Synthesis of diblock copolymers bearing p-methoxyphenacyl side groups
Olivier
Bertrand
,
Jean-François
Gohy
* and
Charles-André
Fustin
*
Institute of Condensed Matter and Nanosciences (IMCN), Bio- and Soft Matter (BSMA), Université catholique de Louvain, Place L. Pasteur 1, Louvain-la-Neuve, Belgium. E-mail: charles-andre.fustin@uclouvain.be; jean-francois.gohy@uclouvain.be
First published on 8th July 2011
Abstract
This contribution reports on the synthesis of light-responsive diblock copolymers containing para-methoxyphenacyl photocleavable side groups. The synthetic strategy consists in the direct polymerization of p-methoxyphenacyl methacrylate by Atom Transfer Radical Polymerization. Several catalytic systems have been screened to synthesize poly(p-methoxyphenacyl methacrylate) in a controlled way. In a second step, diblock copolymers containing a poly(p-methoxyphenacyl methacrylate) block and a block composed of either polystyrene, poly(tert-butyl acrylate) or poly(methyl methacrylate) have been synthesized to evidence the possibility of preparing copolymers with monomers belonging to the three main categories. The cleavage of the p-methoxyphenacyl moieties in solution has been finally demonstrated.
Introduction
For years now polymers have demonstrated their great potential in nanotechnology and life sciences.1,2 Moreover, stimuli-responsive polymers are recognized to be perfect building blocks for the elaboration of “smart” materials. Indeed, those polymers have the ability to change their chemical and/or physical properties in response to slight changes of their environment such as pH, temperature, ionic strength, light, etc.3–6In the last decade, light-sensitive (co)polymers have attracted special attention. Light is indeed a versatile and powerful stimulus because irradiation can be localized in space and time, the process does not need additional reagents, and generates a limited amount of by-products. Moreover, irradiation parameters (wavelength, intensity) can be easily tuned depending on the photosensitive moieties used, insuring selectivity.7 Many examples of block copolymers bearing photosensitive moieties such as azobenzene,8–12coumarin,13,14o-nitrobenzyl,15,16pyrene,17 and spiropyran12,18 derivatives have been previously reported. They were prepared either by controlled radical polymerization (CRP) or by post-modification of polymer precursors, and were exploited in applications such as disruption of micelles to release a cargo, holographic gratings, reversible cross-linking of micelles, morphological transitions in thin films, etc.7
Traditionally used for the photo-release of caged phosphates, phenacyl esters are characterized by a simple chemical structure and an efficient release of the masked acid functions. The side-products are generally biologically inert and transparent at wavelengths longer than 300 nm.19 Moreover, the absence of nitro groups in their chemical structure, which strongly perturb radical polymerization,20 is an undeniable advantage for the synthesis of well-defined polymers. Free radical polymerization (FRP), CRP and polymer modification have been used to produce polymers bearing phenacyl derivatives as side groups. Indeed, FRP was largely used for the synthesis of homopolymers and statistical copolymers.21–24 The grafting of an acetophenone derivative onto poly(methacrylic acid) by a nucleophilic substitution has been used for the formation of statistical copolymers bearing phenacyl derivatives as side groups.26 Only one example of polymerization by a CRP technique has been reported up to now: phenacyl methacrylate was polymerized by atom transfer radical polymerization (ATRP) to produce homopolymers and statistical copolymers.27 However, the “controlled” character of the polymerization was only retained for molar masses lower than 4000 g mol−1. To the best of our knowledge, no high molar mass polymers and no block copolymers bearing photocleavable phenacyl derivatives as side groups were produced by any CRP techniques.
This contribution demonstrates the controlled ATRP of p-methoxyphenacyl methacrylate (MPMA) and the possibility to prepare diblock copolymers with a wide range of monomers. We selected this particular phenacyl derivative because it is known to cleave more efficiently than simple phenacyl upon UV irradiation.20 In the first part of this contribution, the homopolymerization of MPMA by ATRP is reported. Various catalytic systems have been studied to obtain polymers in a controlled way. The second part deals with the synthesis of diblock copolymers composed of one block of PMPMA and another block of polystyrene, poly(tert-butyl acrylate) or poly(methyl methacrylate), evidencing the possibility to use monomers from three main families. Finally the cleavage of the p-methoxyphenacyl side groups is demonstrated in solution.
Experimental
Materials
Styrene (Sty, Aldrich, 99%), tert-butyl acrylate (tBA, Acros, 99%) and methyl methacrylate (MMA, Aldrich, 99%) were passed through activated basic alumina (Acros) columns prior to use. N,N-Dimethylformamide (DMF, Aldrich, 99.8%) was distilled on CaH2 prior to use. Et3N (Fischer Scientific, >98%) was dried over KOH. N-(n-Propyl)-2-pyridylmethanimine (NPPMI) was synthesized as described in ref. 28. CuBr (Aldrich, 99.999%), CuBr2 (Aldrich, 99.999%), CuCl (Aldrich, 99.999%), ethyl 2-bromoisobutyrate (EBiB, Acros, 98%), tosyl chloride (TsCl, Fluka, >99%), anisole (Acros, 99%), N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (PMDETA, Aldrich, 98%), 4,4′-dinonyl-2,2′-dipyridyl (dNbipy, Aldrich, 97%), ethylenediaminetetraacetic acid disodium salt dehydrate (EDTA, Fluka, 97%), 2-bromo-4′-methoxyacetophenone (Aldrich, 97%), methacrylic acid (Acros, 99.5%), and all the other chemicals were used as received.Instrumentation
Proton nuclear magnetic resonance (1H NMR) spectra were acquired on a 300 MHz Bruker Avance II. Molar masses (Mn) and polydispersity indices (PDI) of the polymers were measured on an Agilent gel permeation chromatography (GPC) system equipped with an Agilent 1100/1200 pump (35 °C; eluent: DMF; flow rate of 1 mL min−1), an Agilent differential refractometer and two PSS GRAM columns (Beads 10 μ; porosity of column 1: 1000 Å; porosity of column 2: 100 Å). The calibration was performed using polystyrene standards. Irradiations were performed with 3 Rayonet photochemical reactor lamps (maximum emission wavelength at 300 nm). The intensity received by the samples was equal to 38.5 mW cm−2. UV-visible spectra were recorded on a Varian spectrophotometer (Cary, 50 Conc).Typical procedure for the synthesis of MPMA
Methacrylic acid (3.50 mL; 0.041 mol; 1.1 equiv.) and DMF (103 mL) were placed in a 250 mL flask. Triethylamine (6.30 mL, 0.045 mol; 1.2 equiv) and 2-bromo-4′-methoxyacetophenone (8.59 g, 0.038 mol; 1 equiv) were then added. The reaction mixture was stirred for 1 h at 20 °C, and then water (100 mL) and ethyl acetate (200 mL) were added. The aqueous phase was extracted three times with ethyl acetate, and the organic phases were collected and washed ten times with water. The organic phase was dried with magnesium sulfate, filtered, and the solution was concentrated under reduced pressure. The brownish liquid was filtered on silica (eluent: CH2Cl2) and the solvent was removed under reduced pressure. The slightly yellow solid obtained was purified by recrystallization in hexane (8.65 g, 89%).
1H NMR (300 MHz, CDCl3): δ (ppm) 7.89 (d, 2H, 8.9 Hz, HAr), 6.94 (d, 2H, 8.9 Hz, HAr), 6.25 (t, 1H, 1.5 Hz, CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) ), 5.65 (s, 1H, CH2), 5.35 (s, 2H, O–CH2–CO), 3.86 (s, 3H, O–CH3), 2.00 (s, 3H, CH3–C). 13C NMR (300 MHz, CDCl3): δ (ppm) 190.75 (CO, phenacyl), 166.91 (CO ester), 164.11 (CPh–OMe), 135.71 (C–MeCOO), 130.18 (C–H phenyl), 127.40 (CPh–CO), 126.73 (CH2), 114.13 (C–H phenyl), 66.05 (O–CH2–CO), 55.60 (O–CH3), 18.41 (CH3–C). MS (APCI): m/z = 235 ([M + H]+), 148 ([C9H9O2]+), 127 ([C6H7O3]+), 69 ([C4H5O]+).
), 5.65 (s, 1H, CH2), 5.35 (s, 2H, O–CH2–CO), 3.86 (s, 3H, O–CH3), 2.00 (s, 3H, CH3–C). 13C NMR (300 MHz, CDCl3): δ (ppm) 190.75 (CO, phenacyl), 166.91 (CO ester), 164.11 (CPh–OMe), 135.71 (C–MeCOO), 130.18 (C–H phenyl), 127.40 (CPh–CO), 126.73 (CH2), 114.13 (C–H phenyl), 66.05 (O–CH2–CO), 55.60 (O–CH3), 18.41 (CH3–C). MS (APCI): m/z = 235 ([M + H]+), 148 ([C9H9O2]+), 127 ([C6H7O3]+), 69 ([C4H5O]+).
Typical procedure for the synthesis of PMPMA-Br (Entry 4, Table 1)
Under argon, a round-bottom flask, with a stopcock, containing CuBr (8.1 mg; 0.056 mmol; 1 equiv.) and CuBr2 (1.4 mg; 0.006 mmol; 0.11 equiv.) was filled with a solution containing p-methoxyphenacyl methacrylate (MPMA; 1.32 g; 5.64 mmol; 100 equiv.), EBiB (11 mg; 0.056 mmol; 1 equiv.), NPPMI (27.9 mg; 0.19 mmol; 3.3 equiv.), and anisole (1.33 mL; 50 wt%) previously degassed by three freeze-pump-thaw cycles. The mixture was degassed by three freeze-pump-thaw cycles, filled with argon and stirred in an oil bath at 70 °C for 5 h (MPMA conversion = 42%). The polymerization was quenched by quickly cooling the tube in a water–ice bath and exposing the reaction mixture to air. The reaction mixture was filtered on neutral Al2O3 (eluent: CH2Cl2). The solvent was removed under reduced pressure. The residue was precipitated in Et2O twice, filtered and dried in vacuo at 30 °C for 24 h, affording a white solid (0.43 g, 76%).| Entrya | EBiB/CuBr/CuBr2/ligand/MPMA (molar ratio) | Ligand | T b/°C | t c/h | Convd (%) | M n (th)/g mol−1 | M n e/g mol−1 | PDIe |
|---|---|---|---|---|---|---|---|---|
| a Reactions were performed in anisole (50 wt%). b T = reaction temperature. c t = reaction time. d Conv = conversion of the monomer determined by 1H NMR. e M n and PDI were measured by GPC using a PS calibration. f Performed with 40 wt% of anisole. g Performed with 90 wt% of anisole. | ||||||||
| 1f | 1/1/0/2/100 | dNbipy | 40 | 1.25 | 84 | 19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 700 700 |
34200 |
1.92 |
| 2g | 1/1/0.1/2.2/100 | dNbipy | 60 | 2 | 38 | 9000 | 11500 |
1.21 |
| 3 | 1/1/0.1/3.3/100 | NPPMI | 70 | 9 | 38 | 9000 | 26400 |
1.15 |
| 4 | 1/1/0.1/3.3/100 | NPPMI | 70 | 5 | 42 | 9900 | 19600 |
1.14 |
| 5 | 1/1/0.1/3.3/150 | NPPMI | 70 | 8 | 70 | 24400 |
32900 |
1.36 |
| 6 | 1/1/0.1/3.3/150 | NPPMI | 70 | 6 | 61 | 20000 |
25600 |
1.33 |
| 7 | 1/0.5/0/1.5/150 | NPPMI | 60 | 5.5 | 35 | 12400 |
16100 |
1.25 |
| 8 | 1/0.5/0/1.5/150 | NPPMI | 60 | 5.5 | 46 | 16000 |
54000 |
1.97 |
M
n (GPC) = 19600 g mol−1; PDI (GPC) = 1.14. 1H NMR (300 MHz, CDCl3): δ (ppm) 7.90–7.70 (b, 2H, HAr), 6.90–6.70 (b, 2H, HAr), 5.50–5.10 (b, 2H, O–CH2–CO), 3.90–3.70 (b, 3H, O–CH3), 2.45–1.70 (b, 2H, CH2 backbone), 1.50–1.00 (b, 3H, CH3 backbone).
Typical procedure for the synthesis of PMPMA-Cl (Entry 2, Table 2)
Under argon, a round-bottom flask, with a stopcock, containing CuCl (8.3 mg; 0.084 mmol; 1 equiv.) was filled with a solution containing p-methoxyphenacyl methacrylate (MPMA; 2.75 g; 11.73 mmol; 140 equiv.), tosyl chloride (TsCl; 16 mg; 0.08 mmol; 1 equiv.), dNbipy (68.5 mg; 0.17 mmol; 2 equiv.), and anisole (2.76 mL; 50 wt%) previously degassed by three freeze-pump-thaw cycles. The mixture was degassed by three freeze-pump-thaw cycles, filled with argon and stirred in an oil bath at 60 °C for 5 h (MPMA conversion = 67%). The polymerization was quenched by quickly cooling the tube in a water–ice bath and exposing the reaction mixture to air. The reaction mixture was filtered on neutral Al2O3 (eluent: CH2Cl2). The solvent was removed under reduced pressure. The residue was precipitated in Et2O twice, filtered and dried in vacuo at 30 °C for 24 h, affording a white solid (1.586 g, 82%).| Entrya | MPMA (equiv.) | t b/h | Convc (%) | M n (th)/g mol−1 | M n d/g mol−1 | PDId |
|---|---|---|---|---|---|---|
| a Reaction conditions: TsCl/CuCl/dNbipy = 1/1/2, anisole 50 wt%, T = 60 °C. b t = reaction time. c Conv = conversion of the monomer determined by 1H NMR. d M n and PDI were measured by GPC using a PS calibration. | ||||||
| 1 | 150 | 8 | 61 | 21600 |
19900 |
1.13 |
| 2 | 140 | 5 | 67 | 22200 |
20900 |
1.10 |
M
n (GPC) = 20900 g mol−1; PDI (GPC) = 1.10. 1H NMR (300 MHz, CDCl3): δ (ppm) 7.90–7.70 (b, 2H, HAr), 6.90–6.70 (b, 2H, HAr), 5.50–5.10 (b, 2H, O–CH2–CO), 3.90–3.70 (b, 3H, O–CH3), 2.45–1.70 (b, 2H, CH2 backbone), 1.50–1.00 (b, 3H, CH3 backbone).
Typical procedure for the synthesis of PMPMA-b-PtBA (entry 4, Table 3)
Under argon, a round-bottom flask, with a stopcock, containing CuCl (2.3 mg; 0.023 mmol; 1 equiv.) was filled with a solution containing tert-butyl acrylate (tBA; 0.68 mL; 4.6 mmol; 200 equiv.), PMPMA-Cl (Mn = 19900, PDI = 1.13; 0.46 g; 0.023 mmol; 1 equiv.), dNbipy (19 mg; 0.046 mmol; 2 equiv.), and DMF (1.90 mL; 75 wt%) previously degassed by three freeze-pump-thaw cycles. The mixture was degassed by three freeze-pump-thaw cycles, filled with argon and stirred in an oil bath at 50 °C for 3 h 30 (tBA conversion = 15%). The polymerization was quenched by quickly cooling the tube in a water–ice bath and exposing the reaction mixture to air. The reaction mixture was filtered on neutral Al2O3 (eluent: CH2Cl2). The solvent was removed under reduced pressure. The residue was precipitated in MeOH:H2O 75:25, filtered and dried in vacuo at 30 °C for 24 h, affording a white solid (0.30 g, 55%).
| Entrya | PMPMA Mn (PDI) | Monomer | Monomer (equiv.) | T b/°C | t c/h | Convd (%) | M n (th)/g mol−1 | M n e/g mol−1 | M n (2nd block)e | PDIf (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| a Reaction conditions: PMPMA/CuCl/PMDETA = 1/1/1.1, DMF 70 wt%. b T = reaction temperature. c t = reaction time. d Conv = conversion of the monomer determined by 1H NMR. e M n and Mn (2nd block) were determined by 1H NMR. f PDI were measured by GPC using a PS calibration. g Reaction conditions: PMPMA/CuCl/dNbipy = 1/1.15/2.3, DMF 75 wt%. h Reaction conditions: PMPMA/CuCl/dNbipy = 1/1.15/2.3. | ||||||||||
| 1 | 19900 (1.13) |
tBA | 200 | 70 | 8 | 93 | 43700 |
46000 |
26100 |
1.50 |
| 2 | 19900 (1.13) |
tBA | 300 | 60 | 4 | 65 | 45000 |
40900 |
21000 |
1.32 |
| 3 | 19900 (1.13) |
tBA | 345 | 50 | 5 | 60 | 43000 |
47400 |
27500 |
1.31 |
| 4g | 19900 (1.13) |
tBA | 200 | 50 | 3.5 | 15 | 23700 |
22800 |
2900 | 1.15 |
| 5h | 20900 (1.10) |
MMA | 345 | 50 | 1.5 | 29 | 30900 |
29500 |
8600 | 1.20 |
M
n (GPC) = 24700 g mol−1; PDI (GPC) = 1.15. 1H NMR (300 MHz, CDCl3): δ (ppm) 7.90–7.70 (b, 2H, HAr), 6.90–6.70 (b, 2H, HAr), 5.50–5.10 (b, 2H, O–CH2–CO), 3.90–3.70 (b, 3H, O–CH3), 2.45–0.90 (b, 8H, CH backbone (PMPMA + PtBA) + CH3 (PtBA)).
Typical procedure for the synthesis of PMPMA-b-PMMA (entry 5, Table 3)
Under argon, a round-bottom flask, with a stopcock, containing CuCl (2.8 mg; 0.028 mmol; 1.15 equiv.) and dNbipy (23.6 mg; 0.058 mmol; 2.3 equiv.) was filled with a solution containing methyl methacrylate (MMA; 0.93 mL; 8.7 mmol; 345 equiv.), PMPMA-Cl (Mn = 20900, PDI = 1.10; 0.53 g; 0.025 mmol; 1 equiv.) and DMF (2.4 mL; 70 wt%) previously degassed by three freeze-pump-thaw cycles. The mixture was degassed by three freeze-pump-thaw cycles, filled with argon and stirred in an oil bath at 50 °C for 1 h 30 (MMA conversion = 29%). The polymerization was quenched by quickly cooling the tube in a water–ice bath and exposing the reaction mixture to air. The reaction mixture was filtered on neutral Al2O3 (eluent: CH2Cl2). The solvent was removed under reduced pressure. The residue was precipitated in hexane, filtered and dried in vacuo at 30 °C for 24 h, affording a white powder (0.48 g, 61%).
M
n (GPC) = 29100 g mol−1; PDI (GPC) = 1.20. 1H NMR (300 MHz, CDCl3): δ (ppm) 7.90–7.70 (b, 2H, HAr (PMPMA)), 6.90–6.70 (b, 2H, HAr (PMPMA)), 5.50–5.10 (b, 2H, O–CH2–CO (PMPMA)), 3.90–3.70 (b, 3H, O–CH3 (PMPMA)), 3.70–3.50 (b, 3H, O–CH3 (PMMA)), 2.45–0.60 (b, 10H, CH2 + CH3 backbone (PMPMA + PMMA)).
Typical procedure for the synthesis of PS87-Br
Under argon, a round-bottom flask, with a stopcock, containing CuBr (14.9 mg; 0.104 mmol; 1 equiv.) was filled with a solution containing styrene (4.76 mL; 41.5 mmol; 400 equiv.), EBiB (20.3 mg; 0.104 mmol; 1 equiv.), PMDETA (19.8 mg; 0.11 mmol; 1.1 equiv.), and anisole (1.09 mL; 20 wt%) previously degassed by three freeze-pump-thaw cycles. The mixture was degassed by three freeze-pump-thaw cycles, filled with argon and stirred in an oil bath at 100 °C for 6 h (styrene conversion = 23%). The polymerization was quenched by quickly cooling the tube in a water–ice bath and exposing the reaction mixture to air. The reaction mixture was diluted with CH2Cl2 and washed with an aqueous solution of EDTA (0.04 M). The organic phase was dried over MgSO4, filtered and the solvent was removed under reduced pressure. The residue was precipitated in MeOH twice, filtered and dried in vacuo at 30 °C for 24 h, affording a white solid (0.83 g, 83%).M n (GPC) = 9100 g mol−1; PDI (GPC) = 1.07. 1H NMR (300 MHz, CDCl3): δ (ppm) 7.20–6.10 (b, 5H, HAr), 2.40–1.10 (b, 3H, H backbone).
Typical procedure for the synthesis of PS87-b-PMPMA144 (Entry 2, Table 4)
Under argon, a round-bottom flask, with a stopcock, containing CuCl (2.4 mg; 0.024 mmol; 1 equiv.) was filled with a solution containing p-methoxyphenacyl methacrylate (MPMA; 0.85 g; 3.6 mmol; 150 equiv.), PS-Br (0.22 g; 0.024 mmol; 1 equiv.), dNbipy (19.8 mg; 0.048 mmol; 2 equiv.), and anisole (1.99 mL; 70 wt%) previously degassed by three freeze-pump-thaw cycles. The mixture was degassed by three freeze-pump-thaw cycles, filled with argon and stirred in an oil bath at 60 °C for 4 h 30 (MPMA conversion = 74%). The polymerization was quenched by quickly cooling the tube in a water–ice bath and exposing the reaction mixture to air. The reaction mixture was filtered on neutral Al2O3 (eluent: CH2Cl2). The solvent was removed under reduced pressure. The residue was precipitated in Et2O twice, filtered and dried in vacuo at 30 °C for 24 h. Residual homo-PS was removed by extraction with a selective solvent (Et2O), affording a white solid (0.55 g, 87%).| Entrya | PS Mn (PDI) | t b/h | Convc (%) | M n (th)/g mol−1 | M n d/g mol−1 | M n (2nd block)d | PDIe |
|---|---|---|---|---|---|---|---|
| a Reaction conditions: PS/CuCl/dNbipy/MPMA = 1/1/2/150, anisole 60 wt%, T = 60 °C. b t = reaction time. c Conv = conversion of the monomer determined by 1H NMR. d Determined by 1H NMR. e Determined by GPC. | |||||||
| 1 | 9100 (1.07) | 4.5 | 74 | 35200 |
42300 |
33200 |
1.29 |
| 2 | 9100 (1.07) | 3 | 67 | 33100 |
40200 |
31100 |
1.30 |
M
n (GPC) = 37500 g mol−1; PDI (GPC) = 1.29. 1H NMR (300 MHz, CDCl3): δ (ppm) 7.90–7.70 (b, 288H, HAr, PMPMA), 7.20–6.30 (b, 723H, HAr, PS + PMPMA), 5.60–5.00 (b, 296H, O–CH2–CO), 3.95–3.40 (b, 441H, O–CH3) 2.40–1.10 (b, 981H, H backbone, PS + PMPMA).
Typical procedure for the photocleavage in solution (UV-Vis)
A solution of PMPMA in CHCl3 (C = 5 × 10−3 mg mL−1) was irradiated under stirring at 300 nm. The photocleavage was followed by UV-visible spectroscopy until no change was observed in the shape of the spectra (t(min) = 0, 15, 30, 45, 75, 135, 165).Results and discussion
The monomer was synthesized following the procedure described by Voit et al.23para-Methoxyphenacyl methacrylate is obtained by a nucleophilic substitution which consists in firstly deprotonating the carboxylic acid with triethylamine to generate the carboxylate which substitutes the bromide of 2-bromo-4′-methoxyacetophenone in a second step (Scheme 1).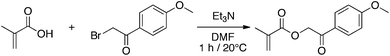 | ||
| Scheme 1 Synthesis of p-methoxyphenacyl methacrylate. | ||
Polymerization of MPMA
Polymers were synthesized using atom transfer radical polymerization (ATRP). Table 1 presents a summary of the results of the ATRP of MPMA catalyzed by copper(I) bromide (Scheme 2a). The parameters that were screened are the ligand (dNbipy or NPPMI), the temperature (40 to 70 °C), the addition of CuBr2, and the monomer concentration (40, 50 or 90 wt% of anisole as solvent). Except for two sets of conditions (Entries 1 and 8), the accordingly obtained polymers exhibit good to reasonable PDI. However, GPC chromatograms (Fig. 1b–d) revealed an asymmetry toward the high elution times (low molar masses), suggesting the occurrence of side reactions and therefore a not fully controlled behavior. The best results were obtained at 70 °C with NPPMI as ligand, and 0.1 equivalent of CuBr2 (Table 1, Entry 3). Under these conditions, a polymer with a PDI of 1.15 and exhibiting a symmetrical chromatogram (Fig. 1a) was obtained. However, problems of reproducibility were met and an important difference between the theoretical and experimental molar masses was observed, indicating a non-controlled behavior. | ||
| Scheme 2 Synthetic approach for the formation of diblock copolymers bearing p-methoxyphenacyl side groups: (a) homopolymerization of MPMA by ATRP, (b) copolymerization of tert-butyl acrylate and methyl methacrylate from a PMPMA-Cl macroinitiator, (c) copolymerization of MPMA from a PS-Br macroinitiator. | ||
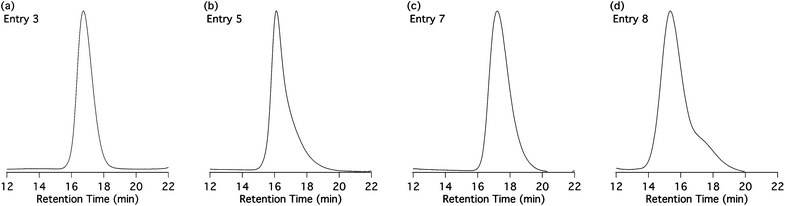
Coskun's group has studied the nature of the end-groups of poly(phenacyl methacrylate) produced by FRP and ATRP.24,27 The side reaction that occurs during the polymerization of PMPMA is the formation of a lactone cycle at the chain end. This lactone is formed either by the removal of phenacyl bromide (Scheme 3a) or by the attack of the chain-end radical onto the carbonyl of a phenacyl side group (Scheme 3b). As the concentration of radicals in the reaction medium is low in ATRP, the probability of the radical lactone cyclization is not considered as the major deactivation reaction. Deactivation of the chains through p-methoxyacetophenone bromide abstraction and cyclisation into a lactone is thus hypothesized in the case of ATRP (Scheme 3a). This side reaction, which occurs rather early in the polymerization process, leads to the formation of dead chains of low molar mass, which explains the observation of the shoulder at high elution times in the chromatograms of Fig. 1.
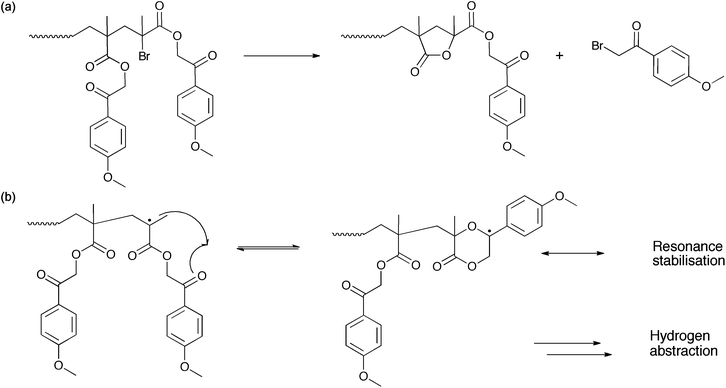 | ||
| Scheme 3 Side reactions occurring during the polymerization of p-methoxyphenacyl methacrylate: (a) removal of p-methoxyphenacyl bromide, (b) radical cyclization. | ||
In an effort to prevent the abstraction of p-methoxyphenacyl halide during polymerization, the halide–polymer bond strength has been increased by replacing the bromide by a chloride.
The catalytic system was thus changed to CuCl/dNbipy and TsCl was used as the initiator (Scheme 2a). The conditions and the results of the polymerizations of MPMA with the CuCl/dNbipy catalytic system are presented in Table 2. Well-defined PMPMA with molar masses ranging from 19000 to 21000 g mol−1 (Mn) and with narrow molar mass distributions (PDI < 1.15) were obtained in this way.
The low PDI gives us a clue that the polymerization of MPMA with CuCl/dNbipy as the catalytic system is controlled. To confirm the controlled behavior of the polymerization, a kinetic study has been realized for the conditions presented in Entry 1 of Table 2. Fig. 2a presents the semi-logarithmic plot of conversion vs. time. A linear variation of the conversion with time is observed, indicating a constant concentration of active species in the medium. Moreover, the molar masses can be predicted since they increase linearly with conversion (Fig. 2b). A typical GPC chromatogram of PMPMA produced with the CuCl/dNbipy system is shown in Fig. 3. A symmetrical peak with a significant narrowing of the molar mass distribution compared to the GPC chromatograms of PMPMA produced with CuBr can be observed. All these observations confirm the controlled character of the polymerization of MPMA with the CuCl/dNbipy catalytic system in the studied conversion range.
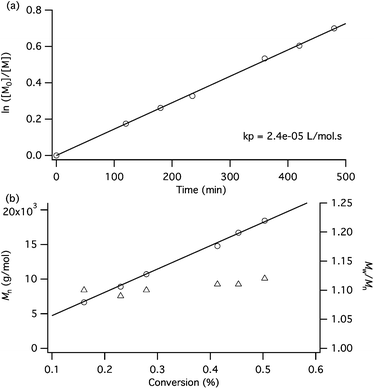 | ||
| Fig. 2 (a) Kinetic plot for the ATRP of MPMA using CuCl/dNbipy as the catalytic system, (b) dependence of molar mass (Mn, ○) and polydispersity index (Mw/Mn, △) on monomer conversion. Reaction conditions: TsCl/CuCl/dNbipy/MPMA = 1/1/2/150 T = 60 °C in anisole (50 wt%). | ||
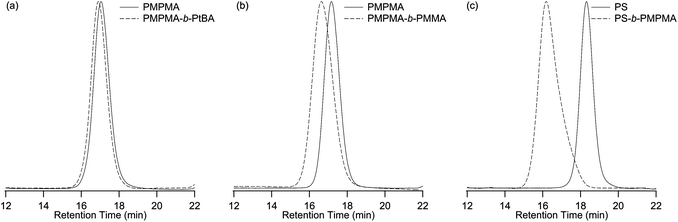 | ||
| Fig. 3 Typical GPC chromatograms of macroinitiators (solid curve) and of diblock copolymers (dashed curve): (a) copolymerization of tBA starting from PMPMA (Entry 4, Table 3), (b) copolymerization of MMA starting from PMPMA (Entry 5, Table 3), (c) copolymerization of MPMA starting from PS (Entry 1, Table 4). | ||
Diblock copolymers
In the first section of this paper, the controlled behavior of the polymerization of MPMA by ATRP has been demonstrated. The second section will focus on the synthesis of well-defined block copolymers containing a poly(p-methoxyphenacyl methacrylate) block. The second block of these copolymers will be composed of polystyrene, poly(tert-butyl acrylate) or poly(methyl methacrylate). These monomers have been selected to demonstrate that a wide range of diblock copolymers containing poly(p-methoxyphenacyl methacrylate) can be produced by ATRP, and that the PMPMA previously synthesized is living and can thus reinitiate a polymerization. Scheme 2b presents the copolymerization reactions starting from a PMPMA macroinitiator. The macroinitiators were prepared with the CuCl/dNbipy catalytic system (Table 2). The experimental conditions and the results for the polymerizations of tert-butyl acrylate and methyl methacrylate are presented in Table 3.The polymerization of tBA was carried out with CuCl/PMDETA in DMF (70 wt%). The effect of temperature was investigated for these conditions by performing the reaction at 70, 60 and 50 °C. We can notice that the decrease of the temperature from 70 °C to 60 °C for the polymerization of tBA leads to a decrease of the PDI (Entries 1 and 2). The decrease of the temperature to 50 °C does not change the PDI (Entries 2 and 3). Polymerization of tBA with CuCl/dNbipy as the catalyst at low temperature (50 °C) yields a PMPMA-b-PtBA with a narrow PDI (Entry 4, Fig. 3a, PDI = 1.15). Similar conditions were applied for the polymerization of MMA, yielding a block copolymer with Mn = 29100 g mol−1 and a PDI of 1.20 (Entry 5, Fig. 3b).
As demonstrated by the polymerization of tBA from a PMPMA macroinitiator (Table 2), polymerization at higher temperature (T > 60 °C) leads to the formation of copolymers with a broad PDI. This was logically more pronounced for the polymerization of styrene at 100 °C (results not shown). Another approach was thus followed for the preparation of copolymers composed of PMPMA and PS (Scheme 2c) to avoid heating PMPMA at too high temperatures. Starting from a PS-Br macroinitiator, MPMA was polymerized with the CuCl/dNbipy catalytic system in anisole (60 wt%) at 60 °C. Table 4 presents the results of the polymerization of MPMA from a PS macroinitiator.
The conditions used for the polymerization of MPMA were the same as for the homopolymerization (Table 2) except for the initiator. The presence of bromide at the chain-end of the macroinitiator has two effects on the polymerization. First, an increase of the polymerization rate is observed. Second, a slight tailing for the GPC chromatogram of the PS-b-PMPMA is observed in Fig. 3c. This tailing indicates that a loss of phenacyl bromide occurs during the polymerization as a mixture of both bromine and chlorine atoms caps the polymer chain during the polymerization.29 Even if side reactions occur during the polymerization, well-defined PS-b-PMPMA block copolymers with PDI ≤ 1.3 are nevertheless obtained.
Photocleavage in solution
Upon irradiation, phenacyl esters undergo a cleavage by homolytic C–O bond scission to give an acryloxy radical and a phenacyl derivative radical (Scheme 4).25 The success of this approach relies on the rapid H atom transfer to the acryloxy radical to yield a carboxylic acid. According to this mechanism, p-methoxyacetophenone is generated as a photoproduct.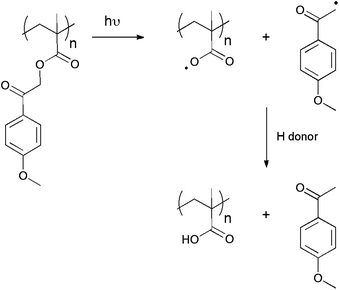 | ||
| Scheme 4 Decomposition mechanism of poly(p-methoxyphenacyl methacrylate) upon irradiation. | ||
The photocleavage (λ = 300 nm) of the PMPMA has been carried out in CHCl3 stabilized by ethanol and followed by UV-Vis spectroscopy. In this experiment, ethanol plays the role of the hydrogen donor.30Fig. 4 illustrates the typical evolution of the UV-Vis absorption spectra of the PMPMA polymer upon irradiation. A decrease of the intensity of the absorption band with irradiation time is observed. This decrease is accompanied by a shift of the band maximum to lower wavelengths (17 nm), probably due to the products or intermediates formed during irradiation.26
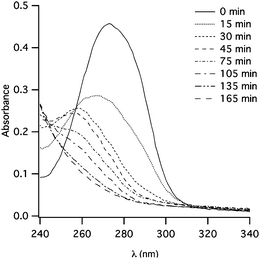 | ||
| Fig. 4 Evolution with irradiation time of the UV-Vis spectra of PMPMA irradiated at 300 nm in CHCl3 (C = 5 × 10−3 g L−1). | ||
In addition to UV-Vis spectroscopy, 1H NMR analysis was realized on the irradiated solution. To this aim, a solution of PMPMA in CHCl3 was irradiated for 40 h. Since the resulting hydrophilic poly(methacrylic acid) is insoluble in CHCl3, the polymer could easily be separated from the photoproduct of low molar mass by filtration. Both fractions were then analyzed by 1H NMR. Their respective spectra are shown in Fig. 5. The spectrum of the polymer fraction is indeed characteristic of PMAA, and the one of the small molecule fraction demonstrates that the major side product is p-methoxyacetophenone, confirming the photocleavage mechanism presented in Scheme 4.
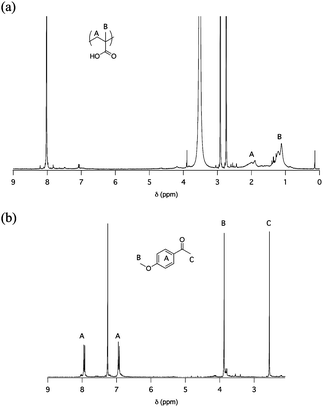 | ||
| Fig. 5 1H NMR spectra of the products generated after 40 h of irradiation of a PMPMA solution: (a) polymer fraction (d7-DMF), (b) small molecule fraction (CDCl3) (Table 2, Entry 1, C = 3 g L−1 in CHCl3). | ||
The photocleavage has also been realized on the block copolymers of the three families: PMPMA-b-PtBA, PMPMA-b-PMMA and PS-b-PMPMA. Fig. 6 presents the evolution with irradiation time of the UV-Vis spectra of the block copolymers. These spectra show an evolution similar to the one observed for the PMPMA homopolymer, evidencing clearly the efficiency of the photocleavage.
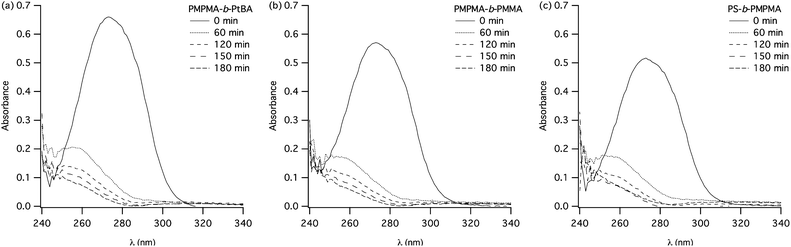 | ||
| Fig. 6 Evolution with irradiation time of the UV-Vis spectra of solutions irradiated at 300 nm in CHCl3 (C = 1 × 10−2 g L−1) of: (a) PMPMA-b-PtBA (entry 4, Table 3), (b) PMPMA-b-PMMA (entry 5, Table 3), (c) PS-b-PMPMA (entry 1, Table 4). | ||
Conclusions
In this contribution, the synthesis by ATRP of different well-defined block copolymers bearing p-methoxyphenacyl side groups was presented for the first time.In the first part the homopolymerization of MPMA was realized by screening different experimental parameters. The best results were obtained by using TsCl/CuCl/dNbipy as the catalytic system, which gave rise to a well-controlled behavior, and led to homopolymers with low PDI. In the second part, copolymerization of (meth)acrylate monomers starting from a PMPMA macroinitiator was studied. Several polymerization parameters were screened and optimized to allow the synthesis of well-defined block copolymers. Diblock copolymers composed of polystyrene and poly(p-methoxyphenacyl methacrylate) were obtained with a halide exchange approach, starting from a PS-Br macroinitiator with CuCl/dNbipy as the catalytic system.
Finally, the successful and quantitative photocleavage of the p-methoxyphenacyl moiety was demonstrated on the homopolymer and on the different block copolymers by UV-Vis spectroscopy. 1H NMR showed that the irradiation leads, as expected, to the formation of PMAA and p-methoxyacetophenone.
The preparation of well-defined block copolymers bearing phenacyl side groups in a direct and controlled way opens up new opportunities for the development of applications in diverse fields exploiting photocleavable polymers. Indeed, fields like nanotechnology and controlled release are in a constant need for well-defined, easily accessible, stimuli responsive copolymers. The proposed synthetic strategy will allow the fine tuning of the photosensitive block copolymer to the targeted application.
Acknowledgements
The authors are grateful to BELSPO for financial support in the frame of IAP 6/27: Functional Supramolecular Systems. C. A. F. is Research Associate of the FRS-FNRS.Notes and references
- C. Park, J. Yoon and E. L. Thomas, Polymer, 2003, 44, 6725 CrossRef CAS.
- Z. L. Tyrrell, Y. Shen and M. Radosz, Prog. Polym. Sci., 2010, 35, 1128 CrossRef CAS.
- N. Rapoport, Prog. Polym. Sci., 2007, 32, 962 CrossRef CAS.
- F. Liu and M. W. Urban, Prog. Polym. Sci., 2010, 35, 3 CrossRef CAS.
- D. Roy, J. N. Cambre and B. S. Sumerlin, Prog. Polym. Sci., 2010, 35, 278 CrossRef CAS.
- M. A. Cohen Stuart, W. T. S. Huck, J. Genzer, M. Mueller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. W. Urban, F. Winnik, S. Zauscher, I. Luzinov and S. Minko, Nat. Mater., 2010, 9, 101 CrossRef CAS.
- J. M. Schumers, C. A. Fustin and J. F. Gohy, Macromol. Rapid Commun., 2010, 31, 1588 CrossRef CAS.
- G. Wang, X. Tong and Y. Zhao, Macromolecules, 2004, 37, 8911 CrossRef CAS.
- Y. P. Wang, M. Zhang, C. Moers, S. L. Chen, H. P. Xu, Z. Q. Wang, X. Zhang and Z. B. Li, Polymer, 2009, 50, 4821 CrossRef CAS.
- F. D. Jochum and P. Theato, Chem. Commun., 2010, 46, 6717 RSC.
- (a) J. C. Barrett, J. I. Mamiya, K. G. Yager and T. Ikeda, Soft Matter, 2007, 3, 1249 RSC; (b) S. K. Yesodha, C. K. S. Pillai and N. Tsutsumi, Prog. Polym. Sci., 2004, 29, 45 CrossRef CAS.
- Y. Zhao, L. Tremblay and Y. Zhao, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 4055 CrossRef CAS.
- J. Babin, M. Lepage and Y. Zhao, Macromolecules, 2008, 41, 1246 CrossRef CAS.
- J. Q. Jiang, B. Qi, M. Lepage and Y. Zhao, Macromolecules, 2007, 40, 790 CrossRef CAS.
- J. Jiang, X. Tong, D. Morris and Y. Zhao, Macromolecules, 2006, 39, 4633 CrossRef CAS.
- X. Jiang, C. A. Lavender, J. W. Woodcock and B. Zhao, Macromolecules, 2008, 41, 2632 CrossRef CAS.
- J. Jiang, X. Tong and Y. Zhao, J. Am. Chem. Soc., 2005, 127, 8290 CrossRef CAS.
- H. Lee, W. Wu, J. K. Oh, L. Mueller, G. Sherwood, L. Peteanu, T. Kowalewski and K. Matyjaszewski, Angew. Chem., Int. Ed., 2007, 46, 2453 CrossRef CAS.
- C. G. Bochet, J. Chem. Soc., Perkin Trans. 1, 2002, 125 CAS.
- J. M. Schumers, C. A. Fustin, A. Can, R. Hoogenboom, U. S. Schubert and J. F. Gohy, Macromol. Rapid Commun., 2009, 47, 6504 CAS.
- C. Soykan, M. Ahmedzade and M. Fatih Coskun, Eur. Polym. J., 2000, 36, 1667 CrossRef CAS.
- M. Fatih Coskun, C. Soykan, M. Ahmedzade and K. Demirelli, Polym. Degrad. Stab., 2001, 72, 69 CrossRef.
- M. Millaruelo, K.-J. Eichhorn, B. Sieczkowska and B. Voit, Langmuir, 2006, 22, 9436 CrossRef CAS.
- M. F. Coskun, K. Demirelli and M. Coskun, J. Macromol. Sci., Part A: Pure Appl.Chem., 2007, 44, 1217 CAS.
- K. Inomataa, S. Kawasakib, A. Kameyamab and T. Nishikubo, React. Funct. Polym., 2000, 45, 1 CrossRef.
- K. Inomataa, S. Kawasakib, A. Kameyamab and T. Nishikubo, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 530 CrossRef.
- M. F. Coskun, K. Demirelli and M. Coskun, J. Macromol. Sci., Part A: Pure Appl.Chem., 2007, 44, 995 CAS.
- D. M. Haddleton, M. C. Crossman, B. H. Dana, D. J. Duncalf, A. M. Heming, D. Kukulj and A. J. Shooter, Macromolecules, 1999, 32, 2110 CrossRef CAS.
- K. Matyjaszewski, D. A. Shipp, J. L. Wang, T. Grimaud and T. E. Patten, Macromolecules, 1998, 31, 6836 CrossRef CAS.
- M. Millaruelo, L. M. Eng, M. Mertig, B. Pilch, U. Oertel, J. Opitz, B. Sieczkowska, F. Simon and B. Voit, Langmuir, 2006, 22, 9446 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |