Some new design strategies for second-order nonlinear optical polymers and dendrimers
Zhen
Li
*,
Qianqian
Li
and
Jingui
Qin
Department of Chemistry and Hubei Key Lab of Organic and Polymeric Opto-Electronic Materials, Wuhan University, Wuhan, 430072, China. E-mail: lizhen@whu.edu.cn; lichemlab@63.com; Fax: +86-27-68755363
First published on 22nd August 2011
Abstract
Second-order nonlinear optical (NLO) materials have played a very important role in laser technology since the invention of the laser. The materials have been developed from inorganic crystals to organic molecules and polymers. The search for new second-order NLO materials with better performance has continuously been in the frontier of materials science and optical engineering. This article reviews some new design strategies for second-order NLO polymers, including dendronized polymers, dendrimers and hyperbranched polymers with the new concept of the “suitable isolation group”.
 Zhen Li | Zhen Li received his B.Sc. and Ph.D. degrees from Wuhan University (WHU) in China in 1997 and 2002, respectively, under the supervision of Prof. Jingui Qin. In 2003–2004, he worked in the Hongkong University of Science and Technology as Research Associate in the group of Prof. Ben Zhong Tang. In 2010, he worked in Georgia Institute of Technology in the group of Prof. Seth Marder. He has been a full professor at WHU since 2006, and his research interests are in the development of organic molecules and polymers with new structure and new functions for organic electronics and photonics. |
 Qianqian Li | Qianqian Li received her B.Sc. degree from Hubei University in China in 2004, and then obtained her Ph.D. degree at Wuhan University in 2009, under the supervision of Prof. Zhen Li. She is now a joint postdoctor in the groups of Prof. Li at WHU and Prof. Jishan Wu at National University of Singapore, and her research interests are in the design and synthesis of new electric and optical functional materials. |
 Jingui Qin | Jingui Qin obtained his B.Sc. and M.Sc. in Department of Chemistry at WHU. After he obtained his D.Phil. degree in Organometallic Chemistry from the University of Oxford, UK under Prof. Malcolm Green FRS in 1987, he returned to WHU and became a full professor in 1993. He has carried out collaboration research in the Institute of Physical and Chemical Research, Japan, the Institute of Molecular Sciences, Japan and California Institute of Technology, USA. His interests are in the design, synthesis, structure and properties of various organic, inorganic and polymeric opto-electronic materials. |
Introduction
Shortly after the invention of the laser, Franken observed an abnormal phenomenon of second-harmonic generation (SHG) in quartz when one beam of laser passed through the crystal in 1961.1 This observation directly gave birth to the research field of nonlinear optics (NLO). Accompanying the appearance of various high intensity lasers and stimulated by their huge potential applications in electronics and photonics, many interests and efforts have been attracted and contributed to the rapid development of this field.When a beam of light propagates through a material, interaction of the electromagnetic field with molecules in the material induces polarization that is charge variation and displacement of associated atoms (or ions in ionic substances). The molecular polarization (p) at the microscopic molecular level, and macroscopic polarization (P) at the macroscopic level, can be expressed as nonlinear equations:
| p = αE + βE2 + γE3 + … | (1) |
| P = χ(1)E + χ(2)E2 + χ(3)E3 + … | (2) |
Generally, the higher order polarizability is much smaller than the lower one (only about 10−7∼10−8 times), that is, α ≫ β ≫ γ, or χ(1) ≫ χ(2) ≫ χ(3). Thus, under the normal illumination light with the small intensity, the induced polarization (p and P) is proportional to the applied field:
| p = αE, P = χ(1)E |
However, the intensity of laser radiation is sufficiently strong, hence the parameters of β and γ or χ(2) and χ(3), cannot be ignored any more, which results in the nonlinear relationship between the polarization (p and P) and the applied field (E). Among them, β and χ(2) are related to the second-order NLO properties, while γ and χ(3) denote the third-order nonlinear optical phenomena, with the two-photon-absorption effect as a typical example. It should be noted that only those molecules and materials lacking centrosymmetry can exhibit a nonzero second-order susceptibility. This requirement for the second-order terms of the above equations is unlike the first- and third-order terms.
Nowadays, the important and active areas in the NLO fields are the second- and third-order NLO effects. For example, as one of the second-order NLO materials, electro-optics lies at the interface of the two important technologies of electronics and photonics (the practical basis of modern information technology), providing the means of converting information from the electronic domain to the photonic domain and vice versa. A typical example of such signal transduction is the encoding of digital (electronic) computer data onto the (optical or photonic) Internet.2i Recently, there has been a resurgence of interest in organic electro-optic materials, motivated by a number of additional factors, such as the chipscale integration, specific material requirements of emerging applications, and the demonstration of new device concepts and unique processing advantages of organic materials.2i
Actually, the study of second-order NLO materials started as early as when the laser was invented, and currently the practically useful NLO materials are mainly inorganic second-order NLO crystals in the visible and UV regions. On the other hand, second-order NLO polymers have shown very promising potential for applications in the field of photonics, especially the greatest bandwidth. However, research of better second-order NLO crystals and polymers is still full of great challenges and attracts great attention from chemists, physicists, material scientists, as well as engineers. Many excellent review articles have continuously been published to summarize the developments in the field.2,3 Based mainly on our recent systematic research of second-order nonlinear optical materials, in this article, we shall present some new design strategies for second-order NLO polymers, including dendronized polymers, dendrimers and hyperbranched polymers.
The concept of the “suitable isolation group” and its application in designing high-performance NLO polymers
For the potential practical applications, organic NLO materials should be processed into thin films to integrate with other ones. At the early stage of NLO research, chromophores were generally doped into polymer matrix to form the guest-host systems.2 Generally, when introduced into polymeric systems as dopants or through covalent bonds, the chromophore moieties with high polar push-pull structure would have centrosymmetric alignment, that is, the donor part of one chromophore moiety would be close to the acceptor part of another chromophore one, due to the strong electronic interactions. However, as mentioned in the introduction part, only those materials lacking centrosymmetry could exhibit a nonzero macroscopic second-order susceptibility. Thus, to achieve the noncentrosymmetric alignment of chromophore moieties with high dipole moment (μ), as demonstrated in Fig. 1, the polymer film should be heated to the temperature (T) higher than the glass temperature (Tg) of the polymer system, then an electric field was applied to the film, leading to the poling-induced noncentrosymmetric alignment of chromophore moieties, which was essential to exhibit the macroscopic NLO effect (d33 or r33). Then, before removing the applied electric field, the film was cooled to room temperature, and the noncentrosymmetric alignment remained.2,4 Actually, during the practical applications or storage, the noncentrosymmetric alignment of the chromophore moieties were apt to relax to the centrosymmetric state, driven by the strong polar-polar electronic interactions among them. Thus, some additional methods such as a crosslinking approach were used. It should be pointed out that both second-harmonic generation (SHG) and electro-optic (EO) effects could be investigated in situ monitoring of the induction of second-order optical nonlinearity by electric field poling, with d33 and r33 often acting as the corresponding coefficients for them respectively (the definition and explanation of these two parameters can be found in detail in ref. 1–3). There was a fixed relationship between these two effects, and in this article, the tested d33 value of our prepared polymers was about 0.61 of the corresponding r33 ones. Usually, the maximum r33 value of lithium niobate (LiNbO3) (32 pm V−1) was utilized as the standard to tell the performance of one polymer.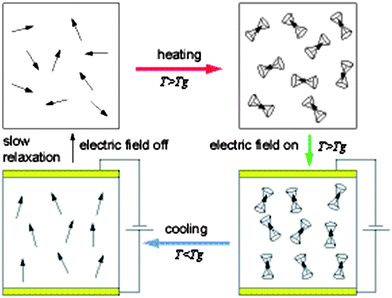 | ||
| Fig. 1 Graphical illustration of the poling procedure for NLO-polymers.2 | ||
The guest-host systems suffered from some defects, especially the crystallization and phase separation at the high concentrations of the doped NLO chromophores, and sublimation during sample preparation. Thus, incorporation of the NLO chromophores into a polymer structure, resulting in chromophore functionalized NLO linear polymers, dendronized polymers, dendrimers and hyperbranched polymers, could allow the introduction of higher concentration of chromophores, and overcome the defects of the guest-host systems. Also, the slow relaxation could be suppressed to a large degree, since the incorporation tethered one or both ends of the chromophore moieties.2,5 And based on the obtained experience of a number of guest-host systems, various kinds of linear NLO polymers, mainly including side-chain and main-chain polymeric systems, have been designed to meet the requirements of practical applications.2,5 Both types of linear polymers could be easily synthesized by different reactions from NLO chromophores bearing one or two reactive groups. Relatively, in the side-chain NLO polymers, the alignment of the chromophore moieties could be easily achieved under the applied electric field, since only one end point was linked to the polymer backbone through a flexible spacer. However, for the same reason, once the good noncentrosymmetric alignment of the chromophore moieties was realized in the main-chain polymers, in which the chromophore moieties were part of the polymer backbone, the stability of the NLO effect after the removal of the applied electric field, was much better than that of side-chain polymers.
With the efforts to obtain NLO polymers with good performance, on the basis of excellent work of other scientists,1–3 we have synthesized a series of linear polymers bearing chromophore moieties in the side chain or main chain.6–8 In this review, we would like to discuss some typical examples. As shown in Chart 1, in 2001, we developed a synthetic strategy for the postfunctionalization of poly(N-vinylcarbazole) (PVK), to yield a PVK-based nonlinear optical (NLO) polymer, P1, with a high density of chromophores (52%). The Tg of P1 was 185 °C, and its NLO activity, which was estimated to be 20 pm V−1 (d33) by in situ second harmonic generation measurement (the experimental errors were about ±15% according to the measuring system), remained unchanged at 120 °C for over 1000 h after a minor initial drop.9a
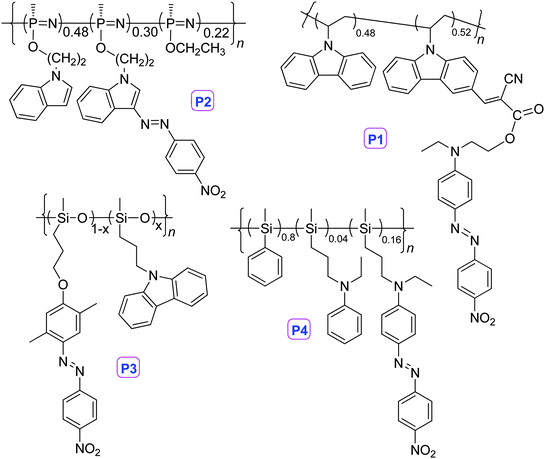 | ||
| Chart 1 The structures of P1–P4. | ||
Generally, NLO polymers with high Tg were suitable for application in devices used at room temperature or even higher temperature. However, under very low temperature, for example, ∼−90 °C in the south pole, polymers with high Tg were very brittle, and could not form thin films with high quality. Thus, NLO polymers with low Tg were needed. From this point, we have introduced chromophores to highly flexible polymeric backbones, i.e., polysiloxane and polyphosphazene. Through the post azo coupling reaction between one reactive polymer and p-nitrobezenediazonium fluoroborate, polyphosphazene P2 was yielded conveniently, which possessed relatively low Tg (86 °C) and good d33 value (32 pm V−1).9bPolysiloxane P3 exhibited even lower Tg (∼50 °C), confirming the good flexibility of the polysiloxane backbone.9c Considering the good photoconductivity of polysilanes due to the delocalization of σ-electrons of the polymeric backbone, we also introduced NLO chromophore moieties to the backbone of polysilane. The obtained P4 demonstrated a d33 value of 19 pm V−1.9d Coupled with the good photoconductivity of polysilanes, P4 might be a potential photorefractive polymeric material.
However, it was found that the macroscopic NLO effects could not be improved much higher. Meanwhile, NLO chromophores were developed rapidly, with their μβ values being improved by up to 250-fold. From their structure of donor-π-acceptor, it was easy to know that NLO chromophores were highly polar molecules. There were strong intermolecular electronic interactions between the chromophore moieties in the polymeric system, which tended to form the centrosymmetric state, making the poling-induced noncentrosymmetric alignment of chromophores a daunting task.2,10,11 This also led to the challenge present in the NLO research field: how to efficiently translate the large β values of the organic chromophores into high macroscopic NLO activities of polymers.
Fortunately, it has been proved that controlling the shape of the chromophore is an efficient approach for minimizing this interaction and enhancing the poling efficiency. In particular, a spherical shape, proposed by Dalton et al.,12 has been considered as the ideal conformation. And applying the site isolation principle, the introduction of some isolation groups (IG) to the chromophore moieties is an effective approach to decrease the interactions and improve the poling efficiency.12,13 Dalton and Jen et al. have done excellent work in this area, and prepared various series of NLO dendrimers and dendronized polymers.14
For example, as shown in Chart 2, when chromophore 2 was encapsulated three dimensionally by three highly fluorinated dendrons from chromophore 1, the value of r33, measured from their corresponding doped polycarbonate films, increased from 10 to 30 pm V−1, confirming that the dendronized moieties in the chromophore gave full scope to the role of isolation.10a
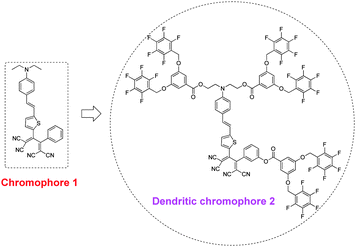 | ||
| Chart 2 The structures of chromophores 1 and 2. | ||
Subsequently, the site isolation principle was introduced to some linear polymer systems to convert them into dendron-substituted polymers, i.e., dendronized polymers. The shape and properties of dendronized polymers could be determined by the size, shape and density of the attached dendritic side-chain moieties, the degree of polymerization and the backbone of polymers. For the side-chain dendronized polymers P5 and P6 (Chart 3), high poling efficiency was achieved, and the values of r33 were 81 and 97 pm V−1, respectively. Furthermore, the dendronized moieties not only attached to the side-chain, but also to the backbone, leading to the cylindrical shape of polymer P7 (Chart 4), and the value of r33 increased to as large as 111 pm V−1.14e
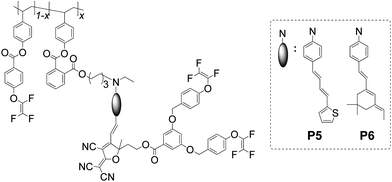 | ||
| Chart 3 The structures of polymers P5 and P6. | ||
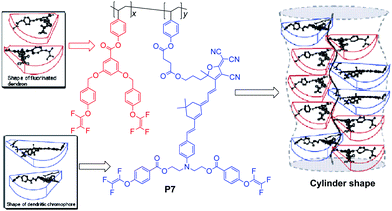 | ||
| Chart 4 The structure of polymer P7. | ||
Their work confirmed that the dendritic structure is a very promising molecular topology for the next generation of highly efficient NLO materials.14 Other groups also have synthesized some NLO dendrimers with good performance succsessfully.15 Therefore, to introduce some IGs to the NLO chromophore moieties was an effective approach to modify the previous linear NLO polymers to achieve much larger macroscopic NLO effects.
According to the one-dimensional rigid orientation gas model:16
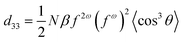 | (3) |
To address the above question, we designed and prepared two series of polyurethanes containing dendronized NLO chromophores as side chains.17Chart 5 showed their structure, and the IGs were labeled with the dashed ring. Conveniently by modifying the synthetic procedure, in P8–P11 and P12–P15, the size of the isolation moieties could be adjusted from small atoms, hydrogen atoms (actually in this case, we could consider there were no IGs present), to much larger groups such as carbazolyl. We further synthesized P16 and P17 for comparison, in which the fluorene moieties were almost perpendicular to the two phenyl rings of the azo moieties, acting as bulky IGs. We also termed this kind of chromophore as “H” type.17
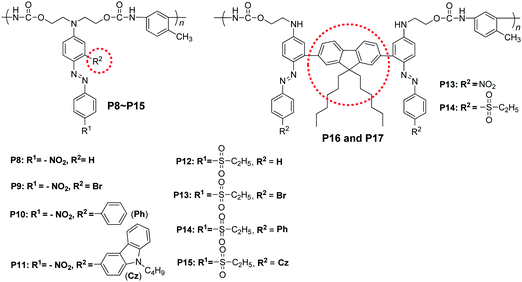 | ||
| Chart 5 The structures of P8–P17. | ||
As determined by the second harmonic generation (SHG) processes characterized by d33, the NLO properties of these polymers did not always increase accompanying the enlargement of the IGs linked to the chromophore moieties, but there was a peak value (Table 1 and Fig. 2) while the IGs were phenyl moieties for the sulfonyl group acceptor moieties, with carbazolyl groups for the nitro ones. This is understandable. As mentioned above, the push-pull NLO chromophores with large dipole moments tend to compactly pack in a centrosymmetric manner due to the strong intermolecular dipole–dipole interactions, making poling-induced noncentrosymmetric alignment of chromophores a daunting task. The purpose of controlling the shape of the chomophores or introducing IGs is to minimize the strong intermolecular dipole–dipole interactions to some degree, in order to improve the d33 values of the polymers. However, the addition of the IGs would increase the bulky of the resultant chromophore moieties, which makes the noncentrosymmetric alignment of the chromophores in the electronic field more difficult. Also, the introduction would dilute the active concentration of the chromophore moieties in the polymers. That is to say, the introduction of the IGs mainly has three effects on the resultant d33 value of the material:
| no. | d 33 a(pm V−1) | d 33 (∞) b(pm V−1) | Φ c |
|---|---|---|---|
| a Second harmonic generation (SHG) coefficient. b The nonresonant d33 values calculated by using the approximate two-level model. c Order parameter Φ = 1 − A1/A0, A1 and A0 are the absorbances of the polymer film after and before corona poling, respectively. | |||
| P8 | 56.4 | 9.5 | 0.15 |
| P9 | 51.9 | 10.1 | 0.18 |
| P10 | 49.4 | 8.3 | 0.20 |
| P11 | 82.3 | 12.0 | 0.34 |
| P12 | 35.9 | 9.7 | 0.14 |
| P13 | 29.1 | 7.5 | 0.14 |
| P14 | 63.0 | 14.9 | 0.31 |
| P15 | 16.1 | 3.8 | 0.23 |
| P16 | 56.0 | 11.0 | 0.31 |
| P17 | 12.1 | 3.3 | 0.18 |
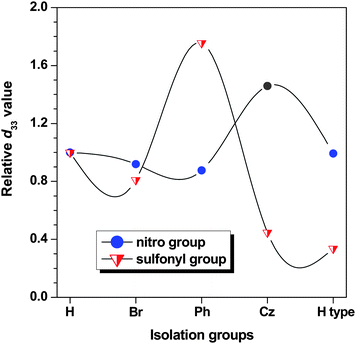 | ||
| Fig. 2 The comparison of the d33 values of the polymers using P8 and P12 as references respectively for the two series of polymers with either nitro or sulfonyl groups as acceptor groups in the chromophore moieties with different IGs. “H type” was the IGs of fluorene moieties. The line is included as a guide to the eye. | ||
To minimize the interaction of the chromopohores to improve the d33 value (named as positive effect, abbreviated as P),
To dilute the active concentration of the chromophore moieties, generally reducing the d33 values (named as negative effect, abbreviated as N);
To restrain the orderly alignment of the chromophores to decrease the d33 value (named as negative effect, abbreviated as N).
So, there should be a balance among the three effects. Here in our case, the only difference between the two series of the polymers is the different acceptor groups, one is nitro groups, another is sulfonyl moieties, and nitro groups are a stronger acceptor than sulfonyl moieties. Then, the intermolecular dipole–dipole interactions between the chromophores containing nitro groups as acceptors should be stronger than that present in the case of sulfonyl moieties. Also, at the same poling conditions, the force for the orderly alignment of the chromophore with sulfonyl moieties as acceptor groups should be weaker than that for the nitro groups. Therefore, to minimize the interaction of the nitro chromophores to the similar degree as in the case of sulfonyl chromophores, the IGs should be larger. And on the other hand, to restrain the alignment of the nitro groups in the electronic field during the poling process to the similar extent as in the case of the sulfonyl chromophore, the introduced IGs also should be larger. Thus, the effects, P and N effects, should be much different when the chromophores are different. When the IGs were very small, such as bromine atoms in the two cases of chromophores and phenyl groups in the case of the nitro chromophores, the P and N effects are not obvious, so the NLO properties are similar as those of hydrogen atoms as IGs (or considering that there are no IGs); when the IGs are moderate, such as the phenyl groups in the case of the sulfonyl chromophores and the carbazolyl groups in the case of the nitro chromophores, the P effect is obvious while the N effect is negligible, so the NLO properties are much better; but when the IGs are even larger, such as in the cases of P15–17, the G effect is still OK or even better, however, the B effect does work now, cancels the G effect to some degree and leads to the worse NLO results.
It should be pointed out that the above discussed NLO properties of polymers are the values tested directly from experiment. If considering the resonant enhancement (the contribution to the tested macroscopic NLO effect from the absorption of the chromophore moieties in the poled films at the wavelength of half of the fundamental wavelength of the laser used) due to the absorption of the chromophore moieties at 532 nm, the NLO properties should be smaller as shown in Table 1 (d33 (∞)), which were calculated by using the approximate two-level model. However, the trends existing in the d33 (∞) values were similar to those we discussed in the test d33 values of the polymers, because the maximum absorption wavelengths of these polymers are similar to the result for the same active chromophore part. Thus, in the following part, we focus the discussion on the d33 values. We further measure their order parameter (Φ, Table 1), a parameter to indicate the extent of the noncentrosymmetric alignment of chromophore moieties in the poled films. Generally, higher Φ value, better alignment.2b Similar as the d33 values, the Φ values increase until a peak point in the IGs enlargement, then decrease while the IGs become even bigger.
Thus, based on the above obtained experimental results, we proposed the concept of “suitable isolation group” (SIG), that is to say, for a given chromophore moiety, there should be an SIG present to balance the G and B effects, and introducing this IG to the chromophore moieties would boost the microscopic β value of the chromophore to possibly better macroscopic NLO properties of the polymers containing this chromophore moieties efficiently.
It was easily seen that in P8–P17, the isolation spacers were introduced to the chromophore moieties near the donor side (the cartoon model of I, Chart 6). Then, how about other positions? Since the function of the isolation group was to decrease the strong dipole–dipole interactions of the chromophore moieties, the linkage position and mode of the IG in the chromophore moieties would affect the actual isolation effect of the introduced IGs. To study the different results of the linking position of IGs, and also to check the above proposed concept of “suitable isolation group”, a series of NLO polymers were designed, in which the IG was introduced to the chromophore moieties at different positions, and the linkage mode of the chromophore moieties to the polymer backbone was different. Chart 6 is a cartoon picture to partially demonstrate the above idea.
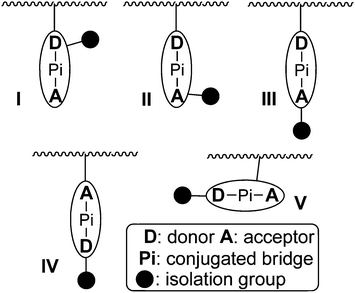 | ||
| Chart 6 A cartoon model of different linkage modes of chromophore moieties to the polymer backbone. | ||
As shown in Chart 7, when the linkage position of the IGs was moved to near the acceptor side (see the cartoon model of II, Chart 6), similar experimental phenomena were observed in their NLO properties. And the phenyl moieties in P19 behaves as the SIG.18 Further changing the bonding position to the acceptor side (see the cartoon model of III, Chart 6), naphthyl (Np) moieties in P23 were proved to be the SIG (Chart 8).19a It should be pointed out that P22–P24 were prepared through facile postfunctional polymer reaction, including the post azo coupling and esterification reactions, from the starting polymeric intermediate, P25 (Chart 8). Actually, these polymers, perhaps, could not be obtained from the direct polymerization process, indicating the synthetic flexibility of the post functional strategy. In particular, the different reacting activity of the amino and hydroxyl groups was rationally utilized to fulfil the whole synthetic process, with the combination of the post azo and esterification reactions. Thus, these preliminary results partially demonstrate the power of the cooperating usage of different kinds of reactions in the post functional strategy, and might attract increasing interests of other scientists to contribute in this area for the further development of the polymer chemistry, in addition to the traditional straightforward synthetic methods of polymers.19
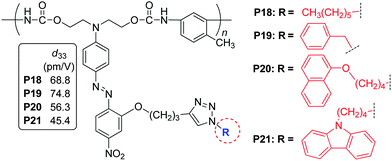 | ||
| Chart 7 The structures of P18–P21. | ||
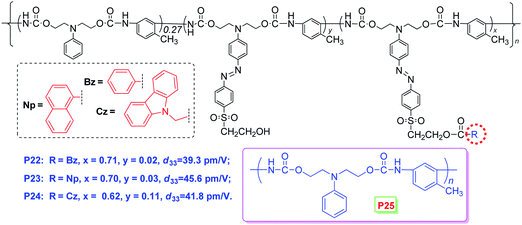 | ||
| Chart 8 The structures of P22–P25. | ||
With the concept of “suitable isolation group” in mind, we tried to find out if there was “suitable isolation group” present in the main-chain NLO polymers, since the above three examples were side-chain NLO polymers. Just simply through the modified synthetic route of P12–P15, main-chain polymers P26–P29 could be easily yielded (Chart 9).20 The d33 value of P27 was about 1.38 times that of P26, while that of P28 being about 1.42, indicating that the introduction of the IGs to the chromophore moieties really benefit the improvement of the NLO properties of the resultant polymeric materials by minimizing the strong intermolecular dipole–dipole interactions between the chromophore groups as in the previously reported cases. However, the d33 value of P29 decreased, confirming that BOP was the SIG for this series of NLO polymers.20 We further utilized another kind of azo chromophores in which the donor group was indole, instead of those aniline-based azo ones, to study if there was some influence of different chromophore moieties on the concept of “suitable isolation group”. As shown in Chart 10, P30–P32 could be obtained with the structure similar to those of P26–P29, by using the similar synthetic procedure.21a As mentioned above, in comparison with their aniline-based analogues, the chromophores containing indole as the donor group showed much blue-shifted absorption (up to 30 nm).22g, 23 Here the blue-shifted absorption of the indole-based chromophore moieties was retained: P30–P32 exhibited much better optical transparency than the corresponding P26, P27 and P29 (larger than 40 nm, Table 2). The NLO test demonstrated that there was SIG present in this series of polyurethanes bearing indole-based chromophore moieties in the main chain.21
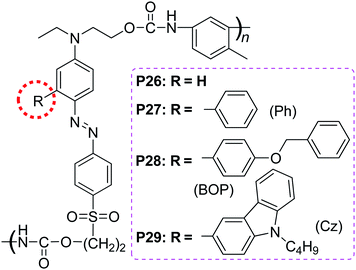 | ||
| Chart 9 The structures of P26–P29. | ||
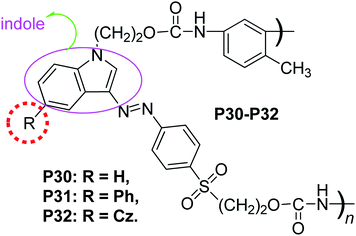 | ||
| Chart 10 The structures of P30–P32. | ||
| no. | λ max a(nm) | d 33 b(pm V−1) |
|---|---|---|
| a The maximum absorption wavelength of polymer solutions in THF (for P26–P29) or DMF (for P30–P32), while the maximum absorption wavelength of the corresponding small chromophore molecules in diluted solutions of the same solvent are given in the parentheses. b Second harmonic generation (SHG) coefficient. | ||
| P26 | 443 (449) | 39.5 |
| P27 | 447 (452) | 54.6 |
| P28 | 448 (452) | 55.9 |
| P29 | 450 (455) | 36.2 |
| P30 | 395 (397) | 20.6 |
| P31 | 401 (403) | 26.2 |
| P32 | 397 (400) | 11.8 |
As all the above cases belonged to the polyurethanes, to examine the concept of “suitable isolation group”, other polymeric systems should be investigated. Accordingly, we have designed other type of polymers, in which different IG in different size were linked to the chromophore moieties.24 For example, Chart 11 demonstrated a series of poly(disubstituted acetylene)s, which contained NLO chromophore moieties in the side chains.24a Also, we attempted to introduce the chromophore moieties to the polymer backbone by different linkage modes: in P34–P37, the chromophore moieties were bonded to the backbone on the donor side (the cartoon model of I, Chart 6); in P38–P40, the linkage position was in the π bridge (the cartoon model of V, Chart 6); while in P41–P43, on the acceptor side (the cartoon model of IV, Chart 6). Actually, P18–P21 discussed above should belong to the cartoon model of II, while P22–P24 to that of III. To study the NLO results of P34–P43 more visually, we compared the tested d33 values of the polymers using that of P34 for P34–P36, P38 for P38–P40, and P41 for P41–P43 as reference (Fig. 3). When the IGs changed from hydrogen atoms to the phenyl groups, the d33 value of P35 increased to 1.8 times that of P34, however, the tested NLO effect decreased dramatically while the size of the isolation moieties (carbazolyl groups here in P36) further increased (Fig. 3a). Similar phenomena were found in the systems of P41–P43: the d33 values will increase and achieve a maximum peak while the size of the IGs enlarges, then decrease quickly as the isolation moieties become even larger. This trend is the same as we observed before, further proving our hypothesis about the effect of the IGs linked in the chromophore moieties.17–21 On the contrary, P38 demonstrates the highest d33 value in the system of P38–P40, and after bonding some IGs to the chromophore moieties at the donor side, the NLO effects of the resultant polymers, P39 and P40, decrease quickly, though the IGs in them are the same as those in P41 and P42, respectively. Therefore, for the first time, our results obtained in the system of P38–P40 indicate that the introduction of the IGs to the chromophore moieties not always benefit the NLO properties of the resultant materials, or we could say, the hydrogen atoms are the best isolation moieties in some special cases. Thus, the NLO properties of P34–P43 further confirmed our previous idea about the effect of the bonded IGs (the concept of suitable isolation group), and the linkage mode could affect the macroscopic NLO effects of the NLO polymers.
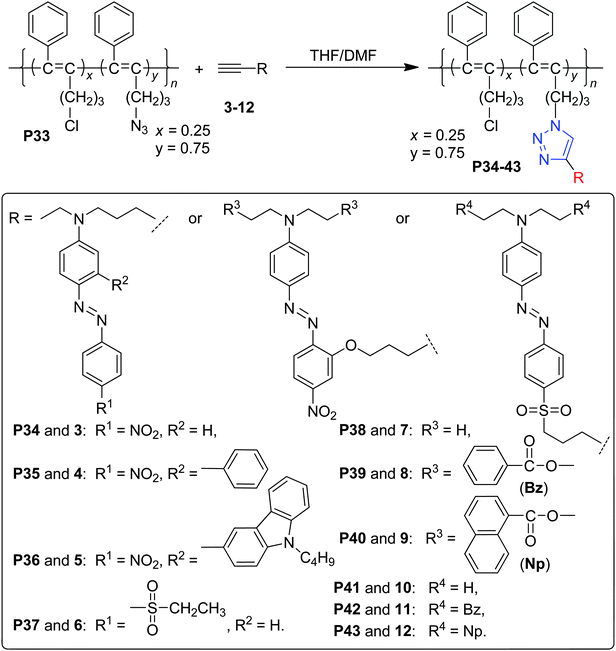 | ||
| Chart 11 The structures of P34–P43. | ||
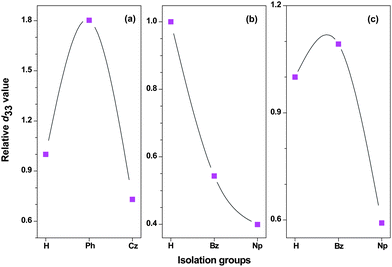 | ||
| Fig. 3 The comparison of the d33 values of the polymers using P34, P38 and P41 as references respectively, for the three series of polymers, (a) P34–P36, (b) P38–P40, and (c) P41–P43. The ine is included as a guide to the eye. | ||
As discussed above, the PVK-based NLO polymer, P1, demonstrated good properties, especially the thermal stability of its NLO effect even at 120 °C for over 1000 h.9a However, the d33 value of the resultant polymer was not very high and its film-forming ability was relatively poor. Thus, we considered that by introducing suitable spacers into the PVK polymeric system, the macroscopic nonlinearity would be enhanced. Similar to the above cases, different IGs in different size were introduced to the chromophore moieties in the PVK polymeric system, as shown in Chart 12.25a As expected, the d33 values of P44–P46 were not always increasing as the IGs enlarged from phenyl ring to carbazole moieties, and there was also a suitable group (BOP moieties in P45) present, a similar phenomenon to the previous work, further proving the concept of “suitable isolation groups”: for a given chromophore moiety and given linkage position, there should be a SIG present to efficiently boost its microscopic β value to possible better macroscopic NLO properties. When the IGs changed from Ph to the BOPgroups, the d33 value of P45 (39.6 pm V−1) was enhanced 1.34 times that of P44. While the size of IGs was further enlarged (Cz groups in P46), the tested d33 value still kept 34.0 pm V−1, 1.16 times that of P44. In general, the d33 value of all the polymers (P44–46), here, increased efficiently in comparison with that of P1 (20 pm V−1), due to the introduced IGs to restrain the intermolecular electrostatic dipole–dipole interactions, indicating that it was an efficient method to enhance the poling efficiency of high-Tgpolymers.
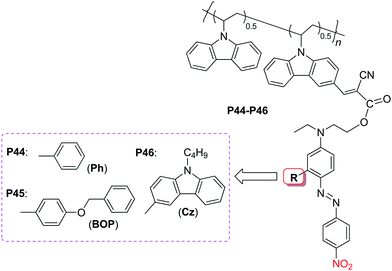 | ||
| Chart 12 The structures of P44–P46. | ||
Excitingly, the solubility and processibility of P44–P46 became much better than that of P1, possibly due to the presence of the linked IGs, which might efficiently suppress the polymer inter-chain entanglements and benefit the polymer chain to extend in solvents. Correspondingly, P44–P46 could form thin films with high quality rather than P1, and this would surely benefit the easy fabrication of photonic devices based on these polymers. Similar phenomena were also observed in other PVK-based NLO polymers.25b Thus, the introduction of the IGs could enhance the NLO effects of the resultant polymers on one hand, also, it could improve the solubility and processibility in a large degree on the other hand.
The concept of “suitable isolation group” was also applied to the polymers bearing DCM (4-(dicyanomethylene)-2-methyl-6-(p-(dimethylamino)styryl)-4H-pyran) as chromophore moieties in the side chains (P47–P50), as reported by Hsiue group (Chart 13).25d In these polymers, the benzene ring seemed to be the best isolation group, and the largest value of d33 reached 68.7 pm V−1 (P49).
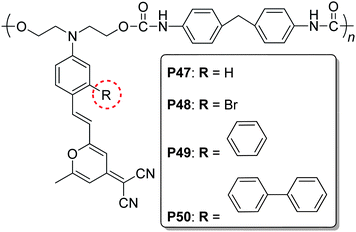 | ||
| Chart 13 The structures of P47–P50. | ||
In all of the above cases, the isolation groups used were non-polar groups to decrease the strong interactions between the chromophore moieties, and did not directly contribute to the macroscopic NLO effect but decrease the effective concentration of the NLO chromophore moieties. Then, how about using one chromophore with lower μβ value as the isolation group for another chromophore group with high μβ value, to achieve high poling efficiency, as the result of the decreased strong electronic interactions? If it works, the macroscopic NLO effect should be further enhanced. To answer this question, P53, constructed from two different chromophore moieties with the regular AB structure (Chart 14) was designed. For comparison, P51 and P52 were also prepared. P53 exhibited the highest d33 value (116.8 pm V−1), while the d33 values of P51 and P52, were only 78.1 and 34.3 pm V−1, respectively. This indicated that the sulfonyl-based chromophore could act as effective isolation group for the nitro-based one, the main chromophore moieties. Thus, the interesting results obtained might open up a new avenue for the further development of the concept of “suitable isolation group”, and it was believed that other very exciting results (i.e. even greater macroscopic NLO effects) could be achieved by the rational design of the molecular structure.
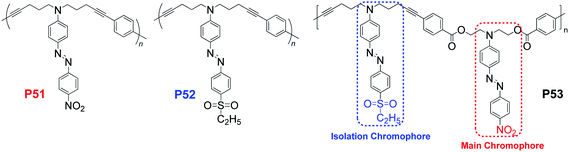 | ||
| Chart 14 The structures of P51–P53. | ||
In all the above dendronized NLO polymers, there was only one IG bonded to the chromophore moieties, and the preliminary experimental results demonstrated that there was an SIG present to boost the μβ value of the chromophore moieties to possibly higher macroscopic NLO effect of the chromophore-containing polymers. Thus, if there were two IGs bonded to the chromophore moieties, the bulk of the SIG should be smaller than that in the above cases of only one IG linked onto the chromophore moieties. This thought was partially proved by the experimental results of P54–P57 (Chart 15), in which the bulk of one IG was fixed while another being changed from small to larger.26P55 demonstrated the highest d33 value (127.7 pm V−1) among them, and the SIG was BOE, much smaller than carbazolyl group in P11, although the chromophore moieties were nearly the same in these polymers. As a deduction, if more than two IGs were linked to the chromophore moieties, the suitable isolation should be even smaller. To prove this idea, also considering the synthetic convenience, we designed a series of NLO dendrimers through the Sharpless “click” reaction, in which the newly formed triazole rings during the click chemistry reaction might act as the SIG to enhance the macroscopic NLO effects, according to the concept of “suitable isolation group”.27
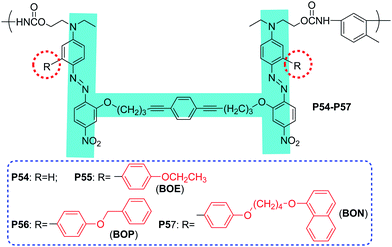 | ||
| Chart 15 The structures of P54–P57. | ||
As shown in Scheme 1, G0–![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) reacted with compound 13via click reaction under room temperature to yield G1. Then, through the efficient azo coupling reaction at 0 °C, another constructing block of the chromophore group with a push–pull structure was formed, resulting in three chromophore moieties in G1–, in comparison with two in G1. Simultaneously, one reactive alkyne group was introduced for the further growing of the dendrimers. Repeating the click reaction, and then azo coupling reaction, G1– could be easily converted to G2, and then G2–. Through another click reaction, G3, which totally contain 14 chromophore moieties, was obtained in good isolated yield (69.8%). So, the successful examples of these NLO dendrimers exhibited the flexible synthetic power with the combination of two efficient chemical reactions, and might give light on the syntheses of other functional dendrimers from more economic preparation routes.
reacted with compound 13via click reaction under room temperature to yield G1. Then, through the efficient azo coupling reaction at 0 °C, another constructing block of the chromophore group with a push–pull structure was formed, resulting in three chromophore moieties in G1–, in comparison with two in G1. Simultaneously, one reactive alkyne group was introduced for the further growing of the dendrimers. Repeating the click reaction, and then azo coupling reaction, G1– could be easily converted to G2, and then G2–. Through another click reaction, G3, which totally contain 14 chromophore moieties, was obtained in good isolated yield (69.8%). So, the successful examples of these NLO dendrimers exhibited the flexible synthetic power with the combination of two efficient chemical reactions, and might give light on the syntheses of other functional dendrimers from more economic preparation routes.
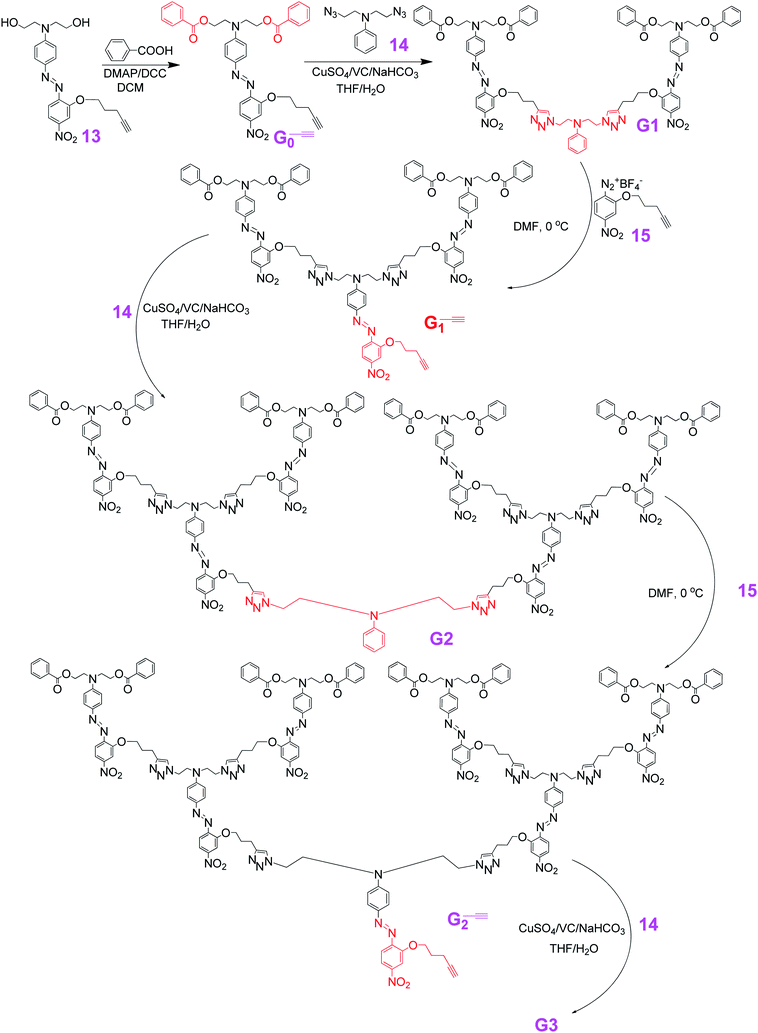 | ||
| Scheme 1 | ||
The NLO effects of G1–G3 were much higher (Table 3) than our other NLO polymers carrying similar azobenzene chromophores including those with SIGs mentioned above, this should be ascribed to the dendritic structure of these NLO dendrimers and the good isolation effect of triazole and benzoic groups. From the first generation (G1) to the third one (G3), the tested NLO values increased more than 20%, partially proving the more apparent effective site isolation effect. Upon the growth of the NLO dendrimers, the loading density of the effective chromophore moieties increased accordingly. In this case, the density was raised from 0.402 for G1 to as high as 0.520 in G3, which really possessed a very high loading density of effective chromophore moieties (Table 3). It is well-known that in theory,16 under identical experimental conditions, d33 is proportional to the density of the chromophore moieties (eqn (3)), however, due to the significantly stronger chromophore–chromophore electrostatic interaction, the relationship between the chromophore density and the NLO effect was not linear any longer, and there was a maximum value of NLO effect at a certain concentration of the chromophore moieties. Further increasing the loading level of chromophore moieties could only decrease the NLO effect. By the introduction of isolation spacers according to the site isolation principle, the loading density of the chromophore moieties could be dramatically increased before the maximum NLO value achieved; however, there was still a critical point and the relationship was not linear. Here, the loading density increased upon the growth of NLO dendrimers, correspondingly, the tested NLO effect became larger, demonstrating a deviation from the dipolar frustration that typically limits the NLO effect in conventional chromophore/polymer composite materials. The tested order parameter (Φ) of G1–G3 (Table 3) also confirmed that the alignment of the chromophore moieties under electric field poling became easier, accompanying with the growth of the generation number. This indicated that triazole groups acted as good IGs to minimize the strong intermolecular dipole–dipole interactions among the chromophore moieties upon the growth of the NLO dendrimers, realizing our thought of the utilization of triazole rings as good isolation spacers. Combined with the recent work of Sullivan, Robinson and Dalton, and their prediction,28 this exciting phenomenon also disclosed that by choosing suitable isolation spacers and through rational design, it was possible to experimentally realize the linear relationship between the loading density of chromophore moieties and the electro-optical activity. G0–G3 were also bonded to polyfluorenes as side chains, and the experimental results further confirmed the above abnormal phenomena again: accompanying with the increasing of the loading density of the chromophore moieties, the macroscopic NLO effects of the dendronized polyfluorenes became even higher.29
| no. | d 33 a(pm V−1) | Φ b | N c |
|---|---|---|---|
| a Second harmonic generation (SHG) coefficient. b Order parameter Φ = 1 − A1/A0, A1 and A0 are the absorbance of the polymer film after and before corona poling, respectively. c the loading density of the effective chromophore moieties. | |||
| G1 | 100.0 | 0.20 | 0.402 |
| G2 | 108.1 | 0.18 | 0.488 |
| G3 | 122.7 | 0.25 | 0.520 |
| G4 | 177.0 | 0.27 | 0.537 |
| G5 | 193.1 | 0.31 | 0.544 |
To further check the exciting results, we also prepared G4 and G5 (Chart 16), bearing 30 and 62 NLO azo-benzene chromophore moieties, respectively, by the double usage of “click chemistry” in the combination strategy of divergent and convergent approaches.30 Excitingly, accompanying the loading concentration of the chromophore moieties changing from 0.520 in G3 to 0.537 in G4, then to 0.544 in G5, the measured NLO coefficient values increased from 122.7 (G3) to 177.0 (G4), then to 193.1 pm V−1 (G5), further confirming that the frequently observed asymptotic dependence of electro-optic activity on the chromophore number density may be overcome through rational design (Table 3). It was noted that although the loading density of the chromophore moieties increased by only 0.017 from G3 to G4, the d33 value was enhanced 55 pm V−1, while only about 14 pm V−1 from G2 to G3, no matter that the increase of the loading density was 0.032. From G4 to G5, the loading density increased only 0.007, however, the d33 value was still enhanced 16 pm V−1. This might indicate that after the loading density of the chromophore moieties increased higher than a critical point, accompanying with the increasing of the loading density, the NLO effect would be enhanced dramatically. Thus, our results could provide some useful information for the rational design of NLO materials with better performance.
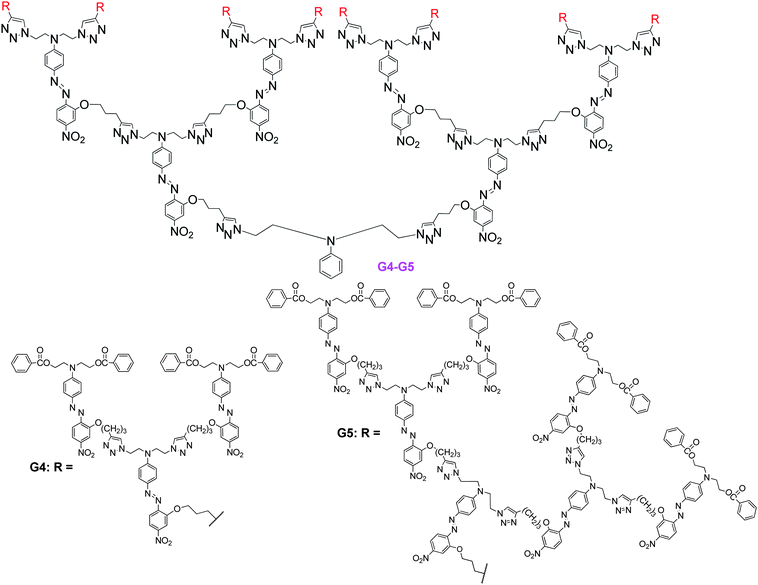 | ||
| Chart 16 The structures of G4 and G5. | ||
New NLO hyperbranched polymers
Similar to dendrimers, hyperbranched polymers possess many unusual properties in comparison to conventional linear polymers. In particular, both of them are globular macromolecules, well suited for the ideal shape modification of NLO chromophores. Also, the three-dimensional spatial separation of the chromophores endows the polymers with a favorable site-isolation effect, and their void-rich topological structure helps to minimize optical loss in the NLO process. Dendrimers are normally tedious to synthesize and are commonly used by doping into film-forming polymer matrixes. In sharp contrast, hyperbranched polymers are easy to synthesize and can be used in pure forms without doping into other matrixes because of their own film-forming ability.31 Earlier in 1997, Zhang et al. reported the synthesis and NLO properties of some new hyperbranched polymers, however, their work did not attract much interest due to the low NLO effect (∼2.8 pm V−1).32 The low d33 value was probably caused by the difficulty in alignment of the NLO chromophore moieties, which were at the cross positions of the branches, like the head-to-tail polymers. In 2004, Li and Tang prepared two azobenzene-containing hyperbranched poly(arylene ethynylene)s (hb-PAEs) (P58 and P59, Chart 17) by palladium-catalyzed coupling of triiodoarenes with a diethynyl azobenzene.33P58 and P59 were soluble, film-forming, and morphologically stable (Tg > 180 °C). Their poled films exhibited high second-harmonic generation coefficients (d33 up to 177 pm V−1), thanks to the chromophore-separation and site-isolation effects of hyperbranched architectural structure of the polymers in the three dimensional space. The optical nonlinearities of the poled films were thermally stable (no drop in d33 when heated up to 152 °C), due to the facile cross-linking of the multiple acetylenic triple bonds in the hb-PAEs at moderate temperatures (down to 88 °C). Then in 2005, Wang et al. designed some hyperbranched oligomeric dopants, when the concentration was about 15 wt% in host polymers, the resultant polymeric system exhibited high NLO effects, with r33 values up to 65 pm V−1.34 And the optical nonlinearities of the poled films remained about 90% of the original effects, even stored at 85 °C for 1200 h under an atmosphere of nitrogen.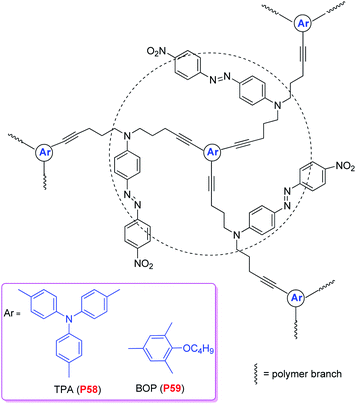 | ||
| Chart 17 The structures of P58 and P59. | ||
Stimulated by these successful examples, we obtained three hyperbranched polymers (P60–P62, Chart 18) containing second-order nonlinear optical chromophores through copolymerization of aromatic dialdehydes (carbazole, triphenylamine or benzene moieties) with sulfonyl-based chromophores bearing three active methelene groups, from “A2 + B3” approach based on simple Knoevenagel reaction. For comparison, their corresponding linear analogue polymers (P63–P65) were also prepared (Chart 19).35 Although the concentrations of the active sulfonyl-based chromophore moieties in hyperbranched polymers were lower than those of their corresponding linear analogues, the tested NLO properties of the hyperbranched polymers were much better than their linear analogues (up to ∼4 times). This indicated that the 3D architectural structure of the hyperbranched polymers and the spatial chromophore isolation in the macromolecular spheres were beneficial to enhance their optical nonlinearities. Thus, hyperbranched NLO polymers might be another choice besides NLO dendrimers and polymers containing dendronized chromophores as side chains, to efficiently translate large molecular first hyperpolarizability (β) value of chromophores into high macroscopic second-harmonic generation (SHG) coefficient of materials.
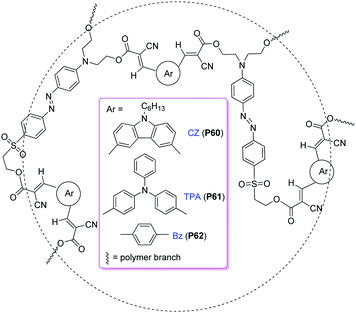 | ||
| Chart 18 The structures of P60–P62. | ||
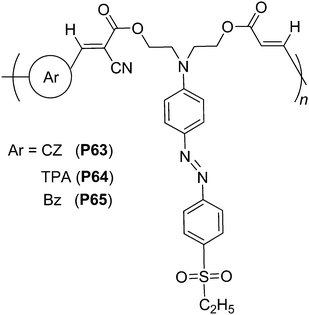 | ||
| Chart 19 The structures of P63–P65. | ||
To construct hyperbranched polymers with the structure more like dendrimers to achieve higher macroscopic NLO effects, and considering the remarkable features of the click chemistry reactions and the good isolation effect of the formed triazole rings in the click chemistry reaction mentioned above,27,29 we have designed a modified synthetic approach with the usage of end-capping azo molecules, so that the self-oligomerization of AB2 monomers reported previously36 could be avoided, to produce new azo chromophore-containing hyperbranched polymers P66 from their corresponding AB2 monomers (Scheme 2).37a These two polymers were easily soluble in common organic solvent, and could form thin films easily. The tested NLO properties demonstrated better macroscopic NLO effects (up to 124.4 pm V−1), in comparison with their linear analogues containing the chromophores with similar structure. Thus, coupled with their convenient synthesis and high thermal stability of the NLO activities, P66 could be good candidates for practical photonic applications.
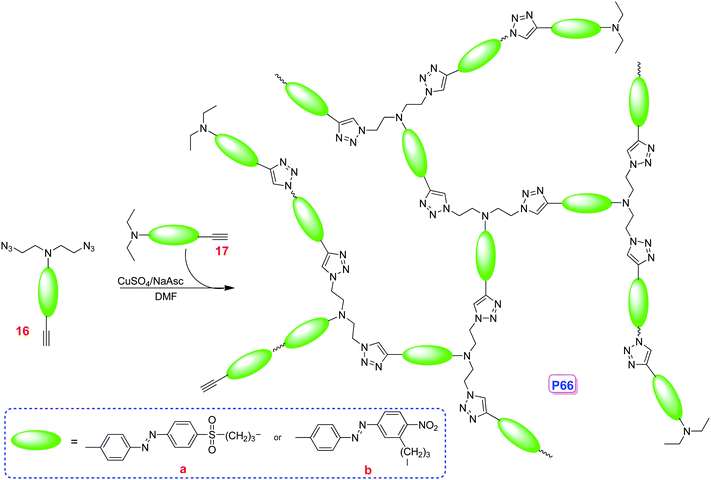 | ||
| Scheme 2 | ||
Through this modified synthetic approach from AB2 monomers, some other hyperbranched polymers with different end-capping groups could be obtained, which exhibited interesting macroscopic NLO effects.37b Also, some pentafluorophenyl could be introduced to the resultant hyperbranched polymers, to further enhance the NLO effects.37c
Recently, Odobel and co-workers designed and synthesized two hyperbranched polymers end-capped with azo chromophores through postfunctionalization (Chart 20). The postfunctionalization of the hyperbranched polymers provided a new way to attach the NLO chromophores to the polymers, which could rapidly tune the properties of the materials (the structure of the linker between the chromophore and the polymer, the concentration and nature of the chromophore). The resulting hyperbranched polymers P67 and P68 exhibited good stability and relatively higher d33 values.38
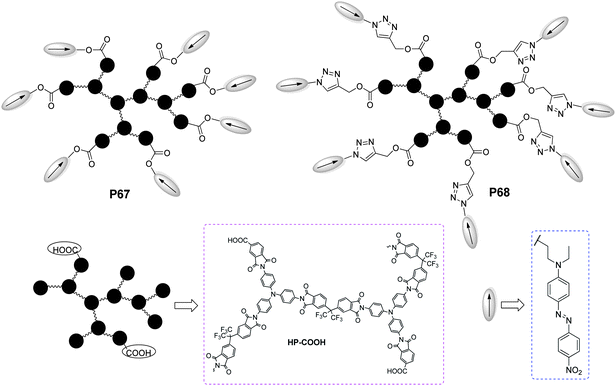 | ||
| Chart 20 The structures of P67 and P68. | ||
New NLO supramolecular self-assembled molecules
In order to exploit new methods to construct the NLO materials, the principles of supramolecular chemistry have been introduced.39 Yu and co-workers reported a new series of NLO supramolecular self assemblies, which were built up through the spontaneous formation of hydrogen bonds between NLO chromophores with mild preparatory conditions (Chart 21), and the resulting NLO supramolecular self assemblies exhibited long-term stabilities, and the largest value of r33 reached 70 pm V−1.39b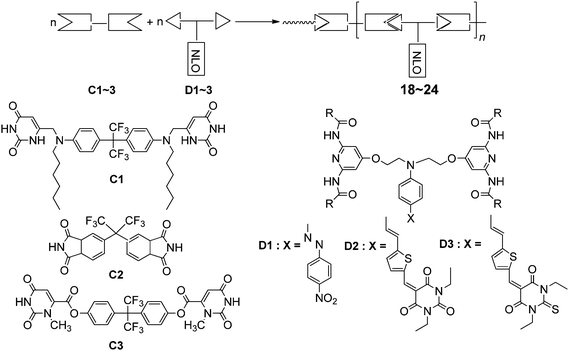 | ||
| Chart 21 The structures of 18–24. | ||
As a new class of supramolecular materials, intricate π–π interactions offer a very effective method to organize dipole chromophores to improve the poling efficiency. Jen’s group developed a new class of molecular glasses based on the reversible self-assembly of aromatic/perfluoroaromatic (Ar–ArF) dendron-substituted NLO chromophores (Chart 22 and 23).40 Because of the interaction of the Ar–ArF interactions, the r33value of chromophore 27 (108 pm V−1) was two times higher than that of chromophore 25 (51 pm V−1) and 26 (52 pm V−1). To further enhance the r33 values of these materials, the molecules were used as hosts in the binary chromophore system, and the ultrahigh r33 value of 327 pm V−1 was achieved in the system (27/28 = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1).
:1).
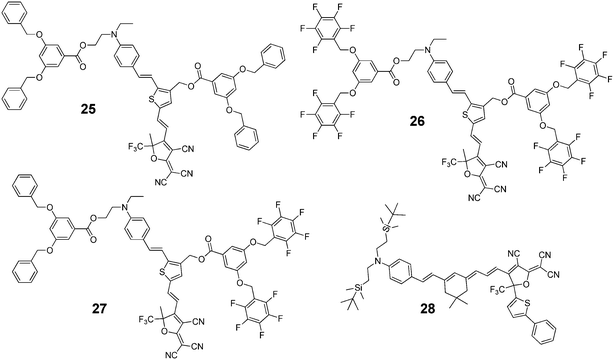 | ||
| Chart 22 The structures of glass forming chromophores 25–28. | ||
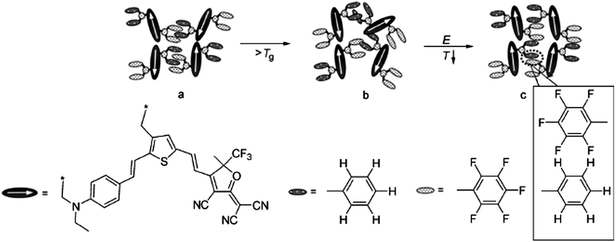 | ||
| Chart 23 Graphical illustration of the alignment formation of self assembled chromophore 27 by Ar–ArF interactions: (a) locked random dipoles (shown as arrows) before poling; (b) unlocked random dipoles before poling; (c) locked acentric dipoles after poling followed by cooling. | ||
Conclusion
This article reviews some new design strategies for the development of second-order nonlinear optical polymers, including dendronized polymers, dendrimers and hyperbranched polymers with the new concept of the “suitable isolation group”. Due to the length limitation, we have mainly focused on our own work. Some related works published by other groups, including some original works and review papers, have been cited in the references for interested readers. It should be pointed out that a large amount of outstanding work has been done by many other scientists in the second order NLO materials field over the past decades, but could not all be included in this article.It is still a big challenge to efficiently translate the large β values of the organic chromophores into high macroscopic NLO activities of polymers, due to the strong intermolecular electronic interactions between the chromophore moieties in the polymeric system. The proposed concept of “suitable isolation group” might provide some useful information for the rational design of new NLO polymers. Especially, the examples of NLO dendrimers demonstrated that after the loading density of the chromophore moieties increased beyond a critical point, the NLO effect would be enhanced dramatically with further increases in loading density, indicating that the frequently observed asymptotic dependence of electro-optic activity on the chromophore number density may be overcome through rational design. Also, the convenient one-pot synthesis of hyperbranched polymers, coupled with their exciting experimental results, demonstrate that they are another kind of promising candidates for practical applications in photonic fields.41 Although the above interesting results were obtained in different polymeric systems containing chromophore moieties with relatively low dipole moments, it was reasonable to predict that the concept of “suitable isolation group” could be extended to the fields of polymeric systems bearing much more polar chromophores. Thus, it is believed that novel NLO polymers with much better macroscopic NLO effects can be achieved through rational design and better research work.
Acknowledgements
We are grateful to the National Science Foundation of China (no. 21034006) for financial support. We acknowledge the contributions from our students and collaborators, whose names are given in the references section.References
- F. A. Franken, A. E. Hill, C. W. Peters and G. Weinreich, Phys. Rev. Lett., 1961, 7, 118 CrossRef.
- For example, see (a) M. J. Cho, D. H. Choi, P. A. Sullivan, A. J. P. Akelaitis and L. R. Dalton, Prog. Polym. Sci., 2008, 33, 1013–1058 CrossRef CAS; (b) D. M. Burland, R. D. Miller and C. A. Walsh, Chem. Rev., 1994, 94, 31 CrossRef CAS; (c) D. R. Kanis, M. A. Ratner and T. J. Marks, Chem. Rev., 1994, 94, 195 CrossRef CAS; (d) J. L. Bredas, C. Adant, P. Tackx, A. Persoons and B. M. Pierce, Chem. Rev., 1994, 94, 243 CrossRef CAS; (e) W. E. Moerner, A. G. Jepsen and C. L. Thompson, Annu. Rev. Mater. Sci., 1997, 32, 585 CrossRef; (f) P. N. Prasad and D. J. Williams, Introduction to Nonlinear Optical Effects in Molecules and Polymers, John Wiley and Sons, New York, 1991 Search PubMed; (g) J. Zyss, Molecular Nonlinear Optics: Materials, Physics and Devices, Academic Press, Boston, 1994 Search PubMed; (h) J. Zyss and I. Ledoux, Chem. Rev., 1994, 94, 77 CrossRef CAS; (i) L. R. Dalton, P. A. Sullivan and D. H. Bale, Chem. Rev., 2010, 110, 25 CrossRef CAS; (j) S. R. Marder, in Inorganic Materials, ed. D. W. Bruce and D. O'Hare, Wiley, New York, 1992, p. 116 Search PubMed; (k) Materials for Nonlinear Optics, Chemical Perspectives, ed. S. R. Marder, J. E. Sohn and G. D. Stucky, ACS Symposium Series 455, American Chemical Society, Washington DC, 1991 Search PubMed; (l) Nonlinear Optics of Organic Molecules and Polymers, ed. H. S. Nalwa and S. Miyata, CRC Press, Boca Raton, FL, 1997 Search PubMed; (m) B. J. Coe, Chem.–Eur. J., 1999, 5, 2464 CrossRef CAS; (n) S. D. Bella, Chem. Soc. Rev., 2001, 30, 355 RSC; (o) C. Chen and G. Liu, Annu. Rev. Mater. Sci., 1986, 16, 203 CrossRef CAS; (p) F. Chaumel, H. Jiang and A. Kakkar, Chem. Mater., 2001, 13, 3389 CrossRef CAS; (q) O. R. Evans and W. Lin, Acc. Chem. Res., 2002, 35, 511 CrossRef CAS.
- For example, see (a) S. R. Marder, Chem. Commun., 2006, 131 RSC; (b) L. R. Dalton, Adv. Polym. Sci., 2002, 158, 1 CAS; (c) F. Kajzar, K. Lee and A. K.-Y. Jen, Adv. Polym. Sci., 2003, 161, 1 CAS; (d) L. Yu, W. K. Chan, Z. Peng and A. Gharavi, Acc. Chem. Res., 1996, 29, 13 CrossRef CAS; (e) T. J. Marks and M. A. Ratner, Angew. Chem., Int. Ed. Engl., 1995, 34, 155 CrossRef CAS; (f) H. M. Kim and B. R. Cho, Chem. Commun., 2009, 153 RSC; (g) Y. Zhang, R. Burzynski, S. Ghosal and M. K. Casstevens, Adv. Mater., 1996, 8, 111 CAS and references therein; (h) M. Lee, H. E. Katz, C. Erben, D. M. Gill, P. Gopalan, J. D. Heber and D. J. McGee, Science, 2002, 298, 1401 CrossRef CAS; (i) Y. Shi, C. Zhang, H. Zhang, J. H. Bechtel, L. R. Dalton, B. H. Robinson and W. H. Steier, Science, 2000, 288, 119 CrossRef CAS; (j) G. S. He, L.-S. Tan, Q. Zheng and P. N. Prasad, Chem. Rev., 2008, 108, 1245 CrossRef CAS; (k) S. R. Marder and J. W. Perry, Science, 1994, 263, 1706 CAS; (l) S. R. Marder, B. Kippelen, A. K. Y. Jen and N. Peyghambarian, Nature, 1997, 388, 845 CrossRef CAS.
- http://artus.et7.tu-harburg.de/%7Eet7www/phpsite/forschung/show.php%3Ffile%3Doptik/index.html .
- W. E. Moerner and S. M. Silence, Chem. Rev., 1994, 94, 127 CrossRef CAS.
- (a) H. Tang, J. Qin, X. Deng and Y. Cao, Organometallics, 2004, 23, 6070 CrossRef CAS; (b) C. Zhan, H. Zeng, J. Qin, D. Liu, N. Cheng and Y. Cui, Synth. Met., 1997, 84, 397 CrossRef; (c) J. Luo, C. Zhan and J. Qin, React. Funct. Polym., 2000, 44, 219 CrossRef CAS; (d) J. Luo, J. Qin, H. Kang and C. Ye, Polym. Int., 2000, 49, 1302 CrossRef CAS; (e) Z. Li, J. Luo, J. Li, C. Zhan and J. Qin, Polym. Bull., 2000, 45, 105 CrossRef CAS; (f) Z. Li, C. Zhan and J. Qin, Chin. Chem. Lett., 2000, 11, 103 CAS; (g) H. Tang and J. Qin, Chin. Chem. Lett., 2000, 11, 675 CAS; (h) J. Li, P. Ren, C. Zhan and J. Qin, Chin. Chem. Lett., 1999, 10, 261 CAS; (i) J. Li, Z. Li, C. Zhan, J. Qin and Y. Li, Synth. Met., 1999, 101, 127 CrossRef CAS; (j) H. Tang, J. Li and J. Qin, React. Funct. Polym., 2001, 48, 193–9 CrossRef CAS.
- (a) Z. Lu, P. Shao, J. Li, J. Hua, J. Qin, A. Qin and C. Ye, Macromolecules, 2004, 37, 7089 CrossRef CAS; (b) Z. Li, J. Li and J. Qin, React. Funct. Polym., 2001, 48, 113–8 CrossRef CAS; (c) Z. Li, J. Qin, Z. Yang and C. Ye, Chin. Chem. Lett., 2004, 15, 489 CAS; (d) Y. Liu, X. Zhang, H. Tang, T. Wang, J. Qin, D. Liu, S. Li and C. Ye, Chin. J. Chem., 2002, 20, 1199 CAS; (e) J. Li, Z. Li, H. Tang, H. Zeng and J. Qin, J. Organomet. Chem., 2003, 685, 258 CrossRef CAS; (f) Z. Li, J. Qin, S. Li and C. Ye, Chin. J. Chem., 2003, 21, 1395 CAS; (g) J. Hua, W. Zhang, J. Qin, S. Li, C. Ye, Z. Lu, X. Wang and M. Gui, Synth. Met., 2003, 135–136, 465 CrossRef CAS; (h) Z. Li, J. Qin, S. Li and C. Ye, Synth. Met., 2003, 135–136, 467 CrossRef CAS; (i) Z. Li, J. Qin, H. Tang and Y. Liu, J. Appl. Polym. Sci., 2003, 89, 2989 CrossRef CAS; (j) J. Hua, J. Luo, K. Long, J. Qin, S. Li, C. Ye and Z. Lu, Eur. Polym. J., 2004, 40, 1193 CrossRef CAS.
- (a) H. Tang, Y. Liu, B. Huang, J. Qin, C. Fuentes-Hernandez, B. Kippelen, S. Li and C. Ye, J. Mater. Chem., 2005, 15, 778 RSC; (b) Z. Lu, J. Li, J. Hua, X. Li, J. Qin, A. Qin and C. Ye, Synth. Met., 2005, 152, 217 CrossRef CAS; (c) J. Li, J. Qin and C. Ye, Synth. Met., 2005, 152, 305 CrossRef CAS; (d) Z. Li, J. Qin, Z. Yang and C. Ye, J. Appl. Polym. Sci., 2004, 94, 769 CrossRef CAS; (e) Z. Lu, Y. Liu, J. Qin, Z. Yang and C. Ye, React. Funct. Polym., 2004, 61, 379 CrossRef CAS; (f) H. Zhen, C. Zhan, J. Qin, D. Liu, N. Cheng and Y. Cui, Chem. J. Chin. Univ., 1995, 16(Nov. Supplement), 206; (g) J. Hua, Z. Li, K. Long, J. Qin, S. Li, C. Ye and Z. Lu, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 1317 CrossRef CAS.
- (a) J. Luo, J. Qin, H. Kang and C. Ye, Chem. Mater., 2001, 13, 927 CrossRef CAS; (b) Z. Li, J. Qin, S. Li, C. Ye, J. Luo and Y. Cao, Macromolecules, 2002, 35, 9232 CrossRef CAS; (c) J. Li, P. Ren, C. Zhan and J. Qin, Polym. Int., 1999, 48, 491 CrossRef CAS; (d) H. Tang, J. Luo, J. Qin, H. Kang and C. Ye, Macromol. Rapid Commun., 2000, 21, 1125 CrossRef CAS.
- (a) J. Luo, H. Ma, M. Haller and R. R. Barto, Chem. Commun., 2002, 8, 888 Search PubMed; (b) T. C. Kowalczyk, T. Kosc, K. D. Singer, P. A. Cahill, C. H. Seager, M. B. Meinhardt, A. J. Beuhler and D. A. Wargowski, J. Appl. Phys., 1994, 76, 2505 CrossRef CAS; (c) F. Wang, A. W. Harper, M. S. Lee and L. R. Dalton, Chem. Mater., 1999, 11, 2285 CrossRef CAS.
- (a) K. Y. Sandhya, C. K. S. Pillai and N. Tsutsumi, Prog. Polym. Sci., 2004, 29, 45 CrossRef CAS; (b) D. Yu, A. Gharavi and L. P. Yu, J. Am. Chem. Soc., 1995, 117, 11680 CrossRef CAS; (c) L. R. Dalton, A. W. Harper and B. H. Robinson, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 4842 CrossRef CAS.
- (a) B. H. Robinson and L. R. Dalton, J. Phys. Chem. A, 2000, 104, 4785 CrossRef CAS; (b) B. H. Robinson, L. R. Dalton, H. W. Harper, A. Ren, F. Wang, C. Zhang, G. Todorova, M. Lee, R. Aniszfeld, S. Garner, A. Chen, W. H. Steier, S. Houbrechts, A. Persoons, I. Ledoux, J. Zyss and A. K.-Y. Jen, Chem. Phys., 1999, 245, 35 CrossRef CAS; (c) L. R. Dalton, W. H. Steier, B. H. Robinson, C. Zhang, A. Ren, S. Garner, A. Chen, T. Londergan, L. Irwin, B. Carlson, L. Fifield, G. Phelan, C. Kincaid, J. Amend and A. K.-Y. Jen, J. Mater. Chem., 1999, 9, 1905 RSC.
- (a) J. M. J. Fréchet, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4782 CrossRef CAS; (b) J. M. J. Fréchet, C. J. Hawker, I. Gitsov and J. W. Leon, J. Macromol. Sci., Part A:Pure. Appl. Chem., 1996, A33, 1399 Search PubMed.
- (a) P. A. Sullivan, A. J.-P. Akelaitis, S. K. Lee, G. McGrew, S. K. Lee, D. H. Choi and L. R. Dalton, Chem. Mater., 2006, 18, 344 CrossRef CAS; (b) S. R. Hammond, O. Clot, K. A. Firestone, D. H. Bale, D. Lao, M. Haller, G. D. Phelan, B. Carlson, A. K.-Y. Jen, P. J. Reid and L. R. Dalton, Chem. Mater., 2008, 20, 3425 CrossRef CAS; (c) H. Ma and A. K.-Y. Jen, Adv. Mater., 2001, 13, 1201 CrossRef CAS; (d) H. Ma, S. Liu, J. Luo, S. Suresh, L. Liu, S. H. Kang, M. Haller, T. Sassa, L. R. Dalton and A. K.-Y. Jen, Adv. Funct. Mater., 2002, 12, 565 CrossRef CAS; (e) J. D. Luo, S. Liu, M. Haller, L. Liu, H. Ma and A. K.-Y. Jen, Adv. Mater., 2002, 14, 1763 CrossRef CAS; (f) T.-D. Kim, J.-W. Kang, J. D. Luo, S.-H. Jang, J.-W. Ka, N. Tucker, J. B. Benedict, L. R. Dalton, T. Gray, R. M. Overney, D. H. Park, W. N. Herman and A. K.-Y. Jen, J. Am. Chem. Soc., 2007, 129, 488 CrossRef CAS; (g) W. Shi, J. Luo, S. Huang, X.-H. Zhou, T.-D. Kim, Y.-J. Cheng, B. M. Polishak, T. R. Younkin, B. A. Block and A. K.-Y. Jen, Chem. Mater., 2008, 20, 6372 CrossRef CAS.
- (a) Y. Okuno, S. Yokoyama and S. Mashiko, J. Phys. Chem. B, 2001, 105, 2163 CrossRef CAS; (b) S. Yokoyama, T. Nakahama, A. Otomo and S. Mashiko, J. Am. Chem. Soc., 2000, 122, 3174 CrossRef CAS; (c) P. Busson, J. Örtegren, H. Ihre, U. W. Gedde, A. Hult, G. Andersson, A. Eriksson and M. Lindgren, Macromolecules, 2002, 35, 1663 CrossRef CAS; (d) O. Varnavski, A. Leanov, L. Liu, J. Takacs and T. Goodson, III, J. Phys. Chem. B, 2000, 104, 179 CrossRef CAS; (e) P. Gopalan, H. E. Katz, D. J. McGee, C. Erben, T. Zielinski, D. Bousquet, D. Muller, J. Grazul and Y. Olsson, J. Am. Chem. Soc., 2004, 127, 1741 CrossRef; (f) V. E. Campbell, I. In, D. J. McGee, N. Woodward, A. Caruso and P. Gopalan, Macromolecules, 2006, 39, 957 CrossRef CAS; (g) J. Y. Do, S. K. Park, J. Ju, S. Park and M. Lee, Macromol. Chem. Phys., 2003, 204, 410 CrossRef CAS.
- C. R. Moylan, R. D. Miller, R. J. Twieg, V. Y. Lee, I. H. McComb, S. Ermer, S. M. Lovejoy and D. S. Leung, Proc. SPIE–Int. Soc. Opt. Eng., 1995, 2527, 150 CAS.
- Z. Li, Z. Li, C. Di, Z. Zhu, Q. Li, Q. Zeng, K. Zhang, Y. Liu, C. Ye and J. Qin, Macromolecules, 2006, 39, 6951 CrossRef CAS.
- Z. Li, Q. Zeng, Z. Li, S. Dong, Z. Zhu, Q. Li, C. Ye, C. Di, Y. Liu and J. Qin, Macromolecules, 2006, 39, 8544 CrossRef CAS.
- (a) Z. Li, P. Li, S. Dong, Z. Zhu, Q. Li, Q. Zeng, Z. Li, C. Ye and J. Qin, Polymer, 2007, 47, 3650 CrossRef; (b) Z. Zhu, Q. Li, Q. Zeng, Z. Li, Z. Li, J. Qin and C. Ye, Dyes Pigm., 2008, 78, 199 CrossRef CAS.
- Z. Li, S. Dong, G. Yu, Z. Li, Y. Liu, C. Ye and J. Qin, Polymer, 2007, 47, 5520 CrossRef.
- (a) Q. Li, Z. Li, F. Zeng, W. Gong, Z. Li, Z. Zhu, Q. Zeng, S. Yu, C. Ye and J. Qin, J. Phys. Chem. B, 2007, 111, 508 CrossRef CAS; (b) Q. Li, Z. Li, C. Ye and J. Qin, J. Phys. Chem. B, 2008, 112, 4928 CrossRef CAS.
- W. Gong, Q. Li, Z. Li, C. Lu, J. Zhu, S. Li, J. Yang, Y. Cui and J. Qin, J. Phys. Chem. B, 2006, 110, 10241 CrossRef CAS.
- (a) Z. Li, C. Huang, J. Hua, J. Qin, Z. Yang and C. Ye, Macromolecules, 2004, 37, 371 CrossRef CAS; (b) H. Moon, J. Hwang, N. Kim and S. Y. Park, Macromolecules, 2000, 33, 5116 CrossRef CAS; (c) Z. Li, J. Qin, S. Li and C. Ye, Synth. Met., 2003, 135–136, 467 CrossRef CAS; (d) Z. Li, J. Qin, A. Qin and C. Ye, Polymer, 2005, 46, 363 CrossRef CAS; (e) Z. Li, J. Qin, Z. Yang and C. Ye, J. Appl. Polym. Sci., 2004, 94, 769 CrossRef CAS; (f) Z. Li, C. Huang, J. Hua, J. Qin, Z. Yang and C. Ye, Acta Chim. Sin., 2004, 62, 410 Search PubMed; (g) Z. Li, J. Qin, S. Li and C. Ye, Chin. J. Chem., 2003, 21, 1395 CAS; (h) Z. Li, W. Gong, J. Qin, Z. Yang and C. Ye, Polymer, 2005, 46, 4971 CAS; (i) Z. Li, J. Hua, Q. Li, C. Huang, A. Qin, C. Ye and J. Qin, Polymer, 2005, 46, 11940 CrossRef CAS.
- (a) Q. Zeng, Z. Li, Z. Li, C. Ye, J. Qin and B. Z. Tang, Macromolecules, 2007, 40, 5634 CrossRef CAS; (b) Z. Li, W. Wu, P. Hu, X. Wu, G. Yu, Y. Liu, C. Ye, Z. Li and J. Qin, Dyes Pigm., 2009, 81, 264 CrossRef CAS; (c) Q. Zeng, G. Qiu, C. Ye, J. Qin and Z. Li, Dyes Pigm., 2010, 84, 229 CrossRef CAS; (d) Z. Li, W. Wu, C. Ye, J. Qin and Z. Li, Macromol. Chem. Phys., 2010, 211, 916–923 CAS; (e) Z. Li, L. Ji, R. Tang, L. Huang, C. Ye, J. Qin and Z. Li, Polym. Adv. Technol., 2011, 22, 675–681 CrossRef CAS; (f) Z. Li, W. Wu, C. Ye, J. Qin and Z. Li, J. Phys. Chem. B, 2009, 113, 14943 CrossRef CAS; (g) Z. Li, P. Hu, G. Yu, W. Zhang, Z. Jiang, Y. Liu, C. Ye, J. Qin and Z. Li, Phys. Chem. Chem. Phys., 2009, 11, 1220 RSC; (h) Q. Li, G. Yu, J. Huang, H. Liu, Z. Li, C. Ye, Y. Liu and J. Qin, Macromol. Rapid Commun., 2008, 29, 798 CrossRef CAS; (i) Z. Li, G. Yu, Z. Li, Y. Liu, C. Ye and J. Qin, Polymer, 2008, 48, 901 CrossRef.
- (a) Z. Li, S. Dong, P. Li, Z. Li, C. Ye and J. Qin, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 2983 CrossRef CAS; (b) Z. Li, G. Yu, S. Dong, W. Wu, Y. Liu, C. Ye, J. Qin and Z. Li, Polymer, 2009, 50, 2806 CrossRef CAS; (c) Z. Li, L. Wang, B. Xiong, C. Ye, J. Qin and Z. Li, Dyes Pigm., 2010, 84, 134–139 CrossRef CAS; (d) P. Chang, J. Chen, H. Tsai and G. Hsiue, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 4937 CrossRef CAS.
- Z. Li, W. Wu, G. Yu, Y. Liu, C. Ye, J. Qin and Z. Li, ACS Appl. Mater. Interfaces, 2009, 1, 856 CrossRef CAS.
- Z. Li, G. Yu, W. Wu, Y. Liu, C. Ye, J. Qin and Z. Li, Macromolecules, 2009, 42, 3864 CrossRef CAS.
- P. A. Sullivan, H. Rommel, Y. Liao, B. C. Olbricht, A. J. P. Akelaitis, K. A. Firestone, J.-W. Kang, J. Luo, J. A. Davies, D. H. Choi, B. E. Eichinger, P. J. Reid, A. Chen, A. K. Y. Jen, B. H. Robinson and L. R. Dalton, J. Am. Chem. Soc., 2007, 129, 7523 CrossRef CAS.
- Z. Li, G. Yu, Y. Liu, C. Ye, J. Qin and Z. Li, Macromolecules, 2009, 42, 6463 CrossRef CAS.
- Z. Li, W. Wu, Q. Li, G. Yu, L. Xiao, Y. Liu, C. Ye, J. Qin and Z. Li, Angew. Chem., Int. Ed., 2010, 49, 2763–2767 CAS.
- (a) M. Jikei and M. A. Kakimoto, Prog. Polym. Sci., 2001, 26, 1233 CrossRef CAS; (b) B. I. Voit, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 2679 CrossRef CAS; (c) C. Gao and D. Yan, Prog. Polym. Sci., 2004, 29, 183 CrossRef CAS.
- Y. Zhang, T. Wada and H. Sasabe, Polymer, 1997, 38, 2893 CrossRef CAS.
- (a) Z. Li, J. W. Y. Lam, Y. Dong, Y. Dong, B. Z. Tang, A. Qin and C. Ye, Polym. Preprint, 2004, 45, 833 Search PubMed; (b) Z. Li, A. Qin, J. W. Y. Lam, Y. Dong, Y. Dong, C. Ye, I. Williams and B. Tang, Macromolecules, 2006, 39, 1436 CrossRef CAS.
- Y. Bai, N. Song, J. P. Gao, X. Sun, X. Wang, G. Yu and Z. Y. Wang, J. Am. Chem. Soc., 2005, 127, 2060 CrossRef CAS.
- Z. Zhu, Z. Li, Y. Tan, Z. Li, Q. Li, Q. Zeng, C. Ye and J. Qin, Polymer, 2006, 47, 7881 CrossRef CAS.
- A. J. Scheel, H. Komber and B. I. Voit, Macromol. Rapid Commun., 2004, 25, 1175 CrossRef CAS.
- (a) Z. Li, G. Yu, P. Hu, C. Ye, Y. Liu, J. Qin and Z. Li, Macromolecules, 2009, 42, 1589 CrossRef CAS; (b) Z. Li, W. Wu, G. Qiu, G. Yu, Y. Liu, C. Ye, J. Qin and Z. Li, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 1977 CrossRef CAS; (c) W. Wu, Y. Fu, C. Wang, C. Ye, J. Qin and Z. Li, Chem. Asian J., 2011 DOI:10.1002/asia.201100379.
- A. Scarpaci, E. Blart, V. Montembault, L. Fontaine, V. Rodriguez and F. Odobel, ACS Appl. Mater. Interfaces, 2009, 1, 1799 CrossRef CAS.
- (a) D. S. Lawrence, T. Jiang and M. Levett, Chem. Rev., 1995, 95, 2229 CrossRef CAS; (b) H. Saadeh, L. Wang and L. Yu, J. Am. Chem. Soc., 2000, 122, 546 CrossRef CAS.
- (a) T. D. Kim, J. Kang, J. Luo, S. Jang, J. Ka, N. Tucker, J. B. Benedict, L. R. Dalton, T. Gray, R. M. Overney, D. H. Park, W. N. Herman and A. K. Y. Jen, J. Am. Chem. Soc., 2007, 129, 488 CrossRef CAS; (b) X. Zhou, J. Luo, S. Huang, T. Kim, Z. Shi, Y. Cheng, S. Jang, Jr, D. B. Knorr, R. M. Overney and A. K. Y. Jen, Adv. Mater., 2009, 21, 1976 CrossRef CAS.
- Z. Li, W. Wu, C. Ye, J. Qin and Z. Li, Polym. Chem., 2010, 1, 78 RSC.
| This journal is © The Royal Society of Chemistry 2011 |