. P. Crawford, Flexible Flat Panel Displays, Wiley, 2005 Search PubMed.
- Y. Yan and J. Huang, Coord. Chem. Rev., 2010, 254, 1072–1080 CrossRef CAS.
- G. Feng, Y. Jia, L. Liu, W. Chang and J. Li, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 5992–6002 CrossRef CAS.
- X. Wang, G. Li, T. Chen, M. Yang, Z. Zhang, T. Wu and H. Chen, Nano Lett., 2008, 8, 2643–2647 CrossRef CAS.
- F. Wen, W. Zhang, G. Wei, Y. Wang, J. Zhang, M. Zhang and L. Shi, Chem. Mater., 2008, 20, 2144–2150 CrossRef CAS.
- N. Yan, J. Zhang, Y. Yuan, G.-T. Chen, P. J. Dyson, Z.-C. Li and Y. Kou, Chem. Commun., 2010, 46, 1631–1633 RSC.
- Y. Wang, G. We, F. Wena, X. Zhang, W. Zhang and L. Shi, J. Mol. Catal. A: Chem., 2008, 280, 1–6 CrossRef CAS.
- Y. Qin, G. Cheng, A. Sundararaman and F. Jäkle, J. Am. Chem. Soc., 2002, 124, 12672–12673 CrossRef CAS.
- F. Jäkle, Chem. Rev., 2010, 110, 3985–4022 CrossRef.
- C. Cui, E. M. Bonder and F. Jäkle, J. Am. Chem. Soc., 2010, 132, 1810–1812 CrossRef CAS.
- F. Cheng and F. Jäkle, Polym. Chem., 2011 10.1039/c1031py00123j.
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here.