Initiators for the stereoselective ring-opening polymerization of meso-lactide
Jean-Charles
Buffet
and
Jun
Okuda
*
Institut für Anorganische Chemie, RWTH Aachen, Landoltweg 1, D-52056, Aachen, Germany. E-mail: jun.okuda@ac.rwth-aachen.de; Fax: +49 241 80 94645
First published on 18th August 2011
Abstract
Poly(lactides) (PLAs), or poly(lactic acid)s, are among the first commercial biodegradable polymers that have the potential to become commodity plastics. meso-Lactide, a by-product of L-lactide production, will become more easily available. Highly stereoregular poly(meso-lactides) should be crystalline and thus interesting as polymeric material. In this short review, initiators capable of inducing the stereoselective ring-opening polymerization of meso-lactide to ideally give syndiotactic or heterotactic PLAs will be discussed. Mechanistic discussions with regard to understanding the reactivity differences between the various lactide monomers are included.
Introduction
Poly(lactic acid)s (PLAs) are biodegradable and biocompatible polymers and have been intensely studied in recent years.1 Applications include their use as biomedical and pharmaceutical materials.1c,d Currently their use as commodity polymers is under scrutiny, since lactide monomer, the cyclic diester of lactic acid, is derived from biomass and can be regarded as “carbon-neutral”. As polycondensation of lactic acid is not practical, ring-opening polymerization (ROP) of lactide monomer evolved as the most efficient method for production. The two stereogenic centers in one lactide (LA) molecule result in three distinct configurational isomers (S,S)-LA (L-LA), (R,R)-LA (D-LA) and (R,S)-LA (meso-LA). A 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of (S,S)-LA and (R,R)-LA is referred to as rac-LA (Scheme 1).2
:1 mixture of (S,S)-LA and (R,R)-LA is referred to as rac-LA (Scheme 1).2
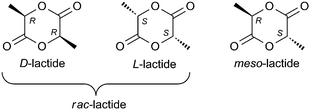 | ||
| Scheme 1 Lactide monomers. | ||
The physical and mechanical properties of any polymeric material critically depend on the microstructure (tacticity) of the main chain. Polymers that have stereocenters in the repeat unit can exhibit two structures of maximum order; isotactic (from L-, D- or rac-LA) and syndiotactic (from meso-lactide). Adjacent stereocenters of isotactic polymers are of the same relative stereochemistry, whereas those of syndiotactic polymers are of opposite relative configuration. Due to their stereoregularity, isotactic (Tm > 180 °C) and syndiotactic polymers (Tm = 152 °C) are crystalline, an important feature for many applications. Heterotactic PLAs (obtained from rac- or meso-LA) have been reported to be amorphous and to show no melting temperature so far. Mixtures of poly(L-lactide), PLLA, and poly(D-lactide), PDLA, forming stereocomplexes (with Tm values up to 220 °C) are currently at the center of focus.3
Stereoregular PLA materials can be prepared from meso-LA by using metal complexes as single-site initiators. Two different mechanisms are conceivable. One is chain-end-control, where the insertion of the incoming meso-LA monomer is determined by the stereogenic center in the last repeating unit in the propagating chain. If the stereogenic center in the last unit is repeated, syndiotactic PLA will be obtained. If the stereogenic center of the last unit favors alternating enchainment through the opposite stereogenic center, heterotactic PLA will result. The other mechanism is enantiomorphic site-control where the insertion of the incoming meso-LA monomer is controlled by the ligand sphere. In this case, syndiotactic PLA will be obtained from meso-LA (Scheme 2).
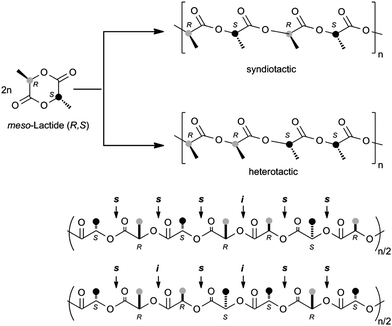 | ||
| Scheme 2 Structures of PLAs synthesized from meso-lactide. | ||
In this review, we will summarize the current status of the synthesis of poly(meso-lactide) by ring-opening polymerization using single-site metal initiators.
Early studies
Kricheldorf et al. were the first to use soluble metal complexes for the ring-opening polymerization of meso-lactide.4Aluminium tri(isopropoxide) Al(OiPr)3, AlEt3/neopentyl alcohol, AlEt3/(+)-menthol and methylalumoxane were shown to polymerize meso-lactide in xylene solutions at 120 °C. meso-Lactide was polymerized at slower rates compared to rac-lactide.4a This was rationalized by suggesting higher stability of meso-lactide with respect to rac-lactide due to the trans disposition of the two methyl groups. When the time of the polymerization of meso-lactide was increased, the polymer stereochemistry was affected. Due to transesterifications (“back-biting”) the iis/siitetrad fraction increased after 48 h.Di(n-butyl)magnesium polymerized meso-lactide in toluene at room temperature with full conversion reached after 8 days resulting in nearly atactic polylactides.4b
Zinc, lead, antimony, or bismuth compounds were also used as initiators in the ring-opening polymerization of meso-lactide at 120 °C in xylene.4c The polymerizations using lead oxide and bismuth(2-ethylhexanoate) showed high conversion but resulted in low molecular weight polymers. When zinc stearate and antimony (2-ethylhexanoate) were used, meso-lactide was polymerized faster than rac-lactide to afford PLAs in similar yields and inherent viscosities.
Aluminium initiators
Inspired by the pioneering work of Spassky et al. on the isospecific polymerization of rac-lactide,5 a breakthrough in the stereoselective polymerization of meso-lactide was reported by Ovitt and Coates in 1999 with the use of an aluminium isopropoxide complex ((R)-1, Scheme 3).6 The enantiopure complex, (R)-1, achieved 94% conversion at 70 °C after 40 h (Mn,exp = 12030 g mol−1 and Mw/Mn = 1.05). Due to its high degree of stereoregularity, the PLA with the highest level of syndiotacticity (96%) exhibited crystallinity with a melting temperature, Tm = 152 °C, following annealing at 95 °C for 60 min (Tg = 38.1 °C). Interestingly, rac-1 showed 98% conversion under similar conditions, but produced an amorphous polymer (Mn,exp = 13600 g mol−1 and Mw/Mn = 1.07) with glass transition temperature Tg = 43.2 °C.7 The most notable feature of the reaction using the racemic catalyst is that the polymer was heterotactic (where 80% of the linkages formed during the polymerization occurred between lactate units of the identical stereochemistry). A polymer exchange mechanism was proposed to account for the formation of this heterotactic microstructure, whereby each individual polymer chain effectively switches between enantiomeric aluminium centers before insertion of a subsequent monomer unit.
 | ||
| Scheme 3 Aluminium initiators for the polymerization of meso-lactide. | ||
Feijen et al. showed that meso-lactide polymerization using chiral aluminium salan complexes in the presence of isopropanol (2a–c, Scheme 3) occurred in a controlled fashion (Mn,exp = 6000–7000 g mol−1 and Mw/Mn = 1.09–1.12), resulting in syndiotactically biased PLAs (Ps = 0.64, 0.70 and 0.69, respectively) (Table 1).8
They also demonstrated that achiral aluminium salen complexes (3a–c, Scheme 3) gave atactic polymers (Ps = 0.56, 0.57 and 0.53, respectively) in the presence of isopropanol.9 The minor difference in the tacticity of these polymers suggests that substituents on the ligand have little effect on the chain-end control during meso-lactide polymerization. In all cases, the polymerization rates for meso-lactide were lower than those for rac-lactide.
Group 3 metal initiators
Carpentier et al. reported 97% conversion of meso-lactide after 0.5 h at 20 °C using the amine bis(phenolate) yttrium complex 4 (Scheme 4).10 Analysis of the polymer microstructure revealed that initiator 4 formed syndiotactically enriched PLA (Ps = 0.75). | ||
| Scheme 4 Group 3 metal initiators for the polymerization of meso-lactide. | ||
Initiators based on group 3 bis(phenolate) complexes 5a–f (Scheme 4), active in heteroselective ROP of rac-LA, were found to polymerize meso-lactide, producing highly syndiotactic PLAs.11 These complexes contain a “temporary” chiral reaction site that apparently allows for selective ring-opening of one of the diastereotopic ester functions.11 The C3-linked complex 5e gave the highest syndiotacticity (Ps = 0.90) when the tert-butylortho-substituents complexes 5a, 5c, and 5f are compared with respect to their ROP activity. Fast conversion and high syndiotacticity were achieved using 5b and 5d, C2-bridged scandium derivative with ortho-cumyl (CMe2Ph) substituents in the phenoxy group (Ps > 0.92). Apparently, complexes with bulkier orthocumyl substituents showed better control over syndiotacticity than those with tert-butyl substituents.
Polymerizations of meso-lactide in melt (60–130 °C) using scandium complex 5f were rapid (50% monomer conversion in less than 10 min) and controlled. Tacticity control (Ps > 0.83) was as high as that observed in solution. The scandium complex 5d showed complete conversion within 30 min (Ps = 0.92), whereas the homologous yttrium complex led to 79% conversion (Ps = 0.71) (Table 2).11
| Initiator | [Initiator]0/mol L−1 | [mon.]0/[init.]0 | % Conv. | P s | Ref. |
|---|---|---|---|---|---|
| a Polymerization conditions: 30 min, THF, 20 °C. b Polymerization conditions: 30 min, toluene, 25 °C. | |||||
| 4 | 0.004a | 100 | 97 | 0.75 | 10 |
| 5a | 0.002b | 100 | 99 | 0.88 | 11 |
| 5b | 0.002b | 100 | 88 | 0.93 | 11 |
| 5c | 0.002b | 100 | 99 | 0.89 | 11 |
| 5d | 0.002b | 100 | 99 | 0.92 | 11 |
| 5e | 0.002b | 100 | 99 | 0.90 | 11 |
| 5f | 0.002b | 100 | 99 | 0.89 | 11 |
| 6 | 0.002b | 100 | 99 | 0.72 | 13 |
| 7 | 0.002b | 100 | 99 | 0.73 | 13 |
The use of scandium amide complexes containing cyclen-derived (NNNN)-type macrocycle ligands 6 and 7 for meso-lactide polymerization was reported (Scheme 4). Complexes 6 and 7 were fast (full conversion at room temperature in less than 30 min), giving syndiotactic PLA (Ps = 0.73) with molecular weight efficiency values12 of 3.81 for 6 (Mn,exp = 46000–55000 g mol−1) and 0.85 for 7 (Mn,exp = 12250–15000 g mol−1).13
Indium initiators
Tolman et al. reported the use of a mixture of InCl3 in the presence of benzyl alcohol and triethylamine for the polymerization of meso-lactide.14 The polymerizations were fast; full conversion reached after 30 h at 0 °C and 5 h at 25 °C. The polylactides obtained at 0 °C were mainly atactic (Ps < 0.62). Using well-characterized indium initiators with a zwitterionic structure [{InCl3(3-diethylamino-1-propanol)(H2O)}2] (8, Scheme 5), they have shown a lack of stereoselectivity in the polymerization of meso-lactide.15 | ||
| Scheme 5 Indium initiators for the polymerization of meso-lactide. | ||
Indium bis(phenolate) complex bearing tert-butylortho-substituent (9, Scheme 5) gave highly syndiotactic PLA with low polydispersity (Ps = 0.93 and Mw/Mn = 1.05) at room temperature, with full conversion reached after 16 h (Table 3).11
| Initiator | [Initiator]0/mol L−1 | [mon.]0/[init.]0 | Time/h | % Conv. | P s | Ref. |
|---|---|---|---|---|---|---|
| a Polymerization conditions: [InCl3]0/[PhCH2OH]0 = 1, dichloromethane and triethylamine, 0 °C. b Polymerization conditions: [InCl3]0/[PhCH2OH]0 = 1, dichloromethane and triethylamine, 25 °C. c Polymerization conditions: [InCl3]0/[PhCH2OH]0 = 1, melt and triethylamine. d Polymerization conditions: toluene, 25 °C. | ||||||
| InCl3 | 0.276a | 100 | 30 | >99 | 0.62 | 14 |
| InCl3 | 0.276b | 100 | 5 | >99 | 0.56 | 14 |
| InCl3 | 0.276c | 50 | 5 | >99 | 0.44 | 14 |
| 9 | 0.520d | 100 | 16 | >99 | 0.93 | 11 |
Group 4 metal initiators
Heteroselective group 4 metal initiators containing a 1,ω-dithiaalkanediyl-bridged bis(phenolato) ligand with bulky groups at the ortho-substituent R were obtained from [M(OR′)4] (M = Ti or Zr, and R′ = iPr or tBu) (10a–e, Scheme 6).16 When complexes 10a, 10d, and 10e with tert-butylortho-substituents are compared with respect to their ROP activity, the C3-linked complex 10e gave the highest conversion, under the same polymerization conditions (38% for 10a, 8% for 10d and 71% for 10e, Table 4), indicating the importance of a flexible backbone. It is noteworthy that the rigid trans-1,2-cyclohexanediyl backbone (complex 10d) gave only 8% conversion after 24 h at 100 °C. | ||
| Scheme 6 Group 4 metal initiators for the polymerization of meso-lactide. | ||
| Initiator | [Initiator]0/mol L−1 | [mon.]0/[init.]0 | Time/h | % Conv. | P s | Ref. |
|---|---|---|---|---|---|---|
| a Polymerization conditions: toluene, 100 °C. b Polymerization conditions: C6D6, 100 °C. c Polymerization conditions: toluene, 50 °C. | ||||||
| 10a | 0.520a | 100 | 24 | 38 | 0.63 | 16 |
| 10b | 0.520a | 100 | 24 | 73 | 0.73 | 16 |
| 10c | 0.520a | 100 | 24 | >99 | 0.71 | 16 |
| 10d | 0.520a | 100 | 24 | 8 | — | 16 |
| 10e | 0.520a | 100 | 24 | 71 | 0.70 | 16 |
| 11a | 0.520b | 50 | 24 | 71 | 0.62 | 16 |
| 11b | 0.520b | 50 | 24 | 94 | 0.71 | 16 |
| 12a | 0.520c | 100 | 48 | 79 | 0.18 | 17 |
| 12b | 0.520c | 100 | 48 | 85 | 0.25 | 17 |
| 12c | 0.520c | 100 | 48 | 93 | 0.62 | 17 |
The syndiotacticity is higher when the ortho-position is a cumyl (2-phenylpropyl) substituent in complex 10b (73% conversion, Ps = 0.73) when compared with complex 10a with tert-butyl substituent in the phenoxy moiety (38% conversion, Ps = 0.63). A change in the metal has a significant effect on the polymerization of meso-lactide. Zirconium complex 10c polymerized meso-lactide faster (>99% conversion) than the homolog titanium complex 10b (73%) with a monomer:initiator ratio of 100. Asymmetric group 4 metal initiators were obtained from [Ti(OiPr)n−xClx] (11a,b, Scheme 6).16
When complex 11a was used as an initiator 71% conversion was attained (Mw/Mn = 1.06 and Ps = 0.62) at 100 °C for 24 h, in C6D6 (monomer:initiator ratio of 50). However, 94% conversion was obtained with 11b (Mw/Mn = 1.07, Ps = 0.71).16 The efficiency values for these initiators were approximately 0.5.
C 2-Symmetric complexes of tetravalent metals containing two 1,ω-dithia-alkanediyl-bridged bis(phenolato) (OSSO)-type ligands polymerized meso-lactide to give heterotactic or syndiotactic polylactides depending on the metal center.17Polymers at 50 °C in toluene obtained by using zirconium complex 12a showed the lowest syndiotacticity (Ps = 0.18) compared with that of polymers obtained by the hafnium complex 12b (Ps = 0.25) and cerium complex 12c (Ps = 0.62), with the latest showing the highest monomer conversion. This was associated with the space available between the two (OSSO)-type ligands in the cerium complex, as indicated the results of single crystal analysis of 12c.17
Zinc initiators
Polymerization of meso-lactide using BDI zinc isopropoxide complex (13a,b, Scheme 7) afforded PLAs with different microstructures.18 Complex 13a provides access to syndiotactic PLAs without the use of chiral ligands. PLA was produced with a molecular weight of 22400 g mol−1 (Mw/Mn = 1.07, Ps = 0.76) after 5 h at 0 °C in dichloromethane (monomer:initiator ratio of 200). However, complex 13b produced moderately heterotactic PLA and suggested chain-end stereocontrol to be operative during polymerization. Complex 13b polymerized meso-lactide at 0 °C with 97% of conversion after 4 h, with a monomer:initiator ratio of 99 (Mn,exp = 13800 g mol−1, Mw/Mn = 1.02 and Ps = 0.37).
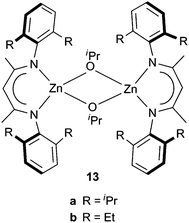 | ||
| Scheme 7 Zinc complexes for polymerization of meso-lactide. | ||
Group 2 metal initiators
Alkaline earth metal amide complexes containing a cyclen-derived (NNNN)-type macrocyclic ligand polymerized meso-lactide (14a,b, Scheme 8) to afford syndiotactically enriched PLAs.19Magnesium complex 14a and calcium complex 14b are fast initiators; full conversions were reached at room temperature, in toluene or in tetrahydrofuran, in less than 30 min. Syndiotacticity Ps was 0.64 for 14a (0.70 in THF) and 0.63 for 14b in toluene. The initiators showed molecular weight efficiency values of 0.60 for 14a (Mw/Mn = 1.50) and 1.18 for 14b (Mw/Mn = 1.41) for meso-lactide polymerization.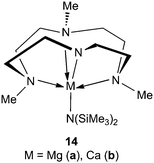 | ||
| Scheme 8 Alkaline earth metal complexes for polymerization of meso-lactide. | ||
Organocatalysts
Waymouth, Hedrick et al. investigated the polymerization of meso-lactide using N-heterocyclic carbenes (15a–c, Scheme 9).20 Compounds 15a and 15b afforded heterotactic PLA (Ps = 0.33 and 0.17, respectively) at −40 °C in dichloromethane (monomer:initiator ratio of 100). Complex 13c produced practically atactic PLAs (Ps = 0.58).
 | ||
| Scheme 9 NHC used for polymerization of meso-lactide. | ||
Mechanistic studies
In the early 1990s, Kricheldorf showed that meso-lactide was polymerized more slowly than rac-lactide when using aluminium and magnesium initiators.4 In 2000, Chisholm et al. analyzed the molecular structure of meso-lactide and its polymerization by single-site initiators.21 They showed preferential polymerization of meso-lactide over L-lactide and rac-lactide. At −40 °C, zinc and magnesium tris(pyrazolyl)hydroborate complexes exclusively polymerized meso-lactide in a 50:50 mixture of meso- and rac-lactide, leaving rac-lactide unreacted. Furthermore, according to a computational study the ground state conformation of meso-lactide is between a twist-boat and a planar structure, and thus the six-membered ring is close to planar. Calculations suggested that the planar structure is favored because the lone pairs of the two oxygen atoms in each ester group repel each other (Fig. 1). This supports the view that meso-lactide is less stable than rac-lactide.22
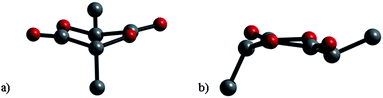 | ||
| Fig. 1 Molecular structures of meso-lactide according to ref. 21: (a) side-on view and (b) side view. | ||
Polymerization of meso-lactide by the chiral aluminium initiator (R)-1 (Scheme 3) proceeded with a slow rate (kobs = 2.0 × 10−3 min−1). Interestingly, this is approximately half the rate of the formation of the heterotactic PLA, prepared from meso-lactide using rac-1 (kobs = 4.4 × 10−3 min−1). This result can be rationalized on the basis that (R)-1 can only attack meso-lactide at one of the two diastereotopic acyl-oxygen bonds (Scheme 10).7
Under the same conditions, aluminium complexes 3a–c (Scheme 3) polymerized meso-lactide with various rates (kobs of 29.4, 4.6 and 13.5 × 10−3 min−1, respectively).9 When the complexes 3a and 3c (R = R′) were used, rac-lactide was polymerized ca. 1.3 times faster than meso-lactide. However, using complex 3b (R ≠ R′), the polymerization of L-lactide is ca. 2.1 fold faster than rac-lactide and 5 times faster than meso-lactide; demonstrating a direct effect of the substituent on the mechanism of the polymerization. This was attributed to the chain-end selection caused by the presence of both stereogenic centers.9
When indium complex 8 (Scheme 5) was used, the polymerization rates for rac-lactide and meso-lactide (kobs = 13.8 × 10−3 min−1) were similar and approximately 10 times higher than that for L-lactide. These findings support a mechanism where both the rate of polymerization and selectivity of monomer enchainment are influenced by multiple stereocenters in the monomer and/or the polymer chain end.15
The polymerization of meso-lactide (kobs = 0.8 × 10−3 min−1) using titanium complex 10e demonstrated a rate comparable to that of rac-lactide which was ca. 2.2 times slower than L-lactide polymerization. This was explained by a reaction site with a preference for L-lactide.16 The asymmetric chlorinated analog titanium complex 11b indicated a polymerization of meso-lactide 4 times slower (kobs = 0.2 × 10−3 min−1) than when complex 10e was used.16
When the tetravalent complexes containing two (OSSO)-type ligand (12a–c) were compared, the cerium complex 12c showed the fastest polymerization rate for meso-lactide (kobs = 3.2 × 10−3 min−1) and ca. 2.1 times faster than rac- and L-lactide. Similar tendency was found using hafnium 12b, meso-lactide (kobs = 2.8 × 10−4 min−1) was polymerized twice as fast compared to rac- and L-lactide. However, zirconium complex 12a polymerized rac-lactide and L-lactide approximately 20 times faster than meso-lactide (kobs = 1.1 × 10−4 min−1).17
At room temperature, zinc complex 13a polymerized rac-lactideca. 1.6 times faster than meso-lactide (kobs = 37.0 × 10−3 min−1) (Table 5).18
| Initiator | [Initiator]0/mol L−1 | [mon.]0/[init.]0 | Solvent | k obs/10−3 min−1 | T/°C | Ref. |
|---|---|---|---|---|---|---|
| R-1 | 0.200 | 100 | Toluene | 2.0 | 70 | 7 |
| rac-1 | 0.200 | 100 | Toluene | 4.4 | 70 | 7 |
| 3a | 0.474 | 96 | Toluene | 29.4 | 70 | 9 |
| 3b | 0.474 | 96 | Toluene | 4.6 | 70 | 9 |
| 3c | 0.474 | 96 | Toluene | 13.5 | 70 | 9 |
| 8 | 0.840 | 100 | CD2Cl2 | 13.8 | 21 | 15 |
| 10e | 0.520 | 100 | C6D6 | 0.8 | 100 | 16 |
| 11b | 0.520 | 100 | C6D6 | 0.2 | 100 | 16 |
| 12a | 0.520 | 100 | C6D6 | 0.1 | 100 | 17 |
| 12b | 0.520 | 100 | C6D6 | 0.3 | 100 | 17 |
| 12c | 0.520 | 100 | C6D6 | 3 | 100 | 17 |
| 13a | 1.023 | 494 | CH2Cl2 | 37.0 | 25 | 18 |
Conclusions and future perspectives
Although meso-lactide has been known for 20 years,23 the number of stereoselective single-site initiators is still small compared to the plethora of well-defined initiators for L- and rac-lactide. Peculiarly, only two examples of melting temperature for poly(meso-lactide) have been reported so far due to the lack of highly syndiotactic PLAs.6,11In contrast, when rac-lactide is polymerized, chirality or rigidity of the backbone of the ligand was not necessary to obtain high tacticity.11,18 In most cases, metal complexes which polymerized rac-lactide to give highly heterotactic PLAs, also yielded highly syndiotactic PLAs from meso-lactide.10,11,18 Trivalent metal complexes appeared to afford higher syndiotacticity than the divalent or tetravalent metal complexes. Furthermore, metals (aluminium6,7 and indium11) having slower rates of polymerization than group 3 metals10,11 resulted in better polydispersities of the poly(meso-lactide).
 | ||
| Scheme 10 Formation of syndiotactic PLA according to ref. 6 and 7. | ||
Since meso-lactide is a by-product of L-lactide synthesis, it will become more easily available with increasing capacity of L-lactide production.23 Therefore, efficient and selective polymerization will be of considerable interest, provided crystalline poly(meso-lactides) can be obtained. This will give large benefits as it will be possible to use L-lactide to form isotactic PLAs and its by-product (meso-lactide) to form syndiotactic PLAs reducing the cost of production and increasing the interest towards biorenewable poly(lactic acid).
Acknowledgements
We thank the Deutsche Forschungsgemeinschaft, the Fonds der Chemischen Industrie for financial support, and Uhde Inventa-Fischer for a generous gift of meso-lactide.Notes and references
- (a) R. E. Drumright, P. R. Gruber and D. E. Henton, Adv. Mater., 2000, 12, 1841 CrossRef CAS; (b) S. Inkinen, M. Hakkarainen, A.-C. Albertsson and A. Södergård, Biomacromolecules, 2011, 12, 523 CrossRef CAS; (c) O. Dechy-Cabaret, B. Martin-Vaca and D. Bourissou, Chem. Rev., 2004, 104, 6147 CrossRef CAS; (d) J. K. Oh, Soft Matter, 2011, 7, 5096 RSC.
- (a) P. J. Dijkstra, H. Du and J. Feijen, Polym. Chem., 2011, 2, 520 RSC; (b) C. M. Thomas, Chem. Soc. Rev., 2010, 39, 165 RSC; (c) M. J. Stanford and A. P. Dove, Chem. Soc. Rev., 2010, 39, 486 RSC; (d) C. A. Wheaton, P. G. Hayes and B. J. Ireland, Dalton Trans., 2009, 4832 RSC; (e) R. H. Platel, L. M. Hodgson and C. K. Williams, Polym. Rev., 2008, 48, 11 CrossRef CAS; (f) A. Amgoune, C. M. Thomas and J.-F. Carpentier, Pure Appl. Chem., 2007, 79, 2013 CrossRef CAS; (g) J. Wu, T.-L. Yu, C.-T. Chen and C.-C. Lin, Coord. Chem. Rev., 2006, 250, 602 CrossRef CAS; (h) B. J. O'Keefe, M. A. Hillmyer and W. B. Tolman, J. Chem. Soc., Dalton Trans., 2001, 2215 RSC.
- K. Fukushima and Y. Kimura, Polym. Int., 2006, 55, 626 CrossRef CAS.
- (a) H. R. Kricheldorf and C. Boettcher, Makromol. Chem., 1993, 194, 1653 CrossRef CAS; (b) H. R. Kricheldorf and S.-R. Lee, Polymer, 1995, 36, 2995 CrossRef CAS; (c) H. R. Kricheldorf and C. Boettcher, J. Macromol. Sci., Part A: Pure Appl. Chem., 1993, 30, 441 CrossRef.
- N. Spassky, M. Wisniewski, C. Pluta and A. Le Borgne, Macromol. Chem. Phys., 1996, 197, 2627 CrossRef CAS.
- T. M. Ovitt and G. W. Coates, J. Am. Chem. Soc., 1999, 121, 4072 CrossRef CAS.
- T. M. Ovitt and G. W. Coates, J. Am. Chem. Soc., 2002, 124, 1316 CrossRef CAS.
- H. Du, A. H. Velders, P. J. Dijkstra, J. Sun, Z. Zhong, X. Chen and J. Feijen, Chem.–Eur. J., 2009, 15, 9836 CrossRef CAS.
- H. Du, A. H. Velders, P. J. Dijkstra, Z. Zhong, X. Chen and J. Feijen, Macromolecules, 2009, 42, 1058 CrossRef CAS.
- A. Amgoune, C. M. Thomas, T. Roisnel and J.-F. Carpentier, Chem.–Eur. J., 2006, 12, 169 CrossRef CAS.
- J.-C. Buffet, A. Kapelski and J. Okuda, Macromolecules, 2010, 43, 10201 CrossRef CAS.
- Molecular weight efficiency values, f, were calculated according to Mn,exp/Mn,theo, with Mn,theo = [mon]0/[initiator]0 × 144.13 × conv.
- J.-C. Buffet and J. Okuda, Dalton Trans., 2011, 40, 7748 RSC.
- A. Pietrangelo, M. A. Hillmyer and W. B. Tolman, Chem. Commun., 2009, 2736 RSC.
- A. Pietrangelo, S. C. Knight, A. K. Gupta, L. J. Yao, M. A. Hillmyer and W. B. Tolman, J. Am. Chem. Soc., 2010, 132, 11649 CrossRef CAS.
- J.-C. Buffet and J. Okuda, Chem. Commun., 2011, 47, 4796 RSC.
- A. Sauer, J.-C. Buffet, T. P. Spaniol, H. Nagae, K. Mashima, and J. Okuda, unpublished results.
- B. M. Chamberlain, M. Cheng, D. R. Moore, T. M. Ovitt, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2001, 123, 3229 CrossRef CAS.
- J.-C. Buffet, J. P. Davin, T. P. Spaniol and J. Okuda, New J. Chem., 2011 10.1039/c1nj20259f.
- A. P. Dove, H. Li, R. C. Pratt, B. G. G. Lohmeijer, D. A. Culkin, R. M. Waymouth and J. L. Hedrick, Chem. Commun., 2006, 2881 RSC.
- M. H. Chisholm, N. W. Eilerts, J. C. Huffman, S. S. Iyer, M. Pacold and K. Phomphrai, J. Am. Chem. Soc., 2000, 122, 11845 CrossRef CAS.
- I. A. Shuklov, H. Jiao, J. Schulze, W. Tietz, K. Kühlein and A. Börner, Tetrahedron Lett., 2011, 52, 1027 CrossRef CAS.
- G. Entenmann, and D. Bendix, Boehringer Ingelheim, Ger. Offen. DE 3820299, 1988; G. Entenmann and D. Bendix, Chem. Abstr., 1989, 110, 213592w Search PubMed.
| This journal is © The Royal Society of Chemistry 2011 |