Synthesis and characterization of biodegradable polyester/polyether resins via Michael-type addition
Stefan
Theiler
a,
Stefanos E.
Diamantouros
b,
Stefan
Jockenhoevel
b,
Helmut
Keul
*a and
Martin
Moeller
*a
aInstitute of Technical and Macromolecular Chemistry, RWTH Aachen and DWI an der RWTH Aachen e.V., Forckenbeckstr. 50, 52062, Aachen, Germany. E-mail: keul@dwi.rwth-aachen.de; moeller@dwi.rwth-aachen.de
bInstitute of Applied Medical Engineering, Helmholtz Institute of RWTH Aachen University and Hospital, Pauwelsstraße 20, 52062, Aachen, Germany
First published on 17th August 2011
Abstract
Biodegradable polyester/polyether resins were prepared by crosslinking star-shaped poly(ε-caprolactone) prepolymers bearing acrylate end groups with amino-telechelic polyethersvia Michael-type addition reaction. The influence of the chemical nature of the crosslinking agent, the molar mass of the prepolymer, and the ratio prepolymer/crosslinker were investigated with respect to the swelling behavior, thermal properties, stress–strain behavior, hydrophilicity, and water sorption of the resulting resins. Resins prepared from prepolymers and/or crosslinking agents of relatively high molar mass and/or exhibiting low degree of crosslinking are semicrystalline materials which show viscoelastic behavior, while resins exhibiting high degree of crosslinking are amorphous materials which show rubbery-elastic behavior. Crosslinking of the polyester prepolymers with PEO-based crosslinkers results in hydrophilic resins with good water sorption properties, while crosslinking with PTHF- and PPO-based crosslinkers results in hydrophobic resins, which show only low water sorption ability. Residual functional end groups in the resins, either present due to incomplete crosslinking or when using molar ratio of amine to acrylate <1 or >1 in the crosslinking step, can be used for post-processing (bio) functionalization.
Introduction
In recent years much attention has been devoted to biodegradable polymers as materials for biomedical applications. Aliphatic polyesters prepared from cyclic monomers such as glycolides, lactones or lactides are the most interesting biodegradable polymers and therefore are gaining more and more interest and importance for many fields of application.1–3 Due to their biocompatibility and biodegradability, in conjunction with good mechanical properties,1,4,5 these polymers are widely used for biomedical applications such as surgical sutures,6drug delivery systems7,8 and tissue engineering scaffolds.9One of the most promising synthetic polyesters is poly(ε-caprolactone) (PCL), since it offers several advantages: (i) an easy synthetic approach viaring-opening polymerization of ε-caprolactone using different nucleophilic initiators10,11 or enzymes as catalysts,12 (ii) a wide processing range between Tm(PCL) ≈ 65 °C and the decomposition temperature of 350 °C, (iii) a rubbery consistency at room temperature due to its semicrystalline character with a low glass transition temperature (Tg(PCL) ≈ −60 °C) and (iv) the ability of hydrolytic degradation under human body conditions, generating natural metabolites, which then can be eliminated from the organism through the Krebs cycle.13,14
Despite the potential of this material several properties of PCL lower its range of application. Up to now the use of PCL is limited to long-term applications due to its low degradation rate, although it can be increased by enzymatic and chemical catalysts such as lipases or acids.15–17 To increase the degradation rate, PCL is usually copolymerized or blended with polymers that degrade faster, such as lactide or glycolide,18–20 polymerized using a hydrophilic macroinitiator such as poly(ethylene oxide)21,22 or its microstructure and/or architecture is adjusted.23,24 Furthermore, for more sophisticated applications PCL is lacking functional groups in the side chain that would allow linking of (bio) molecules for cell adhesion.25,26 Therefore special effort has been devoted to the integration of functional groups into PCL, either along the chain (achieved by copolymerization with functional caprolactone27–29) or at the chain ends. Since latter possibility is limited for linear polymers to two functional groups (the chain ends), the number of functional end groups usually is increased by changing the architecture of the polymer to star-shaped30 or brush-like molecules.23 This change in architecture is accompanied by variation of the crystallinity, melting and glass transition temperatures and, as a consequence, the degradation rate.
A more significant change in these parameters can be achieved by introducing functional groups into the PCL which can be crosslinked either thermally or photochemically. Crosslinking forces the PCL chains into defined conformations and thus crystallization of the material is considerably inhibited or completely suppressed. Resulting networks show rubbery-elastic and homogeneous degradation behavior.31 They are usually achieved by UV-irradiation of PCL bearing acrylate32 or methacrylate groups,33–35 but also crosslinking reactions with peroxides, reactions of triethoxysilane end groups and polycondensation of multifunctional carboxylic acids and polyhydric alcohols are reported.36
So far only little attention has been paid to crosslinking of PCLvia Michael37 or Michael-type addition.38,39 These crosslinking reactions offer several advantages such as tolerance of additional functional groups, mild reaction conditions (preventing the polyester degradation), elimination of solvent and side products40 and no need for metallic catalyst and thus no contamination that could cause problems in biomedical applications.41 The number of crosslinking agents in Michael-type additions with precursors bearing acrylate groups is almost unlimited since every (macro) molecule bearing at least two amine or thiol groups can be used. This allows the adjustment of the network's thermal and mechanical properties as well as the adjustment of their hydrophilicity–hydrophobicity balance and enables a simple access towards copolymers such as poly(ester amine)s42 or polyester/polyether networks.43
Recently we presented the synthesis of functional star-shaped poly(ε-caprolactone) prepolymers bearing acrylate end groups and their crosslinking via Michael-type addition using amino-telechelic poly(tetrahydrofuran), resulting in resins with decreased crystallinity and increased degradation rate.44 The aim of this paper is a systematic investigation of novel biodegradable polyester/polyether resins including a detailed characterization. Special attention is paid to the resins' crystallinity, hydrophilicity and water sorption, which allow drawing conclusions about their degradation behavior.
Poly(ε-caprolactone) prepolymers with different architecture and molar mass were crosslinked with multifunctional hydrophobic (PTHF- or PPO-based) and hydrophilic (PEO-based) crosslinking agents via Michael-type addition. The degree of crosslinking was adjusted by variation of the prepolymer/crosslinker ratio in the feed and the crosslink density by variation of the molar mass of the prepolymer and/or the crosslinking agent. An extensive characterization of the polyester/polyether resins is given, including determination of the degree of crosslinking, swelling behavior, the crystallinity, stress–strain behavior, contact angle, and water sorption of the resins. The possibility of post-processing functionalization of residual amine groups is shown by functionalization with pentafluorobenzaldehyde.
Materials and methods
Materials
ε-Caprolactone (CL, ≥99%, Fluka) was stirred over CaH2 for 24 h, condensed onto molecular sieves (4 Å), and kept in a Schlenk flask under argon. Di(pentaerythritol) (tech., Aldrich) and di(trimethylolpropane) (97%, Aldrich) were purified by sublimation and kept under argon. Zinc(II) 2-ethylhexanoate (Zn(oct)2, 97%, ABCR), acryloyl chloride (96%, Merck), poly(tetrahydrofuran) bis(3-aminopropyl) terminated (Mn = 1100 g mol−1) (Aldrich), Jeffamine ED-2003 (Mn = 2000 g mol−1) (95%, Huntsman Corporation), Jeffamine T-3000 (Mn = 3000 g mol−1) (97%, Huntsman Corporation), PEG-diamines (Mn = 368 g mol−1, 1983 g mol−1 and 6428 g mol−1) (>95%, Iris Biotech) and pentafluorobenzaldehyde (PFBA, 98%, ABCR) were used as received. Star-shaped functional poly(ε-caprolactone)s (sPCL-A 2a–f) used in this work have the following characteristics: 2a is a six-arm star-shaped polymer while 2b–f are four-arm star-shaped polymers. Mn(SEC) = 7400 g mol−1, Mw/Mn = 1.23 (2a), Mn(SEC) = 4300 g mol−1, Mw/Mn = 1.42 (2b), Mn(SEC) = 9000 g mol−1, Mw/Mn = 1.40 (2c), Mn(SEC) = 19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400 g mol−1, Mw/Mn = 1.23 (2d), Mn(SEC) = 40200 g mol−1, Mw/Mn = 1.13 (2e) and Mn(SEC) = 70600 g mol−1, Mw/Mn = 1.11 (2f).44
400 g mol−1, Mw/Mn = 1.23 (2d), Mn(SEC) = 40200 g mol−1, Mw/Mn = 1.13 (2e) and Mn(SEC) = 70600 g mol−1, Mw/Mn = 1.11 (2f).44
Methods
1H and 13C NMR spectra were recorded on a Bruker AV 400 FT-NMR spectrometer at 400 MHz and 100 MHz, respectively. Deuterated chloroform (CDCl3) was used as the solvent, and tetramethylsilane (TMS) served as the internal standard.Size exclusion chromatography (SEC) analyses were carried out using THF (with addition of 250 mg L−12,6-di-tert-butyl-4-methylphenol) as eluting solvents. A high pressure liquid chromatography pump (ERC HPLC model 64200) and a refractive index detector (Jasco RI-2031) were used at 30 °C with a flow rate of 1.0 mL min−1. Four columns with MZ-DVB gel were applied. The length of each column was 300 mm and the diameter 8 mm, the diameter of the gel particles was 5 μm, and the nominal pore width values were 50, 102, 103 and 104 Å. Calibration was achieved using narrow distributed poly(methyl methacrylate) (PMMA) standards.
Raman spectroscopy was carried out on a Bruker FT-Raman-Spectrometer RFS 100/S with a Nd:YAG-laser. The power of the laser was 200 mV at 1064 nm with a spectral resolution of 4 cm−1. For each spectrum 500 scans were recorded.
For determining the swelling ratios 3 resin disks (6 × 6 × 1 mm) were immersed in water and dichloromethane, respectively, and stirred for 15 h, when equilibrium was reached. The swelling ratio was expressed as the volume of the swollen per volume of the dry sample.
Differential scanning calorimetry (DSC) was performed on a Netzsch DSC 204 in aluminium pans which were perforated (2 holes) before being placed. The measurements were performed under nitrogen draft of 20 mL min−1 with a heating rate of 10 K min−1 against an empty aluminium pan. Calibration was achieved using indium and cyclohexane.
Tensile testing was performed on the resins in the axial direction using a mechanical tensile tester (zwicki-Line Z2.5). Shouldered test bars (S 3) were prepared; measurements were carried out at room temperature according to the ISO 37 standard (ISO/DIS 37:1989). Three samples of each material were tested. Stress and strain at fracture were recorded and, if present, as well at yield point. Elastic moduli were determined by calculating the tangent at the elastic region of the stress–strain curves. Fracture strength and elongation were calculated with eqn (1) and (2), toughness was calculated from the area beneath the stress–strain curve.
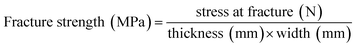 | (1) |
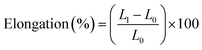 | (2) |
Contact angle measurements were performed following the sessile drop method45 with a contact angle measurement system (Krüss GS 2) on polymer or resin films. Each reported contact angle measurement represents an average value of ten separate drops on different areas of a given film. The size and the volume of the drops were kept constant (5 μL) and contact angle values were recorded (10 determinations of each drop) once the values did not change after drop placement (usually after 10–15 s). Measurements were performed at room temperature and a constant humidity of 60%.
Water sorption measurements were performed on an Isothermal Gravimetric moisture sorption Analyser (IGAsorp) (Hiden Analytical) at room temperature. The resins were powdered (except R:2b-81.001.00 was cut into small pieces) and dried inside the instrument until a relative humidity of <1% was reached. The relative humidity was increased stepwise by 10% up to 95% and back again to 5% with an equilibration time of 60 min per step. Rate constants k of a first order sorption/desorption kinetics are determined by plotting −ln (1 − α) versus sorption time t (α = (mt − mt = 0)/(mmax − mt = 0) with mt being the weight of the resin at time t, mt = 0 the weight of the resin at t = 0 and mmax the weight of the resin at equilibrium).
X-Ray photoelectron spectroscopy measurements were carried out in an Ultra Axis spectrometer (Kratos Analytical). The samples were irradiated with monoenergetic Al Kα1,2 radiation (1486.6 eV) and the spectra were taken at a power of 144 W. The aliphatic carbon (C–C, C–H) at a binding energy of 285 eV (C 1s photoline) was used to determine the charging. The spectral resolution—i.e. the Full Width of Half Maximum (FWHM) of the ester carbon from PET—was better than 0.68 eV for the elemental spectra. The elemental concentration is given in atom%, the lower detection limit is <0.1 atom%. The samples were measured with an information depth of 10 nm from 0 to 1200 eV.
Crosslinking
Solutions (50 wt% in dichloromethane) of star-shaped acrylate functional poly(ε-caprolactone) (2a–f) and different crosslinking agents (3–8, 0.25 to 1.50 eq. amine groups to acrylate groups, in steps of 0.25 eq.) were dispensed in a Teflon master (40 × 10 × 10 mm). The mixture was kept at room temperature in a stream of dry air until the solvent was evaporated (for ∼30 min), heated at 55 °C for 12 h, and carefully released.Nomenclature of the resins
For the resin R:2b-40.250.25, R stands for resin, 2b is the polyester prepolymer and 4 the polyether crosslinking agent from which the resin is prepared. 40.25 indicates that the molar ratio of amine groups of the crosslinking agent to the acrylate groups of the polyester prepolymer is 0.25.Post-processing functionalization
The resins R:2f-40.500.50, R:2f-41.001.00 and R:2f-41.501.50 and the prepolymer 2b were placed in a Schlenk tube 2 cm above 0.05 mL PFBA, the tube was sealed, and heated for 5 h at 60 °C. The resins and the prepolymer were taken out of the tube and dried at 10−3 mbar for 48 h to evaporate non-reacted PFBA. XPS analysis was carried out subsequently.Results and discussion
Star-shaped poly(ε-caprolactone) prepolymers with acrylate end groups and polyester/polyether resins were prepared as previously described in ref. 44. In this paper the successful crosslinking of prepolymers with different numbers of arms and arm lengths and a wide range of polyether crosslinking agents including a comprehensive analysis is presented. The prepolymers used were synthesized viaring-opening polymerization of CL with a multifunctional initiator and end-capping of the PCL star arms with acryloyl chloride. The resulting poly(ε-caprolactone) prepolymers were converted with different amino-telechelic polyethers. The chemical nature, the molar mass and the mass fraction of different crosslinking agents used as well as the molar mass of the prepolymer will influence the resin's properties such as crystallinity, stress–strain behavior, hydrophilicity, and water sorption (Scheme 1).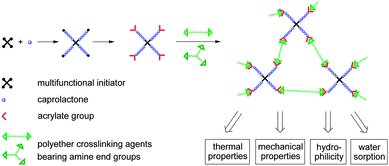 | ||
| Scheme 1 Synthetic concept for the preparation of polyester/polyether resins from functional poly(ε-caprolactone) prepolymers and polyether crosslinking agents. | ||
Synthesis
Star-shaped functional PCLs were synthesized by anionic ring-opening polymerization using di(pentaerythritol) (a) or di(trimethylolpropane) (b–f) as a multifunctional initiator, followed by functionalization with acryloyl chloride (Scheme 2).44 Composition of star-shaped functional prepolymers 2a–f used for processing are listed in Table 1.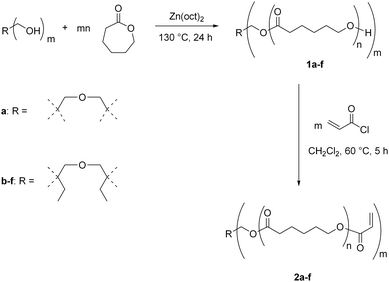 | ||
| Scheme 2 Synthetic pathway to star-shaped functional poly(ε-caprolactone)s (sPCL-A). | ||
| Prepolymer | DF a (%) | M n,NMR b/g mol−1 | M n,SEC c/g mol−1 | M w/Mnc | |
|---|---|---|---|---|---|
| a Degree of functionalization determined by NMR. b Number average molecular weight (Mn) determined by NMR. c Number average molecular weight (Mn) and molecular weight distribution (Mw/Mn) determined by SEC (PMMA standards). d Initiator: di(pentaerythritol). e 6-Arm star-shaped PCL with 5 CL repeating units and an acrylate end group per arm. f Initiator: di(trimethylolpropane). | |||||
| 2a d | s6PCL5-A e | 99 | 3900 | 7400 | 1.23 |
| 2b f | s4PCL5-A | 93 | 2700 | 4300 | 1.42 |
| 2c f | s4PCL10-A | 95 | 4600 | 9000 | 1.40 |
| 2d f | s4PCL20-A | 93 | 9700 | 19400 | 1.23 |
| 2e f | s4PCL50-A | 94 | 23300 |
40200 |
1.13 |
| 2f f | s4PCL100-A | 100 | 46200 |
70600 |
1.11 |
Crosslinking
Biodegradable polyester/polyether resins were prepared by thermal crosslinking of the acrylate functionalized star-shaped poly(ε-caprolactone)s. Crosslinking of the acrylate groups with amino-telechelic polyethersvia Michael-type addition was carried out on polymer films from mixtures of a star-shaped poly(ε-caprolactone) prepolymer (2a–f) and an amino-telechelic PTHF (3), PPO (Jeffamine T-3000) (4), PPO-b-PEO-b-PPO (Jeffamine ED-2003) (5) or PEG (6–8) (Scheme 3, bottom), yielding polyester/polyether resins. Resins from 2b and 3–8 were prepared with molar ratios of amine to acrylate groups (NH2/A) of 0.25 to 1.50, while resins from 2a, c–f and 3 or 5 were prepared with a molar ratio of NH2/A of 1 (Table 2). Scheme 3 (top) exemplifies the chemistry of the crosslinking reaction.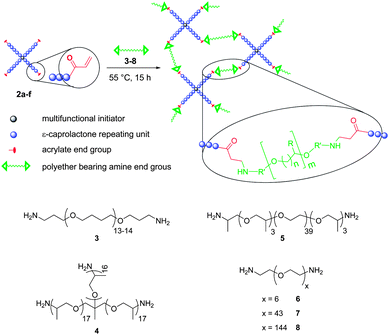 | ||
| Scheme 3 Crosslinking of functional poly(ε-caprolactone) prepolymers with polyether crosslinking agents (bottom) via Michael-type addition (top). | ||
| Prepolymer | Crosslinking agent | Ratio NH2/Aa | Resinb |
|---|---|---|---|
| a Ratio amine (NH2) to acrylate (A) groups. b Crosslinking conditions: 55 °C, 12 h. c In steps of 0.25. d With x = 0.25–1.50. | |||
| s6PCL5-A (2a) | PTHF-diamine (3) | 1.00 | R:2a-31.001.00 |
| s6PCL5-A | Jeffamine ED-2003 (5) | 1.00 | R:2a-51.001.00 |
| s4PCL5-A (2b) | PTHF-diamine (3) | 0.25–1.50c | R: 2b-3 x d |
| s4PCL5-A | Jeffamine T-3000 (4) | 0.25–1.50c | R: 2b-4 x d |
| s4PCL5-A | Jeffamine ED-2003 (5) | 0.25–1.50c | R: 2b-5 x d |
| s4PCL5-A | PEG368-diamine (6) | 0.25–1.50c | R: 2b-6 x d |
| s4PCL5-A | PEG1983-diamine (7) | 0.25–1.50c | R: 2b-7 x d |
| s4PCL5-A | PEG6429-diamine (8) | 0.25–1.50c | R: 2b-8 x d |
| s4PCL10-A (2c) | PTHF-diamine (3) | 1.00 | R:2c-31.001.00 |
| s4PCL10-A | Jeffamine ED-2003 (5) | 1.00 | R:2c-51.001.00 |
| s4PCL20-A (2d) | PTHF-diamine (3) | 1.00 | R:2d-31.001.00 |
| s4PCL20-A | Jeffamine ED-2003 (5) | 1.00 | R:2d-51.001.00 |
| s4PCL50-A(2e) | PTHF-diamine (3) | 1.00 | R:2e-31.001.00 |
| s4PCL50-A | Jeffamine ED-2003 (5) | 1.00 | R:2e-51.001.00 |
| s4PCL100-A (2f) | PTHF-diamine (3) | 1.00 | R:2f-31.001.00 |
| s4PCL100-A | Jeffamine ED-2003 (5) | 1.00 | R:2f-51.001.00 |
The conversion of C,C-double bonds was investigated by means of Raman spectroscopy, comparing the concentration of C,C-double bonds in the functional prepolymers and in the corresponding resins. Representative Raman spectra of 2b and the resins prepared from 2b and 5 with molar ratios of NH2/A of 0.25 to 1.50 are shown in Fig. 1.
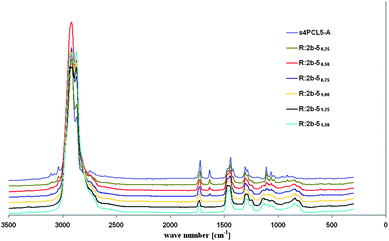 | ||
| Fig. 1 Raman spectra of poly(ε-caprolactone) prepolymer 2b and resins prepared from 2b and 5 with molar ratios of NH2/A of 0.25 to 1.50. | ||
Increasing the amount of crosslinking agent (R:2b-50.250.25 to R:2b-51.501.50) results in a decrease of the signal of the C,C-double bonds at 1636 cm−1 and a slight shift of the ester bond towards higher wavenumbers, since the amount of ester bonds of the acrylate groups decreases. With a molar ratio of NH2/A of 1 (R:2b-51.001.00) most C,C-double bonds are converted, while for the resin R:2b-51.251.25 no signal of residual C,C-double bonds can be detected. Due to the fact that there is no possibility for an exact scaling, no quantitative conclusions can be drawn from the Raman spectra. However, the obtained results prove that the degree of crosslinking (or the amount of residual acrylate or free amine groups) in the resin can be tuned by the variation of the concentrations of the prepolymer and the crosslinking agent in feed. In general all crosslinking agents behave similar, but with hydrophobic ones (3, 4) total conversion of C,C-double bonds can be achieved easier than with hydrophilic ones (5–8), most probably due to a more homogenous mixture of the components. Although the conversion of the amine groups was not determined, it can be assumed that resins prepared with a molar ratio of NH2/A of 1 exhibit the highest degree of crosslinking, since the amount of residual acrylate or amine groups will increase by the addition of an excess of either the prepolymer or the crosslinking agent. Thus the degree of crosslinking first increases from R:2b-X0.250.25 to R:2b-X1.001.00, then decreases again up to R:2b-X1.501.50.
Furthermore, an increase of the molar mass (arm length) of the poly(ε-caprolactone) prepolymers (2a–f) also influences the ability of crosslinking: due to less chain movement the amount of residual C,C-double bonds in the resins slightly increases from R:2a-31.001.00 to R:2f-31.001.00, respectively R:2a-51.001.00 to R:2f-51.001.00.
Swelling properties
The swelling properties of the resins described before were studied in dichloromethane as nonpolar solvent and in water as polar solvent. Since the composition of the resins, their hydrophilicity–hydrophobicity balance (molar mass and mass ratio of hydrophilic and hydrophobic building blocks), and their crosslink density are very different, the swelling behavior is rather complex. The most important factors that influence the swelling behavior are the crosslink density, the mass ratio of hydrophilic and hydrophobic components, and the solvent used.The swelling behavior of the resins prepared from s4PCL5-A (2b) with crosslinking agents 3–8 in dichloromethane and water was investigated. In general, all resins are able to swell in dichloromethane, but only resins prepared with hydrophilic crosslinking agents 5–8 exhibit swelling in water. Due to the complex swelling behavior of the resins no significant conclusions can be drawn when comparing the swelling ratios of resins prepared with different crosslinking agents.
Furthermore, the swelling behavior of resins prepared from hydrophobic crosslinking agent 3 or hydrophilic crosslinking agent 5 and PCL-based prepolymers with increasing molar mass (2a–f) was studied (Fig. 2). An equimolar ratio of amine to acrylate groups was used, leading to decreasing crosslink density with increasing molar mass of the prepolymer. For resins R:2a-31.001.00 to R:2f-31.001.00, composed of only hydrophobic building blocks, the swelling ratio in dichloromethane increases with increasing molar mass of the prepolymer, since the distance between the crosslinks in the resins is increased and thus a greater number of solvent molecules can be inserted. In water no swelling is observed, due to the insolubility of both building blocks in water. Likewise, for resins R:2a-51.001.00 to R:2f-51.001.00, composed of the hydrophobic PCL-based prepolymers and hydrophilic PEO-based crosslinking agent 5 the swelling ratio in dichloromethane increases with increasing molar mass of the prepolymer. Although the hydrophobicity of these resins is lower than that of the analogous resins prepared from 3, they exhibit greater swelling ratios. This is due to the fact that the size of crosslinking agent 5 is twice the size of crosslinking agent 3 and thus the crosslink density is remarkably lower.
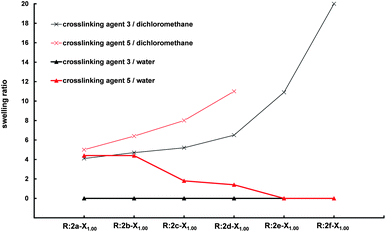 | ||
| Fig. 2 Swelling ratios of resins prepared from PCL prepolymers with increasing molar mass and crosslinking agents 3 and 5 (molar ratio of NH2/A is 1). | ||
Unlike resins prepared from the hydrophobic crosslinking agent 3, those prepared from hydrophilic PEO-based crosslinking agent 5 are able to swell in water as long as the molar mass of the used prepolymer is not too high (up to s4PCL20-A (2d)). By increasing the molar mass of the prepolymer the crosslink density decreases, however, no increase of the swelling ratio is observed, since the resins become more hydrophobic (the mass ratio of PCL/PEO increases), and as a consequence water uptake is hindered.
Thermal analysis
The thermal properties of the poly(ε-caprolactone) prepolymers, the crosslinking agents and the corresponding resins were determined and compared using differential scanning calorimetry (DSC). The influence of the resin composition (crosslinking agent, degree of crosslinking and molar mass of the prepolymer) on the thermal properties was established.All poly(ε-caprolactone) prepolymers 2a–f are semicrystalline polymers. Glass transition temperatures appear between −63 °C and −62 °C. Melting temperatures [Tm,Peak] in the range of 30/41 °C (2a and 2b) to 58 °C (2f) were found, which are typical values for oligo- and poly(ε-caprolactone)s. Increasing the degree of polymerization of the prepolymers, the (longer) chains are able to form larger crystallites resulting in higher melting temperatures (30/41 °C for 2bvs. 58 °C for 2f). Likewise a higher melting enthalpy (59 J g−1 for 2bvs. 79 J g−1 for 2f) is observed, since a higher number of PCL chains compared to the star-shaped initiator leads to the formation of more and larger crystalline domains.
All crosslinking agents are polymers with high crystallinity, except the PPO-based Jeffamine T-3000 (4), which is amorphous. It exhibits a distinctive glass transition (Tg = −69 °C and ΔCp = 0.8 J g−1 K−1). The PTHF-based diamine 3 has a melting point [Tm,Peak] of 22 °C (ΔHm = 113 J K−1), the PEG-based diamines 5–8 exhibit melting points between 17 °C (6) and 65 °C (8) (with ΔHm between 130 J K−1 (6) and 202 J K−1 (8)). As expected, melting temperature of PEG-diamine increases with increasing degree of polymerization.
Polyester/polyether resins can be prepared with various thermal properties. Most of the resins prepared are semicrystalline, but there are also completely amorphous ones. Two facts influence the resin's thermal properties the most: the underlying prepolymer and crosslinking agent with their thermal properties and the degree of crosslinking. Resins prepared with high amounts of PCL prepolymer (e.g.R:2b-40.25 or R:2b-60.250.25 and R:2b-60.500.50) show basically the properties (Tm, ΔHm) of 2b, while thermal properties of resins prepared with high amounts of crosslinking agent (e.g.R:2b-41.25 and R:2b-41.501.50 or R:2b-61.501.50) approach the thermal properties of 4 (Tg) and 6 (Tm), respectively (Fig. 3). The degree of crosslinking is highly responsible for the architecture in the resin. The architecture influences the resin's thermal properties the most when the degree of crosslinking is high (molar ratio of NH2/A close to 1, e.g.R:2b-40.75 and R:4b-31.001.00, R:2b-60.750.75 and R:2b-61.001.00). The crosslinking significantly hinders the lamellar arrangement of the polyester or polyether chains, therefore the amount of crystalline domains in the resins is decreased, or resins are even completely amorphous. Similar trends can be observed for the resins prepared with crosslinking agents 3, 5, 7 and 8. In general, the glass transition temperatures of the resins are higher than the ones of the PCL prepolymers used, indicating that crosslinking reduces the mobility of the chain segments in the resins.
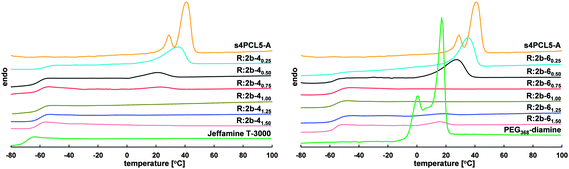 | ||
| Fig. 3 DSC second heating curves of s4PCL5-A (2b), crosslinking agent Jeffamine T-3000 (4) and corresponding resins (left) and of s4PCL5-A (2b), crosslinking agent PEG368-diamine (6) and corresponding resins (right). | ||
Remarkable resins are the ones prepared with the crosslinking agents 4 (amorphous) and 8 (highly crystalline, highest melting enthalpy). Resins prepared from 2b with a high amount of 4 (R:2b-41.001.00, R:2b-41.251.25 and R:2b-41.501.50) are completely amorphous due to the amorphous character of the crosslinking agent in combination with the high degree of crosslinking. When 8 is used in high concentrations for the preparation of polyester/polyether resins (R:2b-81.001.00, R:2b-81.251.25 and R:2b-81.501.50), resins with high crystallinity were obtained, since the high crystallinity of the crosslinking agent exceeds the effect of crosslinking.
The thermal properties of the resins are highly influenced by the crosslinking agent. This influence becomes apparent especially when comparing resins with high concentrations of crosslinking agent (R:2b-X1.501.50), but also resins prepared with a molar ratio of NH2/A of 1 and different crosslinking agents (R:2b-X1.001.00) exhibit remarkable differences in their thermal properties (Fig. 4). Both R:2b-41.001.00 and R:2b-41.501.50, as well as crosslinking agent 4, are completely amorphous, exhibiting glass transition temperatures of −59 and −61 °C. With increasing melting temperature and meting enthalpy of the crosslinking agent (3 < 6 < 5 < 7 < 8), resins prepared with these crosslinking agents become more crystalline, exhibiting increasing melting temperature and enthalpy (R:2b-51.001.00 to R:2b-81.001.00 and R:2b-31.001.00 to R:2b-81.501.50).
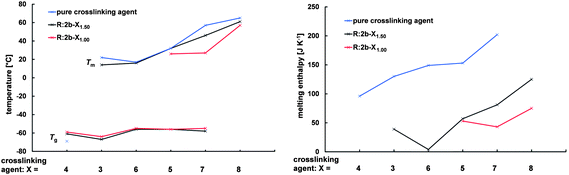 | ||
| Fig. 4 Glass transition and melting temperatures (left) and melting enthalpy (right) of resins R:2b-X1.001.00, R:2b-X1.501.50 and crosslinking agents 3–8. | ||
The influence of the poly(ε-caprolactone) prepolymer on the thermal properties of the resins was investigated by preparing resins from prepolymers with increasing molar mass (2a–f) and 3 or 5 as crosslinking agent. With the exception of R:2a-31.001.00 and R:2b-31.001.00, these resins are semicrystalline materials. The glass transition temperatures of these resins are between −63 °C and −56 °C and thus in the range of the one of the PCL prepolymers, since resins contain mostly of PCL due to their preparation from high molar mass prepolymers. Increasing the molar mass (PCL chain lengths) of the prepolymers, resins with increasing melting temperatures are obtained (e.g. for R:2a-51.001.00: Tm,Peak = 19.0 °C and for R:2f-51.001.00: Tm,Peak = 57.0 °C, Fig. 5). This increase results from the formation of larger crystallites, which is due to the longer PCL chains in the resins (as already seen for the pure PCL prepolymers). With increasing molar mass of the PCL prepolymer the crosslink density (the ratio of crosslinked acrylate groups to CL repeating units) decreases (from 5:1 (2a, 2b) to 100:1 (2f)), suppressing the influence of crosslinking on the resin's crystallinity. Thus increasing melting enthalpies were obtained for resins prepared from prepolymers with increasing molar mass (e.g. for R:2a-51.001.00: ΔHm = 46 J K−1 and for R:2f-51.001.00: ΔHm = 84 J K−1). Similar results were obtained for resins prepared with crosslinking agent 3. Using PCL prepolymers with low molar mass, the resins obtained (R:2a-31.001.00 and R:2b-31.001.00) are amorphous, since crosslinking mostly influences the resin's crystallinity. Decreasing the crosslink density by increasing the molar mass of the PCL prepolymer (R:2c-31.001.00 to R:2f-31.001.00) greatly increases the resins' crystallinity, thermal properties of the resins approximate to the ones of their precursors.
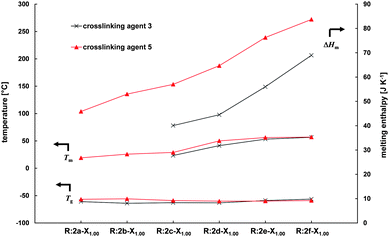 | ||
| Fig. 5 Glass transition and melting temperatures and melting enthalpy of resins prepared from functional PCL prepolymers 2a–f with different PCL chain lengths and 3 (×) or 5 (▲) as crosslinking agent (molar ratio of NH2/A is 1). | ||
In conclusion, the results obtained by thermal analysis indicate that amorphous and semicrystalline resins can be prepared from PCL prepolymers with a wide range of thermal properties, such as glass transition or melting temperature and melting enthalpy. As a result, several properties such as degradation and mechanical properties can be adjusted, expanding the application range for PCL-based materials.
Tensile properties
Crystallinity of a material greatly influences its tensile properties. Semicrystalline polymers show viscoelastic behavior, they are able to undergo large deformations and exhibit moderate elastic moduli. Amorphous resins, in general, show rubbery-elastic behavior, they are ductile materials, but exhibit a low elastic modulus.We determined the effect of resins' composition (prepolymer and crosslinking agent) and degree of crosslinking on the material's tensile properties. Elastic modulus (E), fracture strength (σf) and elongation at fracture (εf) and, if present, yield strength (σy) and elongation at yield point (εy) were evaluated from the stress–strain curves. Toughness at fracture (Wf) and at yield point (Wy) was calculated.
Investigated polyester/polyether resins show a wide range of tensile properties. All resins are either rubbery-elastic or viscoelastic materials, exhibiting elastic moduli from 0.5 ± 0.1 MPa to 148.8 ± 21.6 MPa. Fracture strength is in the range of 0.9 ± 0.1 MPa to 35.4 ± 3.0 MPa, when elongations of 11 ± 1% to 1350 ± 121% were reached. Toughness was in the range of 8.2 ± 2.9 mJ to 5843.7 ± 762.0 mJ. Resins prepared with crosslinking agent 8 or from PCL prepolymers with longer PCL chains (2c–f) exhibit necking.
The stress–strain behavior, elastic modulus and toughness at fracture of the resins prepared from 2b and 4 (top) and 2b and 6 (bottom) are shown in Fig. 6.
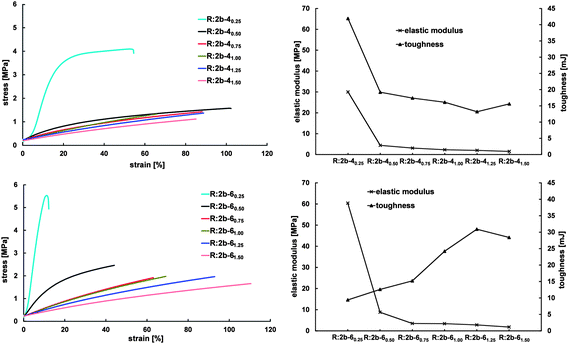 | ||
| Fig. 6 Stress–strain behavior (left) and elastic modulus and toughness at fracture (right) of resins prepared from 2b and 4 (top) and 2b and 6 (bottom). | ||
R:2b-40.250.25 shows viscoelastic behavior while all resins prepared from 2b and 4 show rubbery-elastic behavior. This can be explained with the resins' thermal properties: R:2b-40.250.25 is semicrystalline with Tm,Peak and Tm,Onset above the temperature at which tensile testing was performed, while R:2b-40.500.50 to R:2b-41.501.50 are in the amorphous state at the measuring temperature. Elastic modulus and fracture strength decrease with increasing degree of crosslinking and decreasing concentration of semicrystalline prepolymer from 30.0 ± 2.4 to 1.5 ± 0.1 MPa and 4.0 ± 0.2 to 1.1 ± 0.1 MPa, respectively. An increase of the degree of crosslinking or the amount of amorphous crosslinker in the resins results in the formation of softer resins. Elongation and toughness at fracture are similar for resins with molar ratios of NH2/A of 0.50 to 1.25, most probably due to similar thermal properties of these resins.
Due to its high crystallinity and melting temperature (Fig. 3) R:2b-60.250.25 shows viscoelastic behavior. The resin is hard and brittle, exhibiting an elastic modulus of around 60.4 ± 8.8 MPa and a fracture strength of 4.87 ± 0.7 MPa, coming along with a maximum elongation of 11 ± 1%. Toughness of the material is 9.4 ± 0.1 mJ. As seen for resins with crosslinking agent 4, increasing the degree of crosslinking, achieved by increasing the amount of crosslinking agent, results in the formation of softer and more rubbery-elastic resins. Elastic modulus and fracture strength of the resins decrease up to 1.8 ± 0.2 MPa and 1.6 ± 0.1 MPa, respectively, while elongation and toughness at fracture increase up to 115 ± 9% and 30.9 ± 9.6 mJ, as a consequence of the increasing amorphous character of these resins. Increasing the degree of crosslinking by adding more crosslinking agent exhibiting a Tm below room temperature leads to softer resins. Due to the decrease of crystallinity, these resins show more rubber-like behavior and are less able to withstand mechanical stress.
Similar trends are observed for the resins prepared with the crosslinking agents 3 and 5, since the Tm,Onset of these resins is below room temperature. Resins prepared with crosslinking agent 5 exhibit remarkable ductility (up to 260%), in combination with a low elastic modulus and fracture strength. This might be due to the fact that measurements were performed at temperatures close to the resins' melting interval. In contrast, resins prepared with 7 or 8 as crosslinking agents are hard and exhibit viscoelastic behavior, since they are semicrystalline materials with comparatively high crystallinity and melting temperatures. While resins prepared with 7 are brittle, all resins prepared with 8 are tough and show distinctive necking.
The influence of the crosslinking agent on the resins' stress–strain behavior is shown in Fig. 7. For all resins the molar ratio of NH2/A is 1, thus all differences in the mechanical properties can be directly referred to the crosslinking agent. With large and stiff PEG-blocks with high crystallinity hard resins are formed, which are either brittle (R:2b-71.001.00) or exhibit irreversible deformation (R:2b-81.001.00) already at low strain. Resins prepared with crosslinking agents 3–6 are rubbery-elastic due to their amorphous character. Comparative high strain but only low stress can be applied until breakage of these materials.
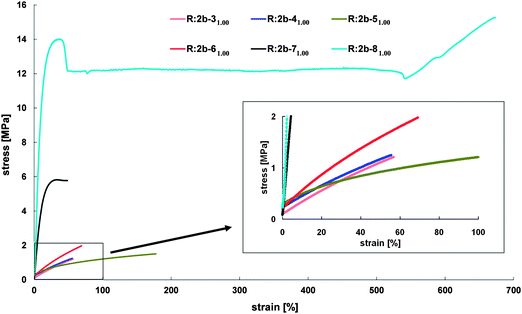 | ||
| Fig. 7 Stress–strain behavior of resins prepared from 2b and crosslinking agents 3–8 (molar ratio of NH2/A is 1). | ||
The influence of the crosslink density (varied by increasing molar mass/the length of PCL chains of prepolymers) on the resins' stress–strain behavior is shown in Fig. 8. Resins prepared from 2a–c and 3 as crosslinking agent (molar ratio of NH2/A is 1) show rubbery-elastic behavior, since the resins are either amorphous networks (R:2a-31.001.00 and R:2b-31.001.00) or exhibit a Tm below room temperature (R:2c-31.001.00). All resins exhibit elastic moduli around 3 MPa. In contrast, resins prepared from 2d–f and 3 show viscoelastic behavior with elastic moduli up to 124 MPa. The change in the tensile properties results from the fact that these resins show high crystallinity in combination with melting temperatures above room temperature. Using crosslinking agent 5 in this test series gives similar results, except for R:2e-51.001.00 and R:2f-51.001.00, which are brittle. Fracture occurs at yield point, thus these resins exhibit no necking. It can be concluded that decreasing crosslink density by increasing the molar mass (the length of PCL chains) of the prepolymer leads to stiffer materials which are able to withstand higher mechanical stress.
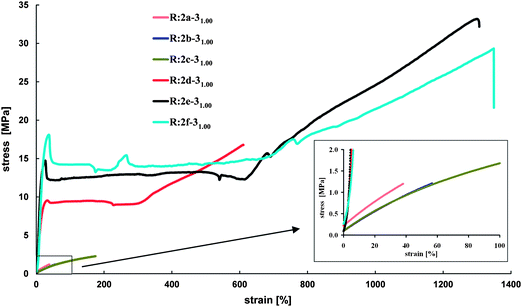 | ||
| Fig. 8 Stress–strain behavior of resins prepared from PCL prepolymers 2a–f and crosslinking agent 3 (molar ratio of NH2/A is 1). | ||
Contact angle measurement
Hydrophilicity is of great importance for biomaterials since it suppresses unspecific cell adsorption and has great influence on the material's degradation behavior. To determine the hydrophilicity of the polyester/polyether resins, contact angle measurements of water were performed. Resins prepared from 2b and different crosslinking agents 3–8 (molar ratio of NH2/A is 1) were investigated to see the influence of the crosslinking agent on the resin's hydrophilicity. Results are shown in Table 3.The PCL prepolymer 2b is a rather hydrophobic material, exhibiting a contact angle of 78.6° ± 1.1°. Using crosslinking agents based on hydrophobic polymers such as PTHF (R:2b-31.001.00) or PPO (R:2b-41.001.00) does not perceptibly change the resin's hydrophilicity compared to the one of the underlying PCL precursors. In contrast, when using more hydrophilic PEO-based crosslinking agent, significantly more hydrophilic resins can be prepared. According to the molar mass of the PEO crosslinking agent, contact angles of up to 41.2° ± 2.1° were obtained (R:2b-81.001.00) (Fig. 9). In summary, material's hydrophilicity–hydrophobicity balance can be adjusted by selection of the crosslinking agent as well as by varying its amount in the resin. This offers e.g. the opportunity to greatly influence the degradation properties of PCL-based materials.
 | ||
| Fig. 9 Contact angle measurements of water on films of s4PCL5-A (2b) (left), R:2b-31.001.00 (middle) and R:2b-81.001.00 (right). | ||
Sorption properties
Water sorption of biomaterials is an important factor when these materials are in contact with surrounding fluids. The water uptake makes the materials more flexible, promotes changes in the dimensions of the material and enhances hydrolysis.46 It is therefore of great importance to study water sorption of biodegradable materials.Since the potential of water sorption is directly linked to the material's hydrophilicity, resins prepared from 2b and different crosslinking agents 3–8 (molar ratio of NH2/A is 1) were investigated to see the influence of the crosslinking agent on the resin's sorption properties (Fig. 10). Up to a humidity of 30% all resins did not uptake almost any water. A first significant difference in water sorption of the investigated resins is obtained at 60% humidity. At this point the water sorption of more hydrophilic resins R:2b-51.001.00, R:2b-61.001.00, R:2b-71.001.00 and R:2b-81.001.00 increases while the one of more hydrophobic resins R:2b-31.001.00 and R:2b-41.001.00 stays remarkably low. After all, due to its large polar PEO segments, R:2b-81.001.00 is able to uptake water up to 53% of its own weight at 95% humidity, while R:2b-31.001.00 and R:2b-41.001.00, consisting of only nonpolar segments, are able to uptake water only up to 4% of their weight. Except for R:2b-81.001.00 the amount of water uptaken and released at a defined humidity is similar for each resin. The great hysteresis in the behavior of R:2b-81.001.00 results from the fact that the period of 60 min was too short to reach the equilibrium state, since the resin's desorption rate is remarkably low (see below).
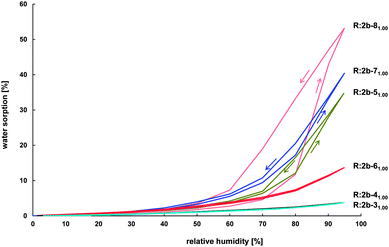 | ||
| Fig. 10 Sorption isotherms for resins prepared from 2b and different crosslinking agents 3–8 (molar ratio of NH2/A is 1) at room temperature, with a stepwise increase of the humidity by 10% and an equilibration time of 60 min per step. | ||
Kinetic measurements of the sorption reveal that adsorption and desorption rates for each resin are in the same range, with the exception of R:2b-81.001.00 (Fig. 11). Although they show the lowest overall water sorption, R:2b-31.001.00 and R:2b-41.001.00 exhibit the highest adsorption and desorption rates, indicating the fastest water uptake and release of all resins. This might be due to the structure of these resins. Sorption of water vapor requires breaking the chain–chain interactions and rearrangement of the chains to mark space for the water molecules. Desorption proceeds from an open structure and requires breaking of the chain–water interactions.47 It can be assumed that both processes run more easily if the investigated materials are amorphous. Thus R:2b-81.001.00, as the resin with the highest crystallinity, shows the lowest sorption rates.
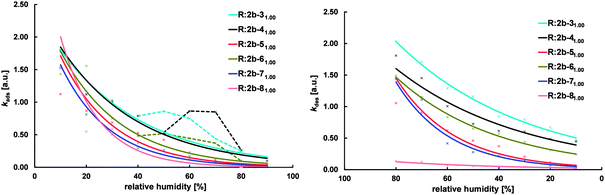 | ||
| Fig. 11 Adsorption (left) and desorption rates (right) of resins prepared from 2b and different crosslinking agents 3–8 (molar ratio of NH2/A is 1) measured at room temperature. | ||
In general, the adsorption rate decreases exponentially with increasing humidity, since increasing saturation of the resins hinders the uptake of further water. However, adsorption rates of R:2b-31.001.00, R:2b-41.001.00 and R:2b-51.001.00 exhibit a significant increase at humidity rates of 50–70%, represented by the dashed lines. It is assumed that at this humidity rates there are changes in the resins' structures, since increasing sorption rate usually refers to a more open structure. As expected, desorption rates of all resins decrease exponentially with decreasing humidity, since the network structures constrict with decreasing amount of embedded water, making further release more difficult.
Post-processing functionalization
Recently we reported the possibility of biofunctionalization of functional PCL prepolymers.48 Next to this possibility of pre-processing functionalization, further functionalization can be performed after crosslinking. Residual amine groups, either present due to incomplete crosslinking or when using a molar ratio of NH2/A >1 in the crosslinking step, can easily be functionalized by nucleophilic addition of any (bio) molecule that carries aldehyde or isothiocyanate groups.49To show the potential of post-processing functionalization, R:2b-60.500.50, R:2b-61.001.00 and R:2b-61.501.50 were derivatized with PFBA (Table 4).
| Materials | Chemical composition (Atom%) | |||
|---|---|---|---|---|
| C | N | O | F | |
| a Resins or prepolymer exposed to PFBA. | ||||
| R:2b-60.500.50 | 73.9 | — | 26.1 | — |
| R:2b-61.001.00 | 74.9 | 1.1 | 24.0 | — |
| R:2b-61.501.50 | 72.5 | 2.9 | 24.6 | — |
| R:2b-60.500.50-F a | 76.5 | 0.7 | 21.5 | 1.3 |
| R:2b-61.001.001.00-F a | 76.4 | 0.7 | 21.0 | 1.9 |
| R:2b-61.501.50-F a | 63.6 | 1.6 | 26.3 | 8.5 |
| s4PCL5-A-F (2b′)a | 73.5 | — | 25.5 | — |
Prepolymer 2b was exposed to PFBA and served as a negative control. Proof of fluorine in the derivatized polyester/polyether resins indicates that post-processing functionalization of these materials is possible. R:2b-60.500.50-F and R:2b-61.001.001.00-F show low concentrations of fluorine, indicating that free amine groups are present, since not all of them were converted during the crosslinking step. R:2b-61.501.50-F shows a remarkable concentration of fluorine, indicating that free amine groups in the resins were available for the functionalization step.
Conclusions
Starting from functional star-shaped poly(ε-caprolactone) prepolymers a variety of novel biodegradable polyester/polyether resins was prepared by crosslinking via Michael-type addition with PTHF-, PPO- and PEO-based crosslinking agents. The composition of the resins (molar mass of the prepolymer, crosslinking agent and degree of crosslinking) strongly affects their properties. Amorphous and semicrystalline resins have been prepared for which it is possible to adjust melting and glass transition temperature as well as crystallinity. Depending on their thermal properties, resins showed viscoelastic or rubbery-elastic behavior, exhibiting elastic moduli from 0.5 to 149 MPa and withstood stress up to 35 MPa. The hydrophilicity–hydrophobicity balance, coming along with swelling behavior and water sorption properties, can be adjusted by selection of the crosslinking agent as well as by varying its amount in the resin. Therefore it can be concluded that the degradation rate of these resins can be tuned in a wide range. In addition, post-processing functionalization can easily be performed due to the presence of residual free amine groups. Adjustment of mechanical and degradation properties in combination with simple processability and the possibility of pre- and post-processing biofunctionalization should greatly increase the field of application to PCL-based biomaterials.Acknowledgements
This research was carried out in the framework of the Transregio 37 project “Mikro- und Nanosystheme in der Medizin—Rekonstruktion biologischer Funktionen” funded by the German Research Foundation.Notes and references
- A. C. Albertsson and I. K. Varma, Biomacromolecules, 2003, 4, 1466–1486 CrossRef CAS.
- A. Duda and S. Penczek, in Biopolymers, ed. A. Steinbüchel and D. Y. Von Cramon, Wiley-VCH, Weinheim, 2002, pp. 371–429 Search PubMed.
- S. Matsumura, in Advances in Polymer Science, ed. S. Kobayashi, H. Ritter and D. Kaplan, Springer, Heidelberg, Berlin, 2006, vol. 194, pp. 95–132 Search PubMed.
- M. E. Gomes and R. L. Reis, Int. Mater. Rev., 2004, 49, 261–273 CrossRef CAS.
- M. E. Gomes and R. L. Reis, Int. Mater. Rev., 2004, 49, 274–285 CrossRef CAS.
- E. Behravesh, A. W. Yasko, P. S. Engel and A. G. Mikos, Clin. Orthop. Relat. Res., 1999, S118–S129 CAS.
- R. Langer, Science, 1990, 249, 1527–1533 CAS.
- J. C. Middleton and A. J. Tipton, Biomaterials, 2000, 21, 2335–2346 CrossRef CAS.
- O. E. Teebken and A. Haverich, Eur. J. Vasc. Endovasc. Surg., 2002, 23, 475–485 CrossRef.
- T. Biela, A. Kowalski, J. Libiszowski, A. Duda and S. Penczek, Macromol. Symp., 2006, 240, 47–55 CrossRef CAS.
- H. Höcker and H. Keul, Adv. Mater., 1994, 6, 21–36 CrossRef.
- M. Hans, P. Gasteier, H. Keul and M. Moeller, Macromolecules, 2006, 39, 3184–3193 CrossRef CAS.
- H. Sah and Y. W. Chien, J. Appl. Polym. Sci., 1995, 58, 197–206 CrossRef CAS.
- Biodegradable Polymers and Plastics, ed. M. Vert, J. Feijen, A. C. Albertsson, G. Scott and E. Chiellini, The Royal Society of Chemistry, Cambridge, 1992 Search PubMed.
- H. Fukuzaki, M. Yoshida, M. Asano, M. Kumakura, T. Mashimo, H. Yuasa, K. Imai and H. Yamanaka, Polymer, 1990, 31, 2006–2014 CrossRef CAS.
- Z. H. Gan, D. H. Yu, Z. Y. Zhong, Q. Z. Liang and X. B. Jing, Polymer, 1999, 40, 2859–2862 CrossRef CAS.
- M. Mochizuki, M. Hirano, Y. Kanmuri, K. Kudo and Y. Tokiwa, J. Appl. Polym. Sci., 1995, 55, 289–296 CrossRef CAS.
- M. E. Gomes, H. S. Azevedo, A. R. Moreira, V. Ella, M. Kellomaki and R. L. Reis, J. Tissue Eng. Regener. Med., 2008, 2, 243–252 CrossRef CAS.
- S. H. Oh, J. Y. Lee, S. H. Ghil, S. S. Lee, S. H. Yuk and J. H. Lee, Biomaterials, 2006, 27, 1936–1944 CrossRef CAS.
- V. R. Sinha and A. Trehan, J. Controlled Release, 2003, 90, 261–280 CrossRef CAS.
- S. Karlsson, M. Hakkarainen and A. C. Albertsson, J. Chromatogr., A, 1994, 688, 251–259 CrossRef CAS.
- S. S. Shah, K. J. Zhu and C. G. Pitt, J. Biomater. Sci., Polym. Ed., 1994, 5, 421–431 CrossRef CAS.
- M. Hans, H. Keul and M. Moeller, Biomacromolecules, 2008, 9, 2954–2962 CrossRef CAS.
- K. Odelius, P. Plikk and A. C. Albertsson, Biomacromolecules, 2005, 6, 2718–2725 CrossRef CAS.
- W. B. Tsai, C. H. Chen, J. F. Chen and K. Y. Chang, J. Mater. Sci.: Mater. Med., 2006, 17, 337–343 CrossRef CAS.
- S. L. Ishaug-Riley, L. E. Okun, G. Prado, M. A. Applegate and A. Ratcliffe, Biomaterials, 1999, 20, 2245–2256 CrossRef CAS.
- M. Trollsas, V. Lee, D. Mecerreyes, P. Lowenhielm, M. Moller, R. Miller and J. Hedrick, Abstr. Pap., Am. Chem. Soc., 2000, 219, U371 Search PubMed.
- D. Mecerreyes, J. Humes, R. D. Miller, J. L. Hedrick, C. Detrembleur, P. Lecomte, R. Jerome and J. San Roman, Macromol. Rapid Commun., 2000, 21, 779–784 CrossRef CAS.
- C. Vaida, P. Mela, H. Keul and M. Moeller, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6789–6800 CrossRef CAS.
- K. R. Gorda and D. G. Peiffer, J. Appl. Polym. Sci., 1993, 50, 1977–1983 CrossRef CAS.
- B. G. Amsden, G. Misra, F. Gu and H. M. Younes, Biomacromolecules, 2004, 5, 2479–2486 CrossRef CAS.
- L. Cai and S. F. Wang, Polymer, 2010, 51, 164–177 CrossRef CAS.
- S. Theiler, P. Mela, H. Keul and M. Moeller, Modern Trends in Polymer Science-Epf 09, 2010, vol. 296, pp. 453–456 Search PubMed.
- M. P. K. Turunen, H. Korhonen, J. Tuominen and J. V. Seppala, Polym. Int., 2002, 51, 92–100 CrossRef CAS.
- A. O. Helminen, H. Korhonen and J. V. Seppala, Macromol. Chem. Phys., 2002, 203, 2630–2639 CrossRef CAS.
- M. Nagata, K. Kato, W. Sakai and N. Tsutsumi, Macromol. Biosci., 2006, 6, 333–339 CrossRef CAS.
- G. Ozturk and T. E. Long, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 5437–5447 CrossRef CAS.
- A. Metters and J. Hubbell, Biomacromolecules, 2005, 6, 290–301 CrossRef CAS.
- M. Heggli, N. Tirelli, A. Zisch and J. A. Hubbell, Bioconjugate Chem., 2003, 14, 967–973 CrossRef CAS.
- B. D. Mather, K. Viswanathan, K. M. Miller and T. E. Long, Prog. Polym. Sci., 2006, 31, 487–531 CrossRef CAS.
- J. Rieger, K. Van Butsele, P. Lecomte, C. Detrembleur, R. Jerome and C. Jerome, Chem. Commun., 2005, 274–276 RSC.
- R. Arote, T. H. Kim, Y. K. Kim, S. K. Hwang, H. L. Jiang, H. H. Song, J. W. Nah, M. H. Cho and C. S. Cho, Biomaterials, 2007, 28, 735–744 CrossRef CAS.
- C. Vaida, P. Mela, K. Kunna, K. Sternberg, H. Keul and M. Moeller, Macromol. Biosci., 2010, 10, 925–933 CrossRef CAS.
- S. Theiler, M. Teske, H. Keul, K. Sternberg and M. Moeller, Polym. Chem., 2010, 1, 1215–1225 RSC.
- R. J. Good, in Contact Angle, Wettability and Adhesion, ed. K. L. Mittal, VSP, Utrecht, 1993 Search PubMed.
- H. S. Azevedo, F. M. Gama and R. L. Reis, Biomacromolecules, 2003, 4, 1703–1712 CrossRef CAS.
- B. Tigges, C. Popescu and O. Weichold, Soft Matter, 2011, 7, 5391–5396 RSC.
- S. Theiler, P. Mela, S. E. Diamantouros, S. Jockenhoevel, H. Keul and M. Moeller, Biotechnol. Bioeng., 2011, 108, 694–703 CrossRef CAS.
- R. Mix, K. Hoffmann, H. J. Buschmann, J. F. Friedrich and U. Resch-Genger, Vak. Forsch. Prax., 2007, 19, 31–37 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |