Catalyst behaviour for 1-pentene and 4-methyl-1-pentene polymerisation for C2-, Cs- and C1-symmetric zirconocenes
Camille
Descour
ab,
Rob
Duchateau
*ab,
Mamoeletsi R.
Mosia
a,
Gert-Jan M.
Gruter
a,
John R.
Severn
c and
Sanjay
Rastogi
d
aDepartment of Polymer Chemistry, Eindhoven University of Technology, 5600 MB, Eindhoven, The Netherlands. E-mail: R.Duchateau@tue.nl; Fax: +31 40 2463966; Tel: +31 40 2474918
bDutch Polymer Institute (DPI), P.O. Box 902, 5600 AX, Eindhoven, The Netherlands
cDSM SRU Performance Materials, P.O. Box 18, 6160 MD, Geleen, The Netherlands
dDepartment of Polymer Technology, Eindhoven University of Technology, 5600 MB, Eindhoven, The Netherlands
First published on 15th August 2011
Abstract
1-Pentene and 4-methyl-1-pentene (4M1P) have been polymerised using several C2-symmetric ansa-zirconocene catalysts rac-X(2-R1,4-R2-Ind)2ZrCl2 [X = C2H4, R1 = R2 = H (1); X = SiMe2, R1 = R2 = H (2); X = SiMe2, R1 = Me, R2 = H (3); X = SiMe2, R1 = H, R2 = Ph (4); X = SiMe2, R1 = Me, R2 = Ph (5)] with MAO as cocatalyst. The effects of polymerisation conditions as well as substituents on the indenyl ligand were studied. Except for the poly-1-pentenes synthesized with 3 and 5 at low temperatures, low molecular weight isotactic polymers were generally obtained. Compared to their behaviour in propylene polymerisation, the relative activity and selectivity of catalysts 1–5 are considerably different for 1-pentene and 4M1P polymerisation. Of the five catalysts, 1 and 4 showed the highest activities for both 1-pentene and 4M1P polymerisation, while 5 resulted in the lowest activities, especially for 4M1P polymerisation. Subsequently, a Cs- and several C1-symmetric zirconocenes, (R1)2C(3-R2-Cp)(2,7,-R3-Flu)ZrCl2 (R1 = Me, R2 = R3 = H (6); R1 = R2 = Me, R3 = H (7); R1 = Me, R2 = t-Bu, R3 = H (8); R1 = Me, R2 = R3 = t-Bu (9); R1 = Ph, R2 = t-Bu, R3 = H (10); R1 = Ph, R2 = R3 = t-Bu (11), were tested in 1-pentene and 4M1P polymerisation with MAO as cocatalyst. The effect of substituents on the bridge, the cyclopentadienyl (Cp) and fluorenyl (Flu) ligand, was studied relative to the polymerisation temperature and type of monomer. The molecular weights of the polymers were considerably higher than those of the poly-1-pentenes and P4M1Ps obtained with C2-symmetric zirconocenes (1–5). The catalytic activities and polymer molecular weights strongly depend on the fluorenyl substituent and the bridge, while the type of substituent on the Cp ligand has a strong influence on the tacticity of the polymers.
Introduction
The stereoselective C2-symmetric ansa-metallocene catalysts available for propylene polymerisation have evolved well beyond the ethylene-bridged bis(indenyl) species originally developed.1 The type of the bridge as well as the nature and position of substituents on the indenyl ligands have an effect on the catalyst activity and selectivity, which finds expression in the properties of the resulting polymer.2 The original rac-Et(Ind)2ZrCl2 (1)1d and rac-Me2Si(Ind)2ZrCl2 (2)3 are only moderately active in propylene polymerisation and afford polypropylenes with relatively low molecular weight and stereoregularity. A methyl group at the 2-position of the indenyl as in rac-Me2Si(2-Me-Ind)2ZrCl2 (3) results in enhanced molecular weight and tacticity of the polypropylenes, though catalyst activity is compromised.3 Substituents on the 4-position, such as the phenyl moiety in rac-Me2Si(4-Ph-Ind)2ZrCl2 (4), have been shown to significantly enhance the stereoregularity of the polypropylenes.4 The combined substitution at the 2- and 4-positions results in a very favourable synergetic effect on activity as well as molecular weight and stereoselectivity.4,5 Spaleck et al. found that using bulky substituents at the 4-position in combination with a methyl at the 2-position resulted in a highly active propylene polymerisation catalyst, rac-Me2Si(2-Me-4-Ph-Ind)2ZrCl2 (5), yielding highly stereoregular, high molecular weight isotactic polypropylene. At the same time, Brintzinger reported on the 2,4,5-trisubstituted rac-Me2Si(2-methylBenz[e]Ind)2ZrCl2, which proved to behave similar to the Spaleck catalyst.6Ewen reported the first bridged cyclopentadienyl-fluorenyl metallocene catalyst Me2C(Cp)(Flu)ZrCl2 (6) (Cp = cyclopentadienyl, Flu = fluorenyl), which yields highly syndiotactic polypropylene.7 Soon after, more catalysts with different bridges and substituents on the fluorenyl moiety have been synthesized and extensively used both in propylene and ethylene/propylene (co)polymerisations to get improved stereoselectivities, activities and molecular weights.2,8 Selective substitution of the Cp ring, resulting in the distortion of catalyst symmetry from Cs to C1, has as expected a prominent effect on the stereoregularity of the polymer. Adding a methyl group on the 3-C of the Cp ring as in Me2C(3-Me-Cp)(Flu)ZrCl2 (7) resulted in a change of stereospecificity of the polypropylene from syndiotactic to hemiisotactic.9 Evidently, the size of the substituents plays a significant role in determining the stereoregularity of the polymer since by merely changing the substituents on the Cp ring polymers with microstructures ranging from syndiotactic, to hemiisotactic, isotactoid and highly isotactic can be obtained.2,10
Compared to propylene polymerisation,1–10 relatively little effort has been directed towards understanding the structure–property relationship of metallocene catalysed homo- and copolymerisation of higher linear and branched α-olefins.11–14 A systematic comparative study on both the substituent effects of catalysts and monomer structure of linear and branched α-olefins on the catalyst activity and polymer properties is still lacking.
Here we report on the behaviour of a series of eleven different C2-, Cs- and C1-symmetric zirconocene catalysts during polymerisation of the higher linear and branched α-olefins: 1-pentene and 4-methyl-1-pentene (4M1P). The effect of ligand substituents on the activity, molecular weight and stereoselectivity will be discussed with respect to the type of monomer used.
Experimental
Materials
All operations were performed under argon using conventional Schlenk techniques. rac-C2H4(Ind)2ZrCl2 (1), rac-Me2Si(Ind)2ZrCl2 (2) and rac-Me2Si(2-Me-Ind)2ZrCl2 (3) were prepared according to reported methods.1,3rac-Me2Si(4-Ph-Ind)2ZrCl2 (4), rac-Me2Si(2-Me-4-Ph-Ind)2ZrCl2 (5) and Me2C(3-t-Bu-Cp)(2,7-t-Bu2-Flu)ZrCl2 (9) were kindly donated by B. Wang and N. Friederichs from Saudi Basic Industries Corporation (Sabic-Europe). Methylaluminoxane (10 wt% in toluene, Chemtura) was used without further purification. Me2C(Cp)(Flu)ZrCl2 (6),7Me2C(3-Me-Cp)(Flu)ZrCl2 (7),9eMe2C(3-t-Bu-Cp)(Flu)ZrCl2 (8),9fPh2C(3-t-Bu-Cp)(Flu)ZrCl2 (10)9l and Ph2C(3-t-Bu-Cp)(2,7-t-Bu2-Flu)ZrCl2 (11)27 were prepared according to reported methods. 4M1P and 1-pentene were purchased from Fischer Scientific and Acros, respectively. The monomers and toluene were purified by passing through a column of activated alumina and were subsequently stored under argon.Polymerisations
All polymerisations were carried out in a 100 mL double wall glass reactor. A thermostat bath and a thermocouple inserted into the reactor were used to regulate the polymerisation temperature. The reactor was charged with toluene (40 mL), MAO and catalyst precursor. For 1–5 (Al![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Zr ratio = 1000), 300 mmol of the monomer and 10 μmol of catalyst precursor was applied. For 6–11 (Al:Zr ratio = 500), 300 mmol of monomer and 5 μmol of catalyst precursor were applied. After 30 minutes, the polymerisations were quenched using ethanol and diluted hydrochloric acid. The precipitated polymer was filtered, then washed with ethanol and 2,6-di-tert-butyl-4-methylphenol dissolved in acetone (as an antioxidant). It was finally dried overnight at 80 °C in a vacuum oven.
:Zr ratio = 1000), 300 mmol of the monomer and 10 μmol of catalyst precursor was applied. For 6–11 (Al:Zr ratio = 500), 300 mmol of monomer and 5 μmol of catalyst precursor were applied. After 30 minutes, the polymerisations were quenched using ethanol and diluted hydrochloric acid. The precipitated polymer was filtered, then washed with ethanol and 2,6-di-tert-butyl-4-methylphenol dissolved in acetone (as an antioxidant). It was finally dried overnight at 80 °C in a vacuum oven.
Measurements
The polymers were analysed by size exclusion chromatography (SEC), nuclear magnetic resonance (NMR) and differential scanning calorimetry (DSC). Thermal analyses were carried out using DSC. To prevent thermal degradation, each sample was used only once and the runs were carried out under a nitrogen purge. Approximately 2 mg of sample was placed in an aluminium pan. Poly-1-pentene samples were heated from 0 to 120 °C at 10 °C min−1, maintained at this temperature for 2 minutes and then cooled to room temperature at 10 °C min−1. The same process was repeated and the melting traces were recorded. The same procedure was used on P4M1P, with the temperature ranging from 20 °C to 270 °C. For poly-1-pentene, SEC measurements were carried out at room temperature using tetrahydrofuran as solvent. Andries Jekel (University of Groningen, The Netherlands) carried out the high temperature SEC measurements (1,2,4-trichlorobenzene, 135 °C) on P4M1P samples. Narrow molecular distribution polystyrene standard samples were used as references. The 13C NMR spectra were recorded on a Varian Inova 500 (125 MHz 13C) spectrometer. Sample solutions were prepared by dissolving 40 mg of P4M1P or poly-1-pentene in 0.4 mL of 1,1,2,2-tetrachloroethane-d2. Chemical shifts are reported in ppm and referenced to deuterium signals of the solvent (13C NMR). Measurements were performed at 110 °C for P4M1P and 25 °C for poly-1-pentene.Results and discussion
C 2-Symmetric zirconocenes
Scheme 1 shows a series of C2-symmetric zirconocenes that have been selected to study the effect of catalyst structure on the polymerisation of 1-pentene and 4M1P. Their performance in propylene polymerisation has been studied in detail, which allows a good comparison.2 Albeit less extensively, 1–3 and 5 have also been studied in the polymerisation of linear or branched α-olefins.13Catalysts 1 and 2 were selected to study the effect of the bridge of the ligand system and catalysts 2–5 were chosen to investigate the effect of different ligand substitutions. The polymerisation results for the 1-pentene and 4M1P polymerisations are given in Table 1, Fig. 1 and 2.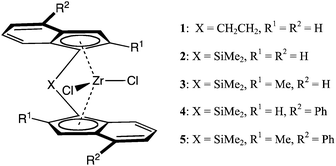 | ||
| Scheme 1 C 2-symmetric zirconocenes used to polymerise 1-pentene and 4M1P: rac-Et(Ind)2ZrCl2 (1), rac-Me2Si(Ind)2ZrCl2 (2), rac-Me2Si(2-Me-Ind)2ZrCl2 (3), rac-Me2Si(4-Ph-Ind)2ZrCl2 (4) and rac-Me2Si(2-Me,4-Ph-Ind)2ZrCl2 (5). | ||
| Exp. | Cat. | Mon. | T/°C | Yield/wt% | Act./g mmol−1 h−1 | M w/103 g per mol | PDI | #b | T m/°C |
|---|---|---|---|---|---|---|---|---|---|
| a Conditions: catalyst = 10 μmol, Al/Zr ratio = 1000, toluene = 40 mL, monomer = 300 mmol, polymerisation time = 30 min. b Calculated number of chains per active site. | |||||||||
| 1 | 1 | 1P | 10 | 48.1 | 2025 | 43.3 | 1.9 | 46.8 | — |
| 2 | 1 | 1P | 40 | 79.7 | 3345 | 25.7 | 2.1 | 130.4 | — |
| 3 | 1 | 1P | 70 | 99.9 | 4206 | 11.1 | 3.4 | 378.9 | — |
| 4 | 1 | 4M1P | 10 | 13.1 | 662 | 26.5 | 1.8 | 25.0 | 224 |
| 5 | 1 | 4M1P | 40 | 38.2 | 1928 | 21.8 | 1.8 | 88.4 | 216 |
| 6 | 1 | 4M1P | 70 | 91.3 | 4610 | 10.7 | 2.0 | 430.7 | 200 |
| 7 | 2 | 1P | 10 | 32.0 | 1345 | 37.1 | 1.7 | 36.3 | 63 |
| 8 | 2 | 1P | 40 | 58.9 | 2478 | 23.2 | 1.7 | 106.8 | — |
| 9 | 2 | 1P | 70 | 99.3 | 4180 | 15.9 | 1.9 | 262.9 | — |
| 10 | 2 | 4M1P | 10 | 6.4 | 323 | 26.7 | 1.6 | 12.1 | 227 |
| 11 | 2 | 4M1P | 40 | 12.2 | 617 | 21.3 | 1.7 | 28.9 | 222 |
| 12 | 2 | 4M1P | 70 | 25.5 | 1287 | 15.0 | 1.8 | 85.9 | 212 |
| 13 | 3 | 1P | 10 | 12.9 | 251 | 280.3 | 1.8 | 0.9 | 68 |
| 14 | 3 | 1P | 40 | 37.3 | 1567 | 101.9 | 2.0 | 15.4 | — |
| 15 | 3 | 1P | 70 | 97.0 | 4082 | 41.5 | 3.3 | 98.4 | — |
| 16 | 3 | 4M1P | 10 | 10.7 | 87 | 42.2 | 1.8 | 12.8 | 230 |
| 17 | 3 | 4M1P | 40 | 17.1 | 173 | 32.9 | 1.9 | 26.3 | 225 |
| 18 | 3 | 4M1P | 70 | 49.6 | 501 | 12.3 | 2.1 | 203.6 | 214 |
| 19 | 4 | 1P | 10 | 42.1 | 1770 | 33.6 | 1.7 | 52.7 | 66 |
| 20 | 4 | 1P | 40 | 74.3 | 3125 | 21.4 | 2.8 | 146.1 | — |
| 21 | 4 | 1P | 70 | 99.7 | 4195 | 14.8 | 1.9 | 283.5 | — |
| 22 | 4 | 4M1P | 10 | 13.1 | 660 | 28.1 | 1.8 | 23.5 | 230 |
| 23 | 4 | 4M1P | 40 | 25.0 | 1259 | 19.7 | 1.7 | 64.0 | 226 |
| 24 | 4 | 4M1P | 70 | 80.1 | 4044 | 14.1 | 1.7 | 286.8 | 215 |
| 25 | 5 | 1P | 10 | 0.9 | 38 | 87.9 | 1.9 | 0.4 | 67 |
| 26 | 5 | 1P | 40 | 26.7 | 1124 | 67.2 | 2.0 | 16.7 | — |
| 27 | 5 | 1P | 70 | 85.4 | 3595 | 55.3 | 1.9 | 65.0 | — |
| 28 | 5 | 4M1P | 10 | 1.3 | 14 | 26.8 | 3.3 | 2.5 | 219 |
| 29 | 5 | 4M1P | 40 | 2.3 | 23 | 15.1 | 2.3 | 7.7 | 215 |
| 30 | 5 | 4M1P | 70 | 11.5 | 116 | 6.1 | 1.7 | 95.1 | 206 |
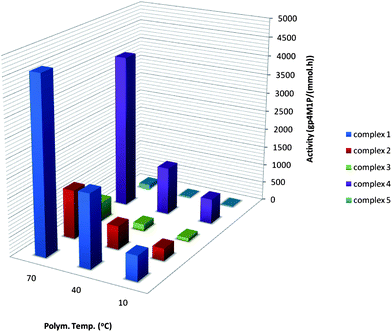 | ||
| Fig. 1 4M1P polymerisation activities obtained for catalysts 1–5 at different temperatures. | ||
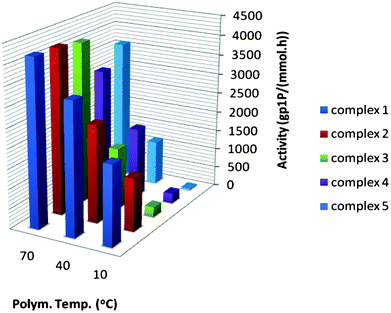 | ||
| Fig. 2 1-Pentene polymerisation activities obtained for catalysts 1–5 at different temperatures. Due to high conversions, the activities at 70 °C are underestimated. | ||
Catalytic activity
Although the optimum Al/Zr ratio might be somewhat different for different catalysts and might also depend on the olefin that is polymerised and type of MAO that is used, a fixed Al/Zr ratio of 1000 was chosen for all the runs.15 We have restricted ourselves to a fixed polymerisation time of 30 minutes. Obviously, this prevented us from drawing any conclusions regarding the kinetic profiles for the various catalysts but that was not the target of the present qualitative study.Fig. 1 shows the activities observed when 4M1P was polymerised with metallocenes 1–5 with MAO as a cocatalyst. Complex 1 outperforms 2–5, even though the activity of 4 is approaching that of 1.4 Worth mentioning is the drastic increase in catalytic activity of both 1 and 4 at elevated polymerisation temperatures. Zirconocene 2 showed a lower activity relative to 1 and no substantial increase in productivity with increasing polymerisation temperature was observed. In fact, the activity of 1 at 70 °C is about 4 times higher than that of 2. The coordination aperture is smaller for 1 than for 2, which seems unfavourable for the polymerisation of sterically hindered monomers.13a This either suggests that the higher flexibility of the ligand system for 1 is more important for higher activity than a more open but rigid ligand system, or that the smaller coordination aperture in combination with the bulky growing chain leads to a better cation—anion separation and therefore a higher activity. As a comparison, the difference in activity of 1 and 2 for the polymerisation of 3-methyl-1-butene (3M1B) was much less pronounced.13a Interestingly, for the polymerisation of allyl benzene, an opposite trend was observed; 2 proved to be up to 6 times more active than 1.13dZirconocene 3 is significantly less active in 4M1P polymerisation than 2, whereas it was found to be somewhat more active than 2 in the polymerisation of 3M1B.13h The only difference between the structures of 2 and 3 is the methyl at the indenyl 2-position. This seemingly small difference clearly has an unexpected effect on the catalyst activity, as was also observed in some extend during propylene polymerisation.2 Possibly, the steric hindrance of the methyl not only hampers chain termination but also leads to an increase in the energy barrier for monomer insertion for this bulky monomer.13a Note that this contradicts the observation made for 3M1B copolymerisations, for which 3 shows a somewhat higher catalytic activity than 2.13h The rather high activity of 4 was surprising. In fact, this catalyst proved to be by far more active than all the other studied silyl-bridged ansa-zirconocenes, whilst its propylene polymerisation activity has been reported to be low.12 A possible explanation for the high activity of 4 during 4M1P polymerisation might be the aforementioned effective cation–anion separation as a result of the bulkiness of the combination of the ligand system and the bulky growing polymer chain, which allows faster monomer insertion.16,17 Although 5 is a very potent propylene polymerisation catalyst, its activity in 4M1P polymerisation is negligible, even at elevated polymerisation temperatures. The positive synergetic effect that has been observed during propylene polymerisation is clearly absent during 4M1P polymerisation. This dramatic loss of performance of 5 in 4M1P polymerisation can be attributed to the excessive steric hindrance induced by the simultaneous substitution at the 2- and 4-position in combination with a bulky growing polymer chain and monomer. For all temperatures, the activities of the studied catalysts decrease in the order 1 > 4 ≫ 2 > 3 > 5, which clearly shows that there is no direct correlation between propylene, 3M1B and 4M1P polymerisation performance. The contrast in reactivity order for propylene and 3M1B polymerisation was rather unexpected. Interesting to note is that a similar deviation from the results with propylene polymerisation was observed by Resconi et al. who found a relative catalytic activity of 2 ≫ 3 > 5 for 1-butene polymerisation.13g
To investigate how the relative catalytic activity of these catalysts would compare with a sterically less demanding linear α-olefin, the 1-pentene polymerisation behaviour of catalysts 1–5 has been studied (Table 1 and Fig. 2). The first observation that can be made is that the activity of all catalysts, including 5, increases significantly with increasing temperature. The activities at 70 °C are underestimated as a result of too high conversions. Hence, the difference in the activity of 1–5 in the polymerisation of 1-pentene can best be compared in the low temperature range (10–40 °C), where conversions are still relatively low. In agreement with the difference in steric bulk of the monomers, the absolute activities of 1–5 were higher than those observed when the same catalyst was used to polymerise 4M1P but lower than those observed for propylene polymerisation under similar conditions.18 As for 4M1P polymerisation, 1 also gives the highest activity for 1-pentene polymerisation. Catalyst 2 displays lower activities compared to 1, but the differences between these two catalysts when used to polymerise 1-pentene are not as dramatic as during 4M1P polymerisation. The activity of 3 at 10 °C is very low, as observed for 4M1P polymerisation. The activity increased significantly with increasing temperature (≥40 °C), which was not the case during 4M1P polymerisation.
Whereas 4 gave the second highest activity for 4M1P polymerisation at all temperatures, the relative activity of 4 in 1-pentene polymerisation is clearly lagging behind and at 70 °C it is even the lowest of all. The rather low activity of 5 is in agreement with the low activity of 5 in 4M1P and 1-butene polymerisation.9f With only two extra methylene units compared to propylene, the increase in steric hindrance seems small, but becomes significant when considering the growing polymer chain as a whole. Consequently, the activity order of the catalysts resembles more that of 4M1P polymerisation than that of propylene polymerisation.
Polymer molecular weight
Like catalyst activity, the molecular weight of polymers is also governed by the catalyst system and the polymerisation conditions used. Fig. 3 depicts the molecular weights of P4M1P synthesized with catalysts 1–5 at different temperatures. The molecular weights of P4M1P synthesized with 1–5 are rather low and similar for all catalysts except 3, which results in a somewhat higher molecular weight at low polymerisation temperatures. Like in propylene and 3M1B polymerisation,2,13h appending a methyl group at the 2-position of the indenyl impedes the chain transfer process. At higher temperatures this positive effect is clearly lost as the molecular weight of the P4M1P synthesized with 3 drops drastically. Although the presence of a methyl at the 2-position (3) results in polymers with high molecular weight—especially at low polymerisation temperatures—adding a phenyl substituent at the 4-position (5) seems to have a negative effect. Possibly the high steric hindrance of the growing chain overrules the ligand substituent effect on molecular weight. Like for 3, the molecular weights of the P4M1P prepared by 5 show strong temperature dependences and at 70 °C 5 produces the lowest molecular weight polymer of all catalysts. The polydispersities are around 2 as expected for a non-living single site catalyst. The high PDI obtained for 5 at 10 °C might be explained by the sluggish catalytic behaviour of this catalyst at low temperature.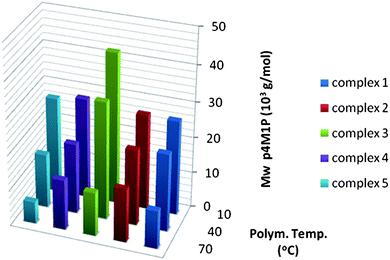 | ||
| Fig. 3 Molecular weights of P4M1P synthesized with MAO activated 1–5 at different temperatures. | ||
The molecular weight of the poly-1-pentene synthesized with 1, 2, 4 and 5 is comparable and rather low. The most striking result from Fig. 4 is the high molecular weight formed by catalyst 3 at low temperatures. Increasing the polymerisation temperature has a detrimental effect on the molecular weight though and at 70 °C the advantage of a methyl at the 2-position is completely lost. Whereas the synergetic effect of the 2-methyl and 4-phenyl substituents in 5 works so well for propylene polymerisation, it is completely absent for 4M1P polymerisation. During 1-pentene polymerisation some of this effect is still visible. In the low temperature range the 2-methyl group in 5 affords higher molecular weights than 2 and at higher temperatures the catalytic activity accelerates rapidly with relative little decrease of molecular weight. At 70 °C the monomer conversion is high which might explain the broadening of the PDI of some of the poly-1-pentenes.
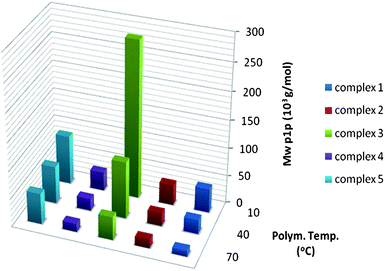 | ||
| Fig. 4 Molecular weights of 1-pentene synthesized with MAO activated 1–5 at different temperatures. | ||
All catalysts show a drastic reduction of the polymer molecular weight and increasing activity with increasing temperature, which results in an increase in the number of chains produced per catalytic site. For 4M1P polymerisation the increase in the number of chains formed at 70 °C compared to 40 °C is striking.
Polymer microstructure
The stereoregularity of polymers is best studied using 13C NMR spectroscopy. While extensive NMR studies on polypropylene9b,19 and for example ethylene/4M1P copolymers have been reported,11 for 4M1P and 1-pentene homopolymers only limited data on regio- and stereo errors are available.14b,20The major problem is that the chemical shifts of the resonances corresponding to the different errors are very similar making the analysis higher than dyad level virtually impossible, which hampers the quantitative evaluation of the stereoregularity. The 13C NMR spectra of all polymers prepared at low temperature show high isotacticity for all catalysts. Of the five catalysts used, 1 shows the strongest dependence of the stereoselectivity on the polymerisation temperature. P4M1P produced with this catalyst display a relatively low stereoregularity compared to the polymers synthesized with catalysts 2–5. P4M1P obtained with both 4 and 5 showed high isotacticity even at high polymerisation temperature. Fig. 5A shows 13C NMR spectra of P4M1P afforded at 70 °C using 1–5. When looking at the resonance at 46 ppm that corresponds to the methylene carbon (3B3) bonded directly to the polymer backbone, there seems to be only little difference in tacticity. In addition to the five main resonances typical of isotactic P4M1P, a number of other signals are observed. These weak resonances can be attributed both to chain ends as well as stereo- and regio-defects. The prevalence of these small resonances differs depending on the catalyst used. Since P4M1P synthesized with 5 at 70 °C has the lowest molecular weight, the resonances associated with chain end carbons (24, 28, 33, 35, 40 and 45 ppm) can be clearly seen from the spectrum.
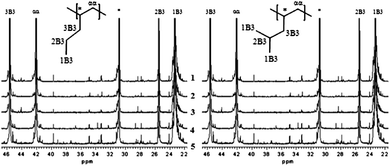 | ||
| Fig. 5 13C NMR spectra of (A) P4M1P and (B) poly-1-pentene synthesized with catalysts 1–5 at 70 °C. | ||
The 13C NMR spectra of poly-1-pentenes synthesized with 1–5 at 70 °C are shown in Fig. 5B. The intensity of resonances associated with chain ends (14, 23, 35 and 39 ppm) can be directly linked to the molecular weight of the polymers. Since poly-1-pentene synthesized with catalyst 5 at 70 °C has the highest molecular weight, the chain end resonances are significantly smaller than for the other samples. The remaining small resonances are attributed to regio-errors. As is evident by the 13C NMR of the polymer, catalyst 1 is more susceptible to regio-misinsertions than for example 5, which is similar to what is observed during propylene polymerisation.2,21 The resonance at 38 ppm corresponds to the methylene carbon (3B3) directly bonded to the poly-1-pentene backbone,20 which is highly sensitive to the relative stereochemistry of the neighbouring monomer units. Whereas Brüll et al. reported the tacticity of poly-1-pentenes obtained with MAO activated 1 to be higher than that of poly-1-pentenes made with 2 and rac-Me2Si(2-Me-Benz[e]Ind)2ZrCl2,20c we see a slightly higher tacticity for the poly-1-pentenes formed by 2 with respect to 1. Catalyst 3 also resulted in poly-1-pentenes with a stereoregularity comparable to that of 2. Poly-1-pentenes produced with 4 and 5 have the highest stereoregularity of all catalysts used. As for propylene polymerisation, substituents on the 4-position have a positive effect on the isotactic microstructure of the polymers of higher α-olefins.2,22
Thermal behaviour
Stereo- and regioregularity, and in low molecular weight polymers also molecular weight, are important parameters affecting the polymer thermal behaviour. Poly-1-pentenes produced with 2–5 at 10 °C show melting endotherms during the second melting run ranging from 63 °C to 68 °C. None of the poly-1-pentenes synthesized above 10 °C show a melting endotherm after the first melting run (Table 1). This is the result of poly-1-pentene being a slow crystallising polymer in combination with the lower isotacticity obtained at higher polymerisation temperatures and a relatively fast cooling rate of 10 °C min−1.The melting temperatures of P4M1P synthesized with different catalysts at different temperatures are shown in Fig. 6. Looking at the plot, the combined effect of molecular weight, tacticity and regioirregularity can be appreciated. Polymers synthesized with 1 have lower melting temperatures compared to polymers made with 2, while they have similar molecular weights. Hence, the lower melting temperatures for P4M1P prepared with 1 reflect the polymers lower stereoregularity. Polymers obtained with 3 have slightly higher melting temperatures compared to those prepared with 2 although their stereoregularity is comparable. Since the molecular weights of the polymers formed by 2 are low, the difference in the melting temperatures of the polymers is likely to be due to differences in molecular weight. Although the molecular weights of the P4M1P obtained with 3 and 4 are rather different, their melting points are virtually identical at all temperatures. According to the 13C NMR spectra of these polymers, P4M1P produced by 3 contains more regio-defects compared to the P4M1P obtained with 4. The low melting temperature of P4M1P formed with 5 can be attributed to the low molecular weight of the polymers, especially those obtained at high temperatures.
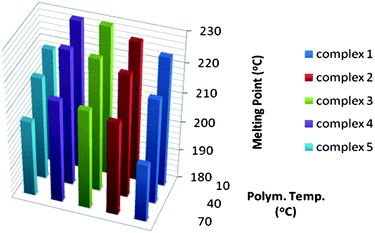 | ||
| Fig. 6 Melting temperatures of P4M1P synthesized with 1–5 at different temperatures. | ||
Whereas the highly stereoregular P4M1P samples display single meting endotherms, the less stereoregular P4M1P samples were found to exhibit multiple melting endotherms in the DSC. This is a common phenomenon and is attributed to the process of melting, recrystallization and remelting: metastable crystallites are recrystallized into a more perfect form following the initial melting.23
C s- and C1-symmetric zirconocenes
The Cs- and C1-symmetric catalyst precursors used to investigate the effect of the catalyst structure and temperature on the polymerisation of 1-pentene and 4M1P are shown in Scheme 2 and the results are given in Table 2 and Fig. 7 and 8. Catalyst 6 was selected to produce syndiotactic polymers. The C1-symmetric catalysts 7–11 were selected to study the effect of different substituents at the bridge, the cyclopentadienyl and the fluorenyl group on the catalysts activity and stereospecificity.8,9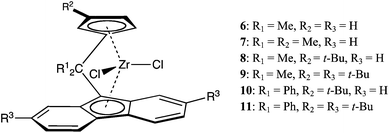 | ||
| Scheme 2 C s- and C1-symmetric zirconocenes used for the polymerisation of 1-pentene and 4M1P: Me2C(Cp)(Flu)ZrCl2 (6), Me2C(3-Me-Cp)(Flu)ZrCl2 (7), Me2C(3-t-Bu-Cp)(Flu)ZrCl2 (8), Me2C(3-t-Bu-Cp)(2,7-t-Bu2-Flu)ZrCl2 (9), Ph2C(3-t-Bu-Cp)(Flu)ZrCl2 (10) and Ph2C(3-t-Bu-Cp)(2,7-t-Bu2-Flu)ZrCl2 (11). | ||
| Exp. | Cat. | Mon. | T/°C | Yield/wt% | Act./g mmol−1 h−1 | M w/103 g per mol | PDI | #d | T m/°C |
|---|---|---|---|---|---|---|---|---|---|
| a Conditions: precatalyst = 5 μmol; Al/Zr ratio = 500; toluene = 40 mL; monomer = 300 mmol, polymerisation time = 30 min. b Three melting endotherms: 189/199/206. c Three melting endotherms: 172/183/191. d Calculated number of chains per active site. | |||||||||
| 1 | 6 | 1P | 10 | 42.0 | 1766 | 37.1 | 2.2 | 23.8 | — |
| 2 | 6 | 1P | 40 | 78.9 | 3320 | 24.0 | 2.1 | 69.2 | — |
| 3 | 6 | 1P | 70 | 29.1 | 1225 | 16.4 | 2.2 | 37.3 | — |
| 4 | 6 | 4M1P | 10 | 4.7 | 235 | 42.7 | 1.9 | 2.8 | 206b |
| 5 | 6 | 4M1P | 40 | 16.9 | 853 | 38.0 | 2.0 | 11.2 | 191c |
| 6 | 6 | 4M1P | 70 | 8.6 | 437 | 20.5 | 2.2 | 10.6 | — |
| 7 | 7 | 1P | 10 | 9.2 | 386 | 98.9 | 1.9 | 2.0 | — |
| 8 | 7 | 1P | 40 | 8.7 | 365 | 61.2 | 2.0 | 3.0 | — |
| 9 | 7 | 1P | 70 | 5.4 | 228 | 36.8 | 2.1 | 3.1 | — |
| 10 | 7 | 4M1P | 10 | 1.3 | 66 | 27.1 | 2.2 | 1.2 | 158 |
| 11 | 7 | 4M1P | 40 | 8.2 | 412 | 24.2 | 2.2 | 8.5 | — |
| 12 | 7 | 4M1P | 70 | 4.8 | 244 | 22.4 | 2.1 | 5.4 | — |
| 13 | 8 | 1P | 10 | 5.8 | 247 | 46.3 | 2.3 | 2.7 | 61 |
| 14 | 8 | 1P | 40 | 33.9 | 1426 | 29.3 | 2.2 | 24.3 | 57 |
| 15 | 8 | 1P | 70 | 30.8 | 1293 | 16.5 | 2.2 | 39.2 | 50 |
| 16 | 8 | 4M1P | 10 | 1.3 | 67 | 39.9 | 1.9 | 0.8 | 231 |
| 17 | 8 | 4M1P | 40 | 12.0 | 603 | 31.2 | 2.2 | 9.7 | 225 |
| 18 | 8 | 4M1P | 70 | 19.3 | 974 | 23.2 | 2.3 | 21.0 | 225 |
| 19 | 9 | 1P | 10 | 12.8 | 537 | 137.5 | 2.1 | 2.0 | 67 |
| 20 | 9 | 1P | 40 | 88.0 | 3702 | 48.3 | 2.2 | 38.3 | — |
| 21 | 9 | 1P | 70 | 98.4 | 4149 | 15.3 | 2.1 | 135.3 | — |
| 22 | 9 | 4M1P | 10 | 12.7 | 63 | 70.0 | 1.9 | 0.4 | 235 |
| 23 | 9 | 4M1P | 40 | 19.9 | 1006 | 64.3 | 2.1 | 7.8 | 232 |
| 24 | 9 | 4M1P | 70 | 45.2 | 2282 | 37.0 | 2.4 | 30.8 | 223 |
| 25 | 10 | 1P | 10 | 2.0 | 83 | 57.9 | 2.2 | 0.7 | — |
| 26 | 10 | 1P | 40 | 58.5 | 2454 | 39.1 | 2.2 | 31.5 | — |
| 27 | 10 | 1P | 70 | 79.9 | 3358 | 12.9 | 2.1 | 130.2 | — |
| 28 | 10 | 4M1P | 10 | 0.8 | 39 | 46.6 | 2.0 | 0.4 | 233 |
| 29 | 10 | 4M1P | 40 | 7.6 | 388 | 40.7 | 2.2 | 4.7 | 230 |
| 30 | 10 | 4M1P | 70 | 29.3 | 1489 | 28.9 | 2.2 | 25.6 | 220 |
| 31 | 11 | 1P | 10 | 18.6 | 781 | 200.4 | 1.9 | 1.9 | 68 |
| 32 | 11 | 1P | 40 | 66.5 | 2796 | 97.6 | 2.1 | 14.3 | — |
| 33 | 11 | 1P | 70 | 100 | 4268 | 27.0 | 2.4 | 78.9 | — |
| 34 | 11 | 4M1P | 10 | 7.9 | 407 | 174.5 | 2.0 | 1.1 | 238 |
| 35 | 11 | 4M1P | 40 | 31.2 | 1574 | 112.3 | 2.2 | 7.0 | 233 |
| 36 | 11 | 4M1P | 70 | 86.7 | 4380 | 35.3 | 2.2 | 62.0 | 223 |
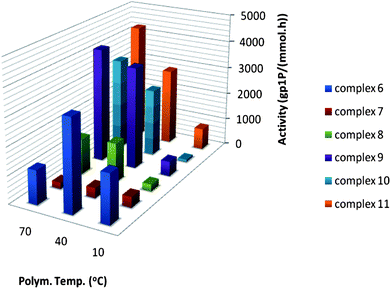 | ||
| Fig. 7 Activities for 1-pentene polymerisation with catalysts 6–11 at different temperatures. | ||
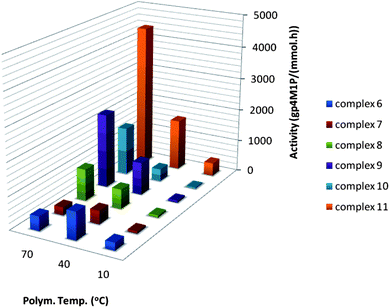 | ||
| Fig. 8 Activities for 4M1P polymerisation with catalysts 6–11 at different temperatures. | ||
Catalytic activity
Fink et al.15 reported on the effect of the Al:Zr ratio on the activity of C1-symmetric catalysts relative to the C2-symmetric ones during the polymerisation of ethylene and propylene and demonstrated that with 6–8 significantly less MAO was needed compared to complex 2 to achieve maximum activity when ethylene was polymerised and even less for propylene polymerisation. Although it is recognised that the optimum Al:Zr ratio differs per catalyst, monomer and type of MAO used, during this study all polymerisations have been carried out using 500 equivalents of MAO. Also for these catalysts we restricted ourselves to polymerization times of 30 minutes to limit the number of experiments.
As expected, the activity for 1-pentene polymerisation is significantly higher than for the branched 4M1P. Asanuma et al. reported differences in activity for 6 depending on the type of monomer in the polymerisation of higher and branched α-olefins.24 The activity of 6 was reported to be low for 1-pentene polymerisation (like for 4M1P polymerisation) while high for 1-butene, 1-hexene and 1-octene polymerisation. Fig. 7 and 8 clearly show the effect of differences in steric hindrance on the catalytic activity and thermal stability. In the lower temperature range, catalyst 6 shows a considerably higher activity than 7 and 8 but is also thermally the least stable one and its optimum in activity for 1-pentene and 4M1P polymerisation is around 40 °C. A similar optimum in activity (40 °C) is seen for 7 although its activity is considerably lower than for 6. Interestingly, 8 shows no loss of activity at higher temperatures and for 4M1P polymerisation the activity increases impressively with increasing temperature. The stability of 8 at elevated temperature is attributed to the steric hindrance offered by the tert-butyl group on the Cp ring. It is not obvious why the activity of 7 falls back compared to the structurally similar but less bulky 6 and sterically more hindered 8. Partly, this could be explained by the dissimilarity in stereo-control induced by the different structures. Whereas the Cs-symmetric 6 affords syndiotactic polymers, the sterically hindered C1-symmetric 8 yields isotactic polymers. Depending on the bulkiness of the monomer and site-epimerisation rate, 7 will either give hemiisotactic or isotactoid polymer (vide infra).
Substituents on the Cp ring were found to have a clear impact on catalyst activity. Substituents on either the bridge or fluorenyl moiety have an impact on the catalytic activity as well. To systematically study the effect of different substituents on the bridge and the 2- and 7-position of the fluorenyl without interference of the Cp substituent, the latter was kept unchanged. Whereas the activity of 8 in 1-pentene polymerisation stays constant between 40 °C and 70 °C, changing the bridge from Me2C to Ph2C25 and/or adding t-Bu substituents to the fluorenyl group invariably results in catalysts with an improved activity, increasing with temperature (Fig. 7). The effect of the bridge is far less prominent for 9 and 11 compared to 8 and 10. The effect of adding fluorenyl t-Bu substituents is obvious when comparing the activity of 8 with that of 9. In the case of 10 and 11 there is only a slight increase in activity for 11 compared to 10. During 4M1P polymerisation an even larger difference in catalytic activity of the catalysts can be observed (Fig. 8). The effect of changing the substituents at the bridge is rather small while the effect of adding tert-butyl substituents to the fluorenyl groups is impressive. Most extraordinary is the activity of 11 in 4M1P polymerisation, which increases significantly with increasing temperature and above 40 °C, is even higher than observed for 1-pentene polymerisation at the same temperature.
As was observed for C2-symmetric zirconocenes (vide supra), seemingly small or distant alterations in the ancillary ligand system can have a pronounced effect on the catalytic activity. The fact that the catalysts with substituents on both the Cp and the fluorenyl moieties are more active than the non-substituted precursors indicate that steric hindrance between the ancillary ligand system and the growing chain is not rate determining. Most probably, the steric hindrance in 11 prevents the formation of a tight ion pair with MAO, which leads to less dormant species and an increase in activity.17 This is supported by the fact that the activity of 11 for polymerisation of 4M1P and 1-pentene is very similar. In spite of its larger steric bulk, the accessibility for 4M1P is similar to 1-pentene, which is not likely in the case of a tight ion pair. Repulsive steric interaction was cited as the main reason behind the low activities of the substituted C2-symmetric catalyst 5 during the polymerisation of 1-pentene and 4M1P. It appears that in the case of C1-symmetric catalysts high steric congestion leads to high activities.
Polymer molecular weight
Not only the activity but also the molecular weight of the polymers is seriously affected by the structure of the catalyst. Fig. 9 shows the molecular weights of poly-1-pentenes synthesized with MAO-activated 6–11 at different temperatures. Surprisingly, the molecular weights of poly-1-pentenes synthesized with 7 are nearly twice as high as those prepared with either 6 or 8 over the whole temperature range. As expected, the molecular weight of the polymer drops with increasing temperatures and at 70 °C the molecular weight is approximately half of that obtained at 10 °C. In contrast to the poly-1-pentenes, the molecular weights of the P4M1Ps produced by 7 are somewhat lower than those prepared with 6 and 8 at the same temperatures and drop only slightly with increasing polymerisation temperature. The difference in molecular weight of poly-1-pentenes synthesized with 8 and 10 is negligible (Fig. 10) and the molecular weight of poly-1-pentenes produced by 9 or 11 at 10 °C are three to four times higher than that of the polymer obtained from 8 and 10, respectively. Although 9 and 11 result in high molecular weight polymers, the decrease in molecular weight with increasing polymerisation temperature is more pronounced for polymers synthesized with these catalysts compared to polymers prepared with 8 and 10. However, the high activity of these catalysts should also be taken into account since the low molecular weight might partly be due to depletion of monomer. The combination of fluorenyl tert-butyl substituents and a Ph2C bridge has a synergetic effect on the molecular weight. Probably, the steric congestion of this catalyst effectively hampers β-H transfer to monomer that requires more space than monomer insertion.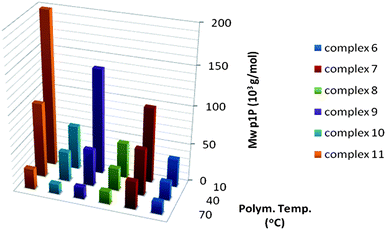 | ||
| Fig. 9 Molecular weights obtained for 1-pentene polymerisation with catalysts 6–11 at different temperatures. | ||
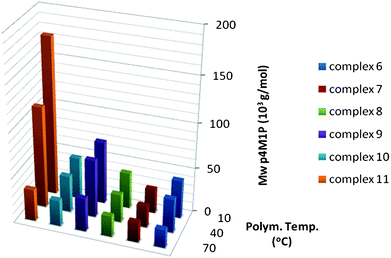 | ||
| Fig. 10 Molecular weights obtained for 4M1P polymerisation with catalysts 6–11 at different temperatures. | ||
The molecular weights of the P4M1Ps synthesized with 6–11 at different temperatures show a similar trend as was observed for poly-1-pentenes (Fig. 10). The effect of phenyl substituents in the bridge is negligible (8versus10) whereas the influence of the fluorenyl tert-butyl substituents is significant, albeit considerably less in the case of 9. Catalyst 9 results in polymers of only slightly higher molecular weights compared to 8. Most interesting is the high molecular weight of the P4M1Ps synthesized with 11 at temperatures up to 40 °C. In fact, the molecular weight of P4M1P produced with 11 at 10 °C is more than four times that of the P4M1P synthesized with 6 under the same conditions. Another point to note is the fact that the decrease in molecular weight with increasing temperature is higher for poly-1-pentene than for P4M1P for all catalysts.
The polydispersities are around 2 as expected for non-living single site catalysts. Like observed for the C2-symmetric catalysts 1–5, with increasing temperature the molecular weight of the polymers obtained by 6–11 decreases dramatically, which especially for 8–11 results in an significant increase in the number of chains produced per catalytic site.
Polymer microstructure
Here we compare the microstructures of poly-1-pentene and P4M1P synthesized with the Cs and C1-symmetric catalysts 6–8 to emphasize the combined effect of catalyst structure and monomer type.
Fig. 11A shows the 13C NMR spectra, and enlarged the 3B3 region, of poly-1-pentene and P4M1P synthesized with 6 at 10 °C. The stereoregularity of both the poly-1-pentene and P4M1P is high, as expected from polymers produced with 6 at low temperature. There is also no evidence for serious regio-errors for both polymers. Increasing the temperature leads to a decline in stereospecificity similar to what was observed for polypropylene produced with 6. Catalyst 7 is known to polymerise propylene to hemiisotactic polypropylene.9 The stereospecificity of 7 is determined by the combined steric hindrance of the ancillary ligand system and the growing chain. As the steric congestion of the system is considerably larger during the polymerisation of higher and branched α-olefins compared to propylene, more isotactoid polymers rather than hemiisotactic polymers are formed. This can be attributed to site-epimerisation of the growing chain as a result of too much steric crowding at the most congested site. Indeed, Herfert and Fink already found that the mmmmpentad content of poly-1-butene and poly-1-hexene synthesized with 7 at 25 °C was higher compared to that of polypropylene synthesized under similar conditions.14e By definition, the methyl pentad content ratio of a hemiisotactic polymer should have the following relationship: mmmm:mmmr:rmmr:mmrr:rrrr:mrrr:mrmr = 3:2:1:4:3:2:1.19k A polymer is therefore said to be hemiisotactic if the mmmmpentad content is about 18.75%. By looking at the 13C NMR spectrum of poly-1-pentene synthesized with 7 (Fig. 11B), it is obvious that the percentage of the mmmmpentad distribution of the polymer is higher than is expected for a hemiisotactic polymer. The microstructure of P4M1P synthesized with 7 under the same conditions is mainly isotactic, which supports the theory of higher frequency of chain-back-skip when sterically more demanding monomers are used. If a rather small substituent like a methyl group on the Cp already influences the mechanism of polymerisation so strongly, an increase in the substituent size should have an even bigger effect. Indeed, poly-1-pentene and P4M1P synthesized with the tert-butyl substituted Cp analogue (8) are highly isotactic. Fig. 11C shows the expected five resonances corresponding to the isotactic polymers, with little evidence of stereo- and regioirregularities. The combination of a tert-butyl group on the Cp ring and the significant steric congestion of the growing poly-1-pentene and especially the P4M1P chain results in rapid site-epimerisation and consequently highly isotactic polymers.
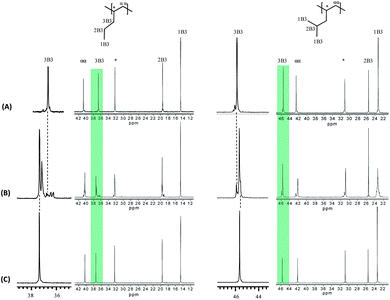 | ||
| Fig. 11 13C NMR spectra and enlarged 3B3 carbon region of poly-1-pentene and P4M1P synthesized at 10 °C with 6 (A), 7 (B), and 8 (C), respectively. | ||
Since 8 already gave highly isotactic poly-1-pentene and P4M1P, it is not surprising that catalysts 9–11 also afford highly isotactic polymers even at 70 °C (Fig. 12). Nevertheless, small differences between the stereoselectivity of the catalysts 8–11 can be observed. For 1-pentene polymerisation the stereospecificity decreases in the order 8 > 9 > 10 > 11. The fact that 9 affords a somewhat more isotactic poly-1-pentene than 10 might suggest that the effect on the tacticity of the phenyl substituents at the bridge is more pronounced than of the fluorenyl tert-butyl substituents, though the influence of both are rather small. For P4MP there is no clear trend except that the most crowded system affords the P4M1P with the lowest tacticity. The lower stereoregularity at high polymerisation temperatures for 11 might be the result of a change in hapticity of the ligand system to relieve the steric congestion around the metal centre, a process that is most likely to happen for the sterically most crowded system.9j
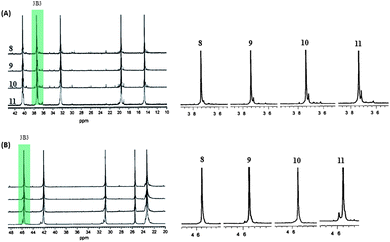 | ||
| Fig. 12 13C NMR spectra and expanded methylene (3B3) region of poly-1-pentene (A) and P4M1P (B) synthesized with catalysts 8–11 at 70 °C. | ||
Thermal behaviour
The use of Cs- and C1-symmetric catalysts has made it possible to synthesize polymers that are highly stereoregular. Whereas syndiotactic poly-1-pentene has been reported to be crystalline,20b the syndiotactic poly-1-pentene formed by 6 does not crystallise from the melt. Like hemiisotactic polypropylene, the poly-1-pentene synthesized with 7 is also amorphous. Unlike the syndiotactic poly-1-pentene produced by 6, syndiotactic P4M1P produced by 6 is crystalline and melts around 200 °C.14 Isotactic P4M1P has a high reported melting temperature of 245 °C.26 The melting temperatures for the P4M1P obtained in this study are rather low, which is probably a consequence of the rather low molecular weight of the polymers (Fig. 13). The melting temperature of P4M1P synthesized at 70 °C with 8 is higher than that of polymers produced with catalysts 9–11 at the same temperature, yet the molecular weight is lower. As seen from the 13C NMR, P4M1P formed with 8 at 70 °C has a higher stereoregularity, which explains the high melting temperature relative to that of the other polymers. Conversely, polymers produced with 11 at low temperatures have the highest melting temperatures. The melting temperatures of all polymers decrease with increasing polymerisation temperature. It appears that although 9 and 11 afford high molecular weight and stereoregularity at low temperatures, the stereoregularity of the polymers produced with 9 and 11 decreases severely with increasing polymerisation temperature relative to the catalysts without substituents on the fluorenyl moiety.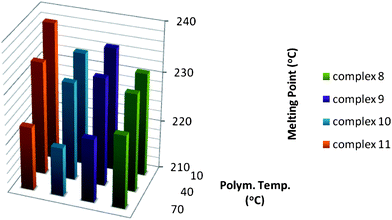 | ||
| Fig. 13 Melting temperature of P4M1P synthesized with catalysts 8–11 at different temperatures. | ||
Conclusions
The activities of the ansa-zirconocenes 1–5 were found to be highly dependent on the combination of the structure of the ligand system and the type of monomer used. The non-substituted ethylene-bridged bis(indenyl) zirconium 1 was found to be quite active in both 1-pentene and 4M1P polymerisation whereas it has proven to be a comparatively poor catalyst in propylene polymerisation. The same can be said for the polymerisation of 4M1P by the silyl-bridged bis(4-phenyl-indenyl) zirconium catalyst 4. Its low catalytic activity in 1-pentene polymerisation was therefore somewhat unexpected. Methyl substituent on the 2-position of the indenyl is detrimental for the catalytic activity for the polymerisation of higher and especially branched α-olefins. The positive synergetic effect of the indenyl 4-phenyl and 2-methyl substituents that has been observed during propylene polymerisation is strongly reduced for 1-pentene polymerisation and completely absent during 4M1P polymerisation. The dramatic loss of performance of 5 is likely due to the excessive steric hindrance induced by the simultaneous substitution at the 2- and 4-position in combination with a bulky growing polymer chain. A similar reduced activity was observed for 1-butene polymerisation.13g Activities for all temperatures, in 1-pentene and 4M1P polymerisation of the studied catalysts, decrease in the order 1 > 2 ≫ 4 > 3 > 5 and 1 > 4 ≫ 2 > 3 > 5, respectively. In both cases complex 5, which performs best for propylene polymerisation, is the poorest catalyst. Although there is a clear deviation from the trend observed in propylene polymerisation, the relative activities compare quite well with the activities of 2, 3 and 5 in 1-butene polymerisation (2 ≫ 3 > 5).13g The relative stereo- (and regio-) selectivity of 1–5 in 1-pentene and 4M1P polymerisation are comparable to that observed for propylene polymerisation.Evidently, even with higher α-olefins, the syndiospecific catalyst 6 adheres to expectations in terms of both regio- and stereoselectivity. For 7, the size of the monomer dictates the rate of site epimerisation such that with higher α-olefins polymers the percentage of mmmmpentad obtained is higher than expected for pure hemiisotactic polymers. Whereas 6 and 7 are thermally labile, the cyclopentadienyltert-butyl substituent renders 8 significantly more stable at high polymerisation temperatures. The presence of tert-butyl substituents on the fluorenyl moiety has both an advantage and a disadvantage. Catalyst activity, polymer molecular weight and regioregularity are enhanced by the presence of the tert-butyl groups. However, the stereoregularity of polymers synthesized with these catalysts at higher temperatures is lower compared to that of polymers synthesized with catalysts without tert-butyl substituents on the fluorenyl moiety. Phenyl substituents on the bridge have limited effect on the polymer molecular weights. However, there seems to be a synergy between phenyl substituents and tert-butyl substituents on the bridge and fluorenyl moiety, as observed with 11. This catalyst resulted in high activity and molecular weight, especially at low polymerisation temperatures. In addition, this catalyst was found to be thermally robust. The activity, molecular weight and stereoregularity of isospecific C1-symmetric catalysts are also dependent on the monomer and yielded unexpected results for both 1-pentene and 4M1P. Unlike the C2-symmetric zirconocenes, isospecific C1-symmetric catalysts 8–11 can result in very high molecular weight poly-1-pentenes and P4M1Ps at low polymerisation temperatures. The activities of these catalysts are also very high compared to the corresponding C2-symmetric catalysts.
Acknowledgements
This research forms part of the research program of the Dutch Polymer Institute (DPI), projects #283 and #708. We thank Dr A. P. Jekel of the Centre for Catalytic Olefin Polymerisation at the Rijksuniversiteit Groningen for the SEC analyses of the polymers. We thank Nic Friederichs and Bing Wang from Saudi Basic Industries Corporation (Sabic-Europe) for providing precatalyst 4, 5 and 9.References
- (a) W. Kaminsky, K. Külper, H. H. Brintzinger and F. R. W. P. Wild, Angew. Chem., Int. Ed. Engl., 1979, 18, 777 CrossRef; (b) F. R. W. P. Wild, L. Zsolnai, G. Huttner and H. H. Brintzinger, J. Organomet. Chem., 1982, 232, 233 CAS; (c) J. A. Ewen, J. Am. Chem. Soc., 1984, 106, 6355 CrossRef CAS; (d) F. R. W. P. Wild, M. Wasiucionek, G. Huttner and H. H. Brintzinger, J. Organomet. Chem., 1985, 288, 63 CrossRef CAS; (e) W. A. Henmann, J. Rohrmann, E. Herdtweck, W. Spaleck and A. Winter, Angew. Chem., Int. Ed. Engl., 1989, 28, 1511 CrossRef.
- L. Resconi, L. Cavallo, A. Fait and F. Piemontesi, Chem. Rev., 2000, 100, 1253 CrossRef CAS.
- W. Spaleck, M. Antberg, J. Rohrmann, A. Winter, B. Bachmann, P. Kiprof, J. Behm and W. Herrmann, Angew. Chem., Int. Ed. Engl., 1992, 31, 1347 CrossRef.
- W. Spaleck, F. Küber, A. Winter, J. Rohrmann, B. Bachmann, M. Antberg, V. Dolle and E. F. Paulus, Organometallics, 1994, 13, 954 CrossRef CAS.
- W. Spaleck, M. Antberg, V. Dolle, R. Klein, J. Rohrmann and A. Winter, New J. Chem., 1990, 14, 499 CAS.
- (a) U. Stehling, J. Diebold, R. Kirsten, W. Röll, H. H. Brintzinger, S. Jüngling, R. Mülhaupt and F. Langhauser, Organometallics, 1994, 13, 964 CrossRef CAS; (b) N. Schneider, M. E. Huttenloch, U. Stehling, R. Kirsten, F. Schaper and H. H. Brintzinger, Organometallics, 1997, 16, 3413 CrossRef CAS.
- (a) J. A. Ewen, R. L. Jones, A. Razavi and J. D. Ferrara, J. Am. Chem. Soc., 1988, 110, 6255 CrossRef CAS; (b) A. Razavi and A. Ferrara, J. Organomet. Chem., 1992, 435, 299 CrossRef CAS; (c) A. Razavi and J. L. Atwood, J. Organomet. Chem., 1993, 459, 117 CrossRef CAS.
- For example see: (a) H. H. Brintzinger, D. Fisher, R. Mülhaupt, B. Rieger and R. M. Waymouth, Angew. Chem., Int. Ed. Engl., 1995, 34, 1143 CrossRef CAS; (b) H. G. Alt and M. Jung, J. Organomet. Chem., 1998, 568, 87 CrossRef CAS; (c) H. G. Alt and R. Zenk, J. Organomet. Chem., 1996, 522, 39 CrossRef CAS; (d) H. G. Alt, R. Zenk and W. Milius, J. Organomet. Chem., 1996, 514, 257 CrossRef CAS; (e) S. A. Miller and J. E. Bercaw, Organometallics, 2004, 23, 1777 CrossRef CAS; (f) L. J. Irwin and S. A. Miller, J. Am. Chem. Soc., 2005, 127, 9972 CrossRef CAS.
- (a) For example see: J. A. Ewen, M. J. Elder, R. L. Jones, L. Haspeslagh, J. L. Atwood, S. G. Bott and K. Robinson, Makromol. Chem., Macromol. Symp., 1991, 48/49, 253 CrossRef; (b) M. Farina, G. Di Silvestro and P. Sozzani, Macromolecules, 1993, 26, 946 CrossRef CAS; (c) B. Rieger, G. Jany, R. Fawzi and M. Steimann, Organometallics, 1994, 13, 647 CrossRef CAS; (d) A. Razavi, L. Napfliotis, L. Peters, D. Vereecke, K. Den Dauw, J. L. Atwood and U. Thewald, Macromol. Symp., 1995, 89, 345 CrossRef CAS; (e) A. Razavi and J. L. Atwood, J. Organomet. Chem., 1995, 497, 105 CrossRef CAS; (f) A. Razavi and J. L. Atwood, J. Organomet. Chem., 1996, 520, 115 CrossRef CAS; (g) U. Dietrich, M. Hackmann, B. Rieger, M. Klingma and M. Leskalä, J. Am. Chem. Soc., 1999, 121, 4348 CrossRef CAS; (h) R. Kleinschmidt, Y. van der Leek, M. Reffke and G. Fink, J. Mol. Catal. A: Chem., 1999, 148, 29 CrossRef CAS; (i) J. Kukral, P. Lehmus, T. Feifel, C. Troll and B. Rieger, Organometallics, 2000, 19, 3767 CrossRef CAS; (j) A. Razavi and U. Thewalt, J. Organomet. Chem., 2001, 621, 267 CrossRef CAS; (k) S. A. Miller and J. E. Bercaw, Organometallics, 2002, 21, 934 CrossRef CAS; (l) A. Hopf and W. Kaminsky, Catal. Commun., 2002, 3, 459 CrossRef CAS; (m) C. Cobzaru, S. Hild, A. Boger, C. Troll and B. Rieger, Coord. Chem. Rev., 2006, 250, 189 CrossRef CAS.
- (a) R. Kleinschmidt, M. Reffke and G. Fink, Macromol. Rapid Commun., 1999, 20, 284 CrossRef CAS; (b) S. A. Miller and J. E. Bercaw, Organometallics, 2002, 21, 934 CrossRef CAS; (c) B. Rieger, G. Jany, R. Fawzi and M. Steimann, Organometallics, 1994, 13, 647 CrossRef CAS; (d) U. Dietrich, M. Hackmann, B. Rieger, M. Klinga and M. Leskelä, J. Am. Chem. Soc., 1999, 121, 4348 CrossRef CAS; (e) C. Cobzaru, S. Hild, A. Boger, C. Troll and B. Rieger, Coord. Chem. Rev., 2006, 250, 189 CrossRef CAS.
- (a) For ethylene based copolymers, see for instance: S. Losio, A. C. Boccia, L. Boggioni, M. C. Sacchi and D. R. Ferro, Macromolecules, 2009, 42, 6964 CrossRef CAS; (b) S. Losio, P. Stagnaro, T. Motta, M. C. Sacchi, F. Piemontesi and M. Galimberti, Macromolecules, 2008, 41, 1104 CrossRef CAS; (c) S. Losio, I. Tritto, G. Zannoni and M. C. Sacchi, Macromolecules, 2006, 39, 8920 CrossRef CAS; (d) C. Lehtinen, P. Starck and B. Loefgren, J. Polym. Sci., Part A: Polym. Chem., 1997, 35, 307 CrossRef CAS; (e) G. Xu and D. Cheng, Macromolecules, 2001, 34, 2040 CrossRef CAS; (f) P. Stagnaro, L. Boragno, S. Losio, M. Canetti, G. C. Alfonso, M. Galimberti, F. Piemontesi and M. C. Sacchi, Macromolecules, 2011, 44, 3712 CrossRef CAS.
- (a) For propylene, see for instance: A. A. Koval'chuk, A. N. Klyamkina, A. M. Aladyshev, P. M. Nedorezova and E. M. Antipov, Polym. Bull., 2006, 56, 145 CrossRef CAS; (b) N. Naga, K. Mizunuma, H. Sadatoshi and M. Kakugo, Macromolecules, 1997, 30, 2197 CrossRef CAS; (c) S. Rulhoff and W. Kaminsky, Macromol. Chem. Phys., 2006, 207, 1450 CrossRef CAS; (d) M. J. Schnieder and R. Mülhaupt, Macromol. Chem. Phys., 1997, 198, 1121 CrossRef; (e) S. Losio, F. Forlini, A. C. Boccia and M. C. Sacchi, Macromolecules, 2011, 44, 3276 CrossRef CAS.
- (a) A. Borriello, V. Busico, R. Cipullo, J. C. Chadwick and O. Sudmeijer, Macromol. Rapid Commun., 1996, 17, 589 CrossRef CAS; (b) M. C. Sacchi, E. Barsties, I. Tritto, P. Locatelli, H. H. Brintzinger and U. Stehling, Macromolecules, 1997, 30, 1267–1271 CrossRef CAS; (c) O. Henschke, J. Knorr and M. Arnold, J. Macromol. Sci., Part A: Pure Appl. Chem., 1998, 35, 473 CrossRef; (d) D.-J. Byun, D.-K. Shin, J. Liu and S. Y. Kim, Polym. Bull., 1999, 42, 265 CrossRef CAS; (e) V. Grumel, R. Brüll, H. Pasch, H. G. Raubenheimer, R. Sanderson and U. M. Wahner, Macromol. Mater. Eng., 2001, 286, 480 CrossRef CAS; (f) F. Grisi, S. Pragliola, C. Costabile and P. Longo, Polymer, 2006, 47, 1930 CrossRef CAS; (g) L. Resconi, I. Camurati and F. Malizia, Macromol. Chem. Phys., 2006, 207, 2257 CrossRef CAS; (h) W. Hu, H. Hagihara and T. Miyoshi, Macromolecules, 2007, 40, 1763 CrossRef CAS; (i) S. Derlin and W. Kaminsky, Macromolecules, 2007, 40, 4130 CrossRef CAS.
- (a) T. Asanuma, Y. Nishimori, M. Ito, N. Uchikawa and T. Shiora, Polym. Bull., 1991, 25, 567 CrossRef CAS; (b) V. Grumel, R. Brüll, H. Pasch, H. G. Raubenheimer, R. Sanderson and U. M. Wahner, Macromol. Mater. Eng., 2002, 287, 559 CrossRef CAS; (c) M. Hoff and W. Kaminsky, Macromol. Chem. Phys., 2004, 205, 1167 CrossRef CAS; (d) D. Coevoet, H. Cramail and A. Deffieux, Macromol. Chem. Phys., 1999, 200, 1208 CrossRef CAS; (e) N. Herfert and G. Fink, Makromol. Chem., Macromol. Symp., 1993, 66, 157 CrossRef CAS; (f) C. R. Baar, C. J. Levy, E. Y. -J Min, L. M. Henling, M. W. Day and J. E. Bercaw, J. Am. Chem. Soc., 2004, 126, 8216 CrossRef CAS; (g) U. M. Wahner, I. Tincul, D. J. Joubert, E. R. Sadiku, F. Forlini, S. Losio, I. Tritto and M. C. Sacchi, Macromol. Chem. Phys., 2003, 204, 1738 CrossRef CAS; (h) M. C. Sacchi, F. Forlini, S. Losio, I. Tritto, U. M. Wahner, I. Tincul, D. J. Joubert and E. R. Sadiku, Macromol. Chem. Phys., 2003, 204, 1643 CrossRef CAS.
- R. Kleinschmidt, Y. van der Leek, M. Reffke and G. Fink, J. Mol. Catal. A: Chem., 1999, 148, 29 CrossRef CAS.
- (a) G. Erker, M. Albrecht, S. Werner and C. Z. Kruger, Naturforsch. B, 1990, 45, 1205 CAS; (b) M. Bochmann and S. L. Lancaster, Angew. Chem., Int. Ed. Engl., 1994, 33, 1634 CrossRef; (c) P. Montag, Y. van der Leek, Angermund and G. Fink, J. Organomet. Chem., 1995, 497, 201 CrossRef CAS; (d) S. Beck, M. H. Prosenc, H. H. Brintzinger, R. Goretzki, N. Herfert and G. Fink, J. Mol. Catal. A: Chem., 1996, 111, 67 CrossRef CAS; (e) I. Tritto, R. Donetti, M. C. Sacchi, P. Locatelli and G. Zannoni, Macromolecules, 1997, 30, 1247 CrossRef CAS; (f) J. Wei, W. Zhang, R. Wickman and L. R. Sita, Angew. Chem., Int. Ed., 2010, 49, 9140 CrossRef CAS; (g) L. Li, M. V. Metz, H. Li, M.-C. Chen, T.-J. Marks, L. Liable-Sands and A. L. Rheingold, J. Am. Chem. Soc., 2002, 124, 12725 CrossRef CAS.
- C. J. Price, H.-Y. Chen, L. M. Launer and S. A. Miller, Angew. Chem., Int. Ed., 2009, 48, 956 CrossRef CAS.
- (a) See for example: I. Kim, J.-M. Zhou and H. Chung, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 1687 CrossRef CAS; (b) R. Brüll, H. Pasch, H. G. Raubenheimer, R. Sanderson and U. M. Wahner, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 2333 CrossRef.
- (a) For example see: F. Heatley and A. Zambelli, Macromolecules, 1969, 2, 618 CrossRef CAS; (b) M. Farina, G. Di Silvestro and P. Sozzani, Macromolecules, 1982, 15, 1452 CrossRef; (c) J. A. Ewen, J. Am. Chem. Soc., 1984, 106, 6355 CrossRef CAS; (d) K. Soga, T. Shiono, S. Takemura and W. Kaminsky, Makromol. Chem. Rapid Commun., 1987, 8, 305 CrossRef CAS; (e) A. Grassi, A. Zambelli, L. Resconi, E. Albizzati and R. Mazzocchi, Macromolecules, 1988, 21, 617 CrossRef CAS; (f) T. Tsutsui, N. Ishimaru, A. Mizuno, A. Toyota and N. Kashiwa, Polymer, 1989, 30, 1350 CrossRef CAS; (g) H. N. Cheng and J. A. Ewen, Makromol. Chem., 1989, 190, 1931 CrossRef CAS; (h) A. Mizuno, T. Tsutsui and N. Kashiwa, Polymer, 1992, 33, 254 CrossRef CAS; (i) L. Resconi, L. Abis and G. Franciscono, Macromolecules, 1992, 25, 6814 CrossRef CAS; (j) L. Resconi, A. Fait, F. Piemontesi, M. Colonnesi, H. Rychlicki and R. Zeigler, Macromolecules, 1995, 28, 6667 CrossRef CAS; (k) M. Farina, G. Di Silvestro and P. Sozzani, Macromolecules, 1993, 26, 946 CrossRef CAS; (l) V. Busico, R. Cipullo, G. Monaco, M. Vacatello and A. L. Segre, Macromolecules, 1997, 30, 6251 CrossRef CAS; (m) J. L. MaciejewskiPetoff, M. D. Bruce, R. M. Waymouth, A. Masood, T. K. Lal, R. W. Quan and S. J. Behrend, Organometallics, 1997, 16, 5909 CrossRef; (n) F. Grisi, P. Longo, A. Zambelli and J. A. Ewen, J. Mol. Catal. A: Chem., 1999, 140, 225 CrossRef CAS; (o) V. Busico, R. Cipullo, A. L. Segre, G. Talarico, M. Vacatello and V. Van Axel Castelli, Macromolecules, 2001, 34, 8412 CrossRef CAS; (p) V. Busico, V. Van Axel Castelli, P. Aprea, R. Cipullo, A. L. Segre, G. Talarico and M. Vacatello, J. Am. Chem. Soc., 2003, 125, 5451 CrossRef CAS.
- (a) T. Asakura, M. Demura and Y. Nishiyama, Macromolecules, 1991, 24, 2334 CrossRef CAS; (b) M. Galimberti, G. Balbontin, I. Camurati and G. Paganetto, Macromol. Rapid Commun., 1994, 15, 633 CrossRef CAS; (c) R. Brüll, D. Kgosane, A. Neveling, H. Pasch, H. G. Raubenheimer, R. Sanderson and U. M. Wahner, Macromol. Symp., 2001, 165, 11 CrossRef; (d) M. R. Mosia, PhD thesis, Eindhoven University of Technology, 2004, ISBN 90-386-2766-1, http://alexandria.tue.nl/extra2/200413170.pdf; (e) H. Gao, X. Liu, Y. Tang, J. Pan and Q. Wu, Polym. Chem., 2011, 2, 1398–1403 RSC.
- (a) M. Lahelin, E. Kokko, P. Lehmus, P. Pitkänen, B. Löfgren and J. Seppälä, Macromol. Chem. Phys., 2003, 204, 1323 CrossRef CAS ; (b) J. F. van Baar, A. D. Horton, K. P. de Kloe, E. Kragtwijk, S. G. Mkoyan, I. E. Nifant'ev, P. A. Schut and I. V. Taidakov, Organometallics, 2003, 22, 2711 CrossRef CAS.
- L. A. Castonguay and A. K. Rappé, J. Am. Chem. Soc., 1992, 114, 5832 CrossRef CAS.
- (a) D. C. Bassett and D. Patel, Polymer, 1994, 35, 1855 CrossRef CAS; (b) C. Silvestre, S. Cimmino, E. Di Pace, M. L. Di Lorenzo, G. Orsello, F. E. Karasz and J. S. Lin, J. Mater. Sci., 2001, 36, 2865 CrossRef CAS.
- T. Asanuma, Y. Nishimori, M. Ito, N. Uchikawa and T. Shiora, Polym. Bull., 1991, 25, 567 CrossRef CAS.
- A. Razavi and J. L. Atwood, J. Organomet. Chem., 1993, 459, 117 CrossRef CAS.
- R. B. Isaacson, I. Kirshenbaum and W. C. Feist, J. Appl. Polym. Sci., 1964, 8, 2789 CrossRef CAS.
- A. Yano, S. Hasegawa, T. Kaneko, T. Sone, M. Sasto and A. Akimoto, Macromol. Chem. Phys., 1999, 200, 1542 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |