DOI:
10.1039/C1PY00250C
(Paper)
Polym. Chem., 2011,
2, 2334-2340
Facile synthesis of agarose-L-phenylalanine ester hydrogels†
Received
1st June 2011
, Accepted 5th July 2011
First published on 29th July 2011
Abstract
Hydrogel forming agarose-L-phenylalanine ester (Ag-PAEst) was synthesized in a facile microwave mediated method involving the reaction of agarose with fluorenyl methyloxycarbonyl (Fmoc) protected amino acidL-phenylalanine (PA) in the presence of dicyclohexylcarbodiimide/4-dimethylaminopyridine (DCC/DMAP) followed by deprotection. Subsequently, the ester was cross-linked with the natural cross-linker genipin to yield a blue hydrogel (G-Ag-PAEst). Both the ester and cross-linked hydrogels had comparable gelling characteristics with agarose. These hydrogels were highly stable in all pH media (pH 1.2, 7.0 and 12.5) under ambient conditions. Physicochemical characterizations of the hydrogels were done by GPC, UV spectrophotometry, FT-IR, 1H- and 13C-NMR spectra. These hydrogels may have potential applications in microbiology, biomedical and pharmaceuticals fields.
Introduction
Hydrogels are three-dimensional water-insoluble cross-linked networks, which are able to absorb and retain water or aqueous solutions (e.g. body fluids) many times over their weight without getting dissolved.1–5 They are in increasing demand in the biomedical and pharmaceutical applications because of their biocompatibility.6–8 Natural polymers have been receiving a great deal of attention because of their biodegradability and availability at low cost. In the past, most of these applications have made use of synthetic water soluble polymers (WSP) such as polyacrylic acids, polyacrylamides, polyethylene oxide, polyvinyl alcohols, and polyvinyl pyrrolidones. Subsequently, these hydrogels were modified by blending some natural polymers with WSP.9 Several published studies investigated modification of natural polymers such as starch, chitosan cellulose, gelatin, agarose, carrageenan, alginate and dextrin employing blending, cross-linking techniques with synthetic polymers to form well-characterized hydrogels.10–18 The resultant hydrogels exhibit different properties than those of the original polymers.19,20
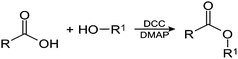
Agarose, the red seaweed polysaccharide is widely used in biomedical and bioengineering applications. The basic disaccharide repeating units of agarose consists of (1,3) linked β-D-galactose (G) and (1,4) linked α-L-3,6-anhydrogalactose (A) (Fig. 1a).21 The aromatic amino acidL-phenylalanine is widely used in human nutrition, pharmaceuticals and food industries (Fig. 1b). Microwave assisted esterification is one of the facile ways to modify polysaccharides. This technique has been used in synthesis to prepare polysaccharide based materials.22 Synthesis of polysaccharide ester conjugates using N,N′-dicyclohexylcarbodiimide (DCC) and 4-dimethylaminopyridine (DMAP) as activating agents for carboxylic acid has been studied widely.23,24
 |
| | Fig. 1 Chemical structures of (a) repeating unit of agarose, (b) L-phenylalanine and (c) genipin. | |
Amino acids containing synthetic polymers have assumed substantial prominence as a new class of bioactive polymeric materials.25–27 However, no significant research endeavors concerning the esterification of the natural polymer agarose with amino acid have been reported so far. The amino acid reacts with the hydroxyl groups of agarose polymer leading to the formation of an amino containing ester derivative, which would be biocompatible, and the free amino group would be available for further modification reactions.
Genipin (Fig. 1c) is a naturally occurring compound extracted from the gardenia fruit, which has been widely used in herbal medicine, and recently it has emerged as a cross-linking agent for different polymeric materials containing free amino groups.28–31 We have also employed genipin as a cross-linker for modifying commercially important hydrocolloids.5,15–17
As part of a continuing program of modification of polysaccharides with nitrogenous substrates in our laboratory,32,33 we report herein, for the first time, a microwave mediated facile synthesis and characterization of agarose ester of L-phenylalanine and its genipin cross-linked product.
Experimental
Materials and instruments
Agarose was extracted from red seaweed Gracilaria dura of Indian waters following the method reported.34Fmoc-L-phenylalanine (Fmoc-PA) was purchased from M/s. Spectrochem Chemicals Ltd., Mumbai, India. The naturally occurring cross-linker genipin was purchased from M/s. Challenge Bioproducts Co. Ltd., Taiwan. A Milestone Start-S (Italy) programmable microwave reactor (Model Start-S; Terminal T260; Line Voltage 230 V; Magnetron S.N. 131528; Frequency 2450 Hz) was used for the reaction. The characterizations were done by FT-IR analysis using a Perkin-Elmer FT-IR machine (Spectrum GX, USA) on a KBr disc (2 mg sample in 600 mg KBr). 13C-NMR spectra were recorded on a Bruker Avance-II 500 (Ultra shield) Spectrometer, Switzerland, at 125 MHz. Samples (agarose, Ag-PAEst) were dissolved in D2O (60 mg mL−1) and spectra were recorded at 70 °C, using DMSO as internal standard (ca. δ 39.5 ppm).35a The 13C NMR spectra of L-phenylalanine were recorded at ambient temperature. The 1H NMR spectra of agarose, genipin and derivatives (Ag-PAEst, G-Ag-PAEst) were recorded (10 mg mL−1 of D2O) at ambient temperature. The UV-Vis absorption spectra of the modified and non-modified agarose were recorded on a Varian CARY 500 UV-VIS-NIR Spectrophotometer. The molecular weights (Mn, Mw) and polydispersity index (PDI) were measured by gel permeation chromatography (GPC) on a Waters Alliance 2695 machine with a RI 2414 detector. Columns (Ultra hydrogel 120, Ultra hydrogel 500) were eluted with 0.1 M NaNO3 at a flow rate of 0.5 mL min−1. Oven and flow cell temperatures were maintained at 45 °C for all measurements. Dextrans of different molecular weights, e.g. 4.4 × 103, 4.3 × 104, 1.96 × 105, and 4.01 × 105 Da, were used as standards for calibration. Gel strengths of agarose, Ag-PAEst (DSEst 0.62) and G-Ag-PAEst were measured in 0.5% (w/v) gel at 20 °C on a Nikkansui-type gel tester (Kiya Seisakusho Ltd., Japan). The gelling and melting temperatures of gel samples were recorded as reported earlier.36 For measurement of the gelling temperature, 10 mL (1.5% w/v, 90 °C) hot sol of the sample was taken in a test tube and it was allowed to cool gradually under ambient conditions while immersing a thermometer in the sol, holding the test tube at an inclined position to have a greater surface area on the top. During the cooling process the thermometer was pulled out of the sol slowly and reintroduced intermittently, noting the temperature, until an impression of the thermometer carving out a channel on the sol/gel surface was left while taking the thermometer out that temperature was considered as the gelling point. For the melting temperature, the gel in an upright test tube was heated on a boiling water bath having placed a steel ball (weight ca. 1.0 g) on the surface of the gel. The temperature at which the steel ball suddenly penetrated the gel body to reach the bottom of the test tube was recorded, which was the melting temperature of the gel. Apparent viscosity was measured (in 0.5% w/v sample) on a Brookfield viscometer (DV-II +Pro), using SC4-18 spindle at 60 rpm and 80 °C. The specific rotations were measured in 0.25% (w/v) solution at 45 °C on a Jasco J-815 CD spectrometer.
Synthesis of Ag-PAEst
In a typical batch, the dried agarose (306 mg, 0.2 mol) was dissolved in N,N-dimethylformamide (DMF) at 80 °C for 3 min in a microwave reactor under stirring. To the solution, a pre-solubilized mixture of Fmoc-L-phenylalanine (387 mg, 0.2 mol), DCC (412 mg, 0.4 mol), and DMAP (30.5 mg, 0.05 mol) in DMF was added and the reaction was carried out under microwave irradiation at 80 °C for 10 min applying 400 W power. After the completion of reaction, the product was isolated by precipitation using isopropyl alcohol (reaction mixture–IPA, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 v/v). The precipitated product was washed with IPA (10 mL × 4) under stirring (20 min for each wash) to remove excess of the reagents followed by drying under vacuum. The dried product was treated with 10% piperidine solution in DMF (10 mL) for 2 h under stirring at ambient conditions for removing the protecting group Fmoc. Finally, the product was isolated by precipitation with IPA followed by washing and drying as described above.
:2 v/v). The precipitated product was washed with IPA (10 mL × 4) under stirring (20 min for each wash) to remove excess of the reagents followed by drying under vacuum. The dried product was treated with 10% piperidine solution in DMF (10 mL) for 2 h under stirring at ambient conditions for removing the protecting group Fmoc. Finally, the product was isolated by precipitation with IPA followed by washing and drying as described above.
Cross-linking Ag-PAEst (DSEst 0.62)
The cross-linking reaction of Ag-PAEst (DSEst 0.62) with genipin was carried out following the method reported earlier.15–17 The Ag-PAEst (DSEst 0.62, 100 mg) was dissolved in 10 mL distilled water at 120 °C for 20 min using an autoclave followed by the addition of genipin 1% (w/v) solution in methanolic water. The reaction mixture was allowed to stand under ambient conditions, which formed a gel on cooling, for 48 h. The reaction mixture started assuming light blue color after 120 min and the color intensified with the passage of time becoming deep blue in color after 48 h. The gelled reaction mixture was worked-up to obtain the genipin cross-linked Ag-PAEst hydrogel by dewatering the gel with IPA (1:2 v/v) for 24 h, followed by air-drying at 50 °C for 2 h.
The gelation degree (G) of Ag-PAEst and G-Ag-PAEst hydrogels was calculated adopting the method reported in the literature,5 using eqn (1).where md is the dry weight of the gel; miso is the weight of the isolated gel.
A known weight of the dried Ag-PAEst or G-Ag-PAEst hydrogel was taken, which was swelled in different pH media (e.g., pH 1.2, 7.0 and 12.5) for 10 h in separate experiments. The weight of the isolated gel samples was determined (miso). The isolated gel samples were dewatered with a hundred-fold excess of IPA overnight, washed with IPA carefully and dried at room temperature under reduced pressure, and the sample was weighed again (md).
Equilibrium swelling (ES)
The equilibrium swelling of agarose, Ag-PAEst and G-Ag-PAEst dried hydrogels in aqueous media having different pHs e.g. 1.2, 7.0 and 12.5 was measured in this study. In the swelling measurements, the dry hydrogel was weighed (W0) and immersed in aqueous media having different pHs separately. After the desired soaking time had elapsed, the wet samples were wiped with filter paper to remove excess liquid, and weighed (Wt). In this study, the Wt had become constant after 1 h of soaking in all pH media. The hydrogel of the products Ag-PAEst and G-Ag-PAEst remained stable for more than a week in all pH media, while agarose hydrogel started losing weight in acidic medium after 24 h. Equilibrium swelling (ES) was calculated using eqn (2).5
Characterization
Characteristic IR bands of Ag-PAEst.
ν
max (cm−1) 3430br (–OH, stretching vibration), 2919w (–CH2, stretching vibration), 1730s (–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, stretching vibration of ester), 1645w (H–O–H, stretching vibration of bound water), 1420w (C–C, bending vibration), 1377w (methylene group), 1255w (covalent sulfate group), 1160, 1072br (C–O–C, stretching vibration of glycosidic linkage), 931m (3,6-anhydro galactose), 891s (1,4-substituted ring), 772w (–CH2, rocking vibration), 688w,br (benzene ring) [band intensities: br = broad; m = medium; s = sharp; w = weak] (Fig. 2a).
O, stretching vibration of ester), 1645w (H–O–H, stretching vibration of bound water), 1420w (C–C, bending vibration), 1377w (methylene group), 1255w (covalent sulfate group), 1160, 1072br (C–O–C, stretching vibration of glycosidic linkage), 931m (3,6-anhydro galactose), 891s (1,4-substituted ring), 772w (–CH2, rocking vibration), 688w,br (benzene ring) [band intensities: br = broad; m = medium; s = sharp; w = weak] (Fig. 2a).
 |
| | Fig. 2
FT-IR spectra of (a) Ag-PAEst, (b) agarose and (c) L-phenylalanine. | |
Characteristic IR bands of agarose.
ν
max (cm−1) 3434br (–OH, stretching vibration), 2924w (–CH2, stretching vibration), 1630w (H–O–H, stretching vibration of bound water), 1461w (C–C, bending vibration), 1378w (methylene group), 1244w (covalent sulfate group), 1154, 1077br (C–O–C, stretching vibration of glycosidic linkage), 930m (3,6-anhydro galactose) [band intensities: br = broad; m = medium; w = weak]37 (Fig. 2b).
Characteristic IR bands of L-phenylalanine.
ν
max (cm−1) 3649w (–NH, stretching vibration), 3034br (–CH, stretching vibration), 2118w (CC, stretching vibration), 1624w (–NH3+ deformation), 1560br (CO, anti-symmetrical stretching vibration), 1495w (–COO−, stretching vibration), 1408 (–CO, symmetric stretching vibration), 1307m (–OH bending vibration), 1224w (–CH2, wagging vibration), 1157m (–C–C, stretching vibration), 1073m (aryl group), 1026m (–C–N, stretching vibration), 848s (substituted ring 1,4 distribution), 745s (–CH2, rocking vibration), 696br (benzene ring) [band intensities: br = broad; m = medium; s = sharp; w = weak]38 (Fig. 2c).
13C NMR of agarose (125 MHz, D2O, DMSO-d6 as internal standard).
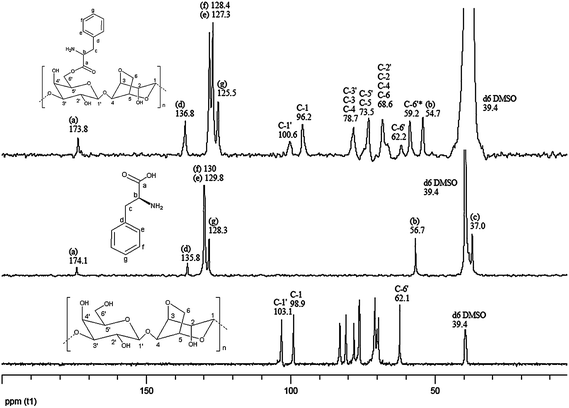
δ 103.1 [C-1′], 98.9 [C-1], 82.9 [C-3′], 80.8 [C-3], 78.0 [C-4], 76.3 [C-5], 76.08 [C-5′], 71.0 [C-2′], 70.6 [C-2], 70.0 [C-6], 69.5 [C-4′], 62.1 [C-6′] (Fig. 3). These values were in good agreement with those reported in our previous work.35a
 |
| | Fig. 3
13C-NMR spectra of agarose, L-phenylalanine, and Ag-PAEst. | |
13C NMR of L-phenylalanine (125 MHz, D2O, DMSO-d6 as internal standard).
δ 174.1 [C-a], 135.8 [C-d], 130 [C-f], 129.8 [C-e], 128.3 [C-g], 56.7 [C-b], 37.0 [C-c] (Fig. 3).
13C NMR of Ag-PAEst (125 MHz, D2O, DMSO-d6 as internal standard).
δ 173.8 [C-a], 136.8 [C-d], 128.4 [C-f], 127.3 [C-e], 125.5 [C-g], 100.6 [C-1′], 96.2 [C-1], 78.7 [C-3′, C-3, C-4], 73.5 [C-5, C-5′], 68.6 [C-2′, C-4′, C-2, C-6], 62.2 [C-6′ linked with L-phenylalanine], 59.2 [C-6′ linked with –OH], 54.7 [C-b] (Fig. 3).
Total nitrogen and degree of substitution
Total nitrogen was estimated by the Kjeldahl method on a KEL PLUS-KES 201 Digestion unit attached to a KEL PLUS-CLASSIC DX Distillation unit (M/s PELICAN equipments, Chennai, India). The degree of substitution of L-phenylalanine on the agarose backbone was calculated on the basis of nitrogen contents of the products, hence, it may be noted that isolated agarose did not contain nitrogen.
Results and discussion
Physicochemical properties
Optimization studies revealed that microwave irradiation for 10 min at 80 °C led to the formation of ester bonds between carboxy termini of Fmoc-L-phenylalanine and hydroxyl groups of agarose in good yield. Ester derivatives of L-phenylalanine with agarose having different molar ratios, 1:0.5, 1:1, and 1:2, were obtained in 90%, 86%, and 85% yields. The respective degrees of substitution (DSEst) and total nitrogen contents were 0.30, 0.62, 1.52 and 0.91 ± 0.1%, 1.87 ± 0.1%, 4.54 ± 0.1% (Table 1). At DSEst 1.52, the agarose ester (Ag-PAEst) became insoluble in water but was soluble in DMSO and DMF at 80 °C with difficulty, apparently due to the greater degree of hydrophobization of the agarose backbone, while the products having DSEst, 0.30 and 0.62, were found to be soluble in water at 80–90 °C (Table 1). The water soluble product with greater DSEst (0.62) was allowed to react with genipin to produce the cross-linked hydrogel. The physicochemical properties of Ag-PAEst (DSEst 0.62) and those of the cross-linked product (G-Ag-PAEst) were studied. The gel strength, gelling and melting temperature and apparent viscosities of Ag-PAEst (DSEst 0.62) and the cross-linked product (G-Ag-PAEst) were 960 ± 20 g cm−2; 34° ± 1, 98° ± 1; 8.6 ± 1 cP and 940 ± 20; 34° ± 0.5, 98° ± 0.5; and 8.5 ± 1cP, respectively. No significant variations were observed in these parameters of these two derivatives when compared with the parent polysaccharide agarose (Table 2). Both the products Ag-PAEst (DSEst 0.62) and G-Ag-PAEst exhibited a strong gelling behavior like agarose, which may be due to the limited presence of -O-phenylalanine group on the agarose backbone facilitating formation of hydrogel networks through hydrogen bonding as well as increased occurrence of “junction zones” in the network.35a The weight average molecular mass (Mw), number average molecular mass (Mn) and polydispersity index (PDI) of agarose and Ag-PAEst (DSEst 0.62) are given in Table 1. The Mw and PDI data of Ag-PAEst indicated that the agarose biopolymer backbone remained largely intact during the esterification by microwave irradiation. The specific rotation [α]45 °C589 nm values of Ag-PAEst (DSEst 0.62) and G-Ag-PAEst were −10° and −54°, respectively, while the value for agarose was −22°. In pH media (pH 7.0 and 12.4), the gelation degrees (G) of the products [Ag-PAEst (DSEst 0.62) and G-Ag-PAEst] were greater than those of agarose, while in pH media (pH 1.2) gelation degrees (G) decreased (Table 2). Equilibrium swelling (ES) values at pH 1.2 had the sequence Ag-PAEst > G-Ag-PAEst > agarose while at pH 7.0 and 12.5 these had a sequence G-Ag-PAEst < Ag-PAEst < agarose. Agarose swelling is the greatest at pH 7.0 and 12.5, while it is the lowest at pH 1.2. This appears to be a direct consequence of acid lability of agarose polymer. The swelling of Ag-PAEst is relatively higher at pH 1.2, presumably because of preferential protonation of the free –NH2 moiety facilitating intensive solvation with water molecules, leading to a stable ionic hydrogel. The genipin cross-linked product (G-Ag-PAEst) on the other hand, swelled comparatively less in all pH media since the –NH2group is engaged in a cross-linking reaction resulting in a structure restricting transport of water molecule into the hydrogel network. In other words, reduced solvation in the absence of the ionic character of the cross-linked molecule35b (Fig. S1–S3†).
Table 1 Yield (%) and physico-chemical properties of agarose and agarose ester derivativese
| Samples |
Ag-Fmoc-PA ratio |
Yield (%) |
Degree of esterification (DSEst) |
Nitrogen contents (%) |
Molecular weights/kDa |
Polydispersity index (PDI) |
Solubilitya |
|
M
w
|
M
n
|
|
Soluble in respective solvents at 80–90 °C.
Water.
Dimethyl formamide (DMF).
Dimethylsulfoxide (DMSO).
NA = not applicable; NM = not measured; Ag = agarose; PA = phenylalanine].
|
|
Agarose
|
NA |
NA |
NA |
0% |
117.571 |
44.570 |
2.638 |
Solubleb |
| Ag-PAEst |
1:0.5 |
90 |
0.30 |
0.91 ± 0.1% |
NM
|
NM
|
NM
|
Solubleb |
| Ag-PAEst |
1:1 |
86 |
0.62 |
1.87 ± 0.1% |
117.152 |
48.987 |
2.392 |
Solubleb |
| Ag-PAEst |
1:2 |
85 |
1.52 |
4.54 ± 0.1% |
NM
|
NM
|
NM
|
Solublec,d |
Table 2 Comparative physico-chemical properties of agarose ester derivatives
| Products |
Gel strengtha/g cm−2 |
Apparent viscosity/cPa |
Gelling temp./°C |
Melting temp./°C |
Gelation degree (G) |
Specific rotationb [α]45 °C589 nm |
| pH 1.2 |
pH 7.0 |
pH 12.5 |
|
Gel strength and apparent viscosity were measured in 0.5% (w/v) at 20 °C and 80 °C, respectively.
Specific rotation was measured in 0.25% (w/v) at 45 °C.
|
|
Agarose
|
950 ± 20 |
8.2 ± 1 |
33 ± 0.5 |
98 ± 0.5 |
0.141 |
0.132 |
0.105 |
−22° |
| Ag-PAEst (DSEst 0.62) |
960 ± 20 |
8.6 ± 1 |
34 ± 0.5 |
98 ± 0.5 |
0.101 |
0.160 |
0.156 |
−10° |
| G-Ag-PAEst (DSEst 0.62) |
940 ± 20 |
8.5 ± 1 |
34 ± 0.5 |
98 ± 0.5 |
0.121 |
0.174 |
0.172 |
−54° |
Spectral characterization of PAEst-Ag
The Ag-PAEst was characterized by FT-IR and 13C-NMR spectrometry. Formation of ester bonds as a result of the reaction between hydroxyl groups of agarose and carboxy termini of L-phenylalanine was confirmed by the appearance of the band at 1740 cm−1 in the FT-IR spectrum of Ag-PAEst. The main characteristic absorption bands of agarose (3430, 1645, 1160, 1072 and 931 cm−1) appeared in the FT-IR spectrum of Ag-PAEst indicating that the backbone of agarose remained intact during esterification (Fig. 2). The 13C-NMR spectra of Ag-PAEst exhibited a peak at 173.9 ppm, confirming the presence of ester carbonyl groups (Fig. 3). The genipin cross-linked product (G-Ag-PAEst) had a distinctive blue color, which was further characterized by UV-Vis spectrophotometry (Fig. 4) as well as by 1H NMR spectrometry (Fig. S4†). The appearance of a new proton signal at 2.69 ppm indicated that the genipin moiety was present in G-Ag-PAEst (Fig. S4c and d†). Besides this, there were changes in the proton NMR spectrum profile of Ag-PAEst, especially in the regions 3.09–3.40 ppm and 7.32–7.44 ppm after cross-linking (Fig. S4b and d†).
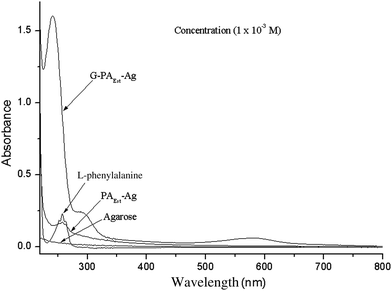
The non-modified agarose did not exhibit any absorption maxima in the UV-Vis region, while L-phenylalanine, genipin33 and Ag-PAEst exhibited absorption at λmax (in distilled water, nm): 258 (ε 288.8 L mol−1 cm−1), λmax 240 nm (ε 5000 L mol−1 cm−1), λmax 258 (ε 171.2 L mol−1 cm−1), respectively. The genipin cross-linked product G-Ag-PAEst showed absorption bands at λmax (in distilled water, nm): 241 nm (ε 1591.4 L mol−1 cm−1), 290 nm (ε 244 L mol−1 cm−1), and 590 nm (59.11 L mol−1 cm−1). The appearance of absorption maxima at λmax 258 nm in Ag-PAEst and at 241, 290 and 590 nm in G-Ag-PAEst indicated insertion of L-phenylalanine and subsequent cross-linking of genipin involving the free amine group of Ag-PAEst, on the agarose backbone (Fig. 4). The new maxima at 590 nm had appeared due to the generation of an extended conjugation system as a result of genipin cross-linking manifesting the blue-color (Scheme 1, step-2). This is in good agreement with those reported in the literature.30,33
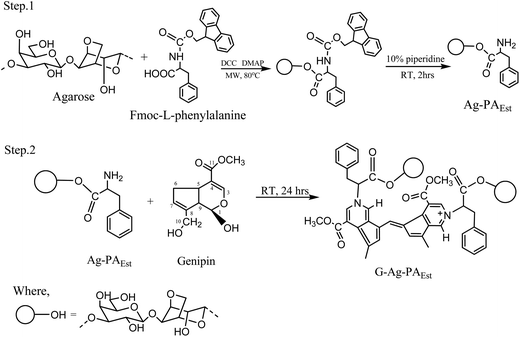 |
| | Scheme 1 Synthesis of hydrogel forming genipin cross-linked agarose ester hydrogel. | |
Formation of Ag-PAEst and G-Ag-PAEst
The carboxyl terminus of the Fmoc-L-phenylalanine was engaged in the ester formation leaving behind a free amino group after the removal of the Fmoc protecting group with 10% piperidine in DMF.23 The amino group later participated in the genipin cross-linking reaction and produced the cross-linked product, the reaction mixture turned dark blue from light blue in 48 h (Scheme 1). The reaction possibly followed a similar mechanism to that described in the literature (Scheme S5†).33
Conclusions
A facile method was developed for the synthesis of an agarose-L-phenylalanine ester hydrogel, which was subsequently cross-linked with the natural cross-linker genipin to afford a blue hydrogel. These hydrogels exhibited similar strong gelling characteristics as those of the agarose hydrogel. Phenylalanine containing hydrogels are amenable to form a hydrophilic interpenetrating network and may be used in biomedical devices e.g. contact lenses, scaffolding and targeted delivery applications (cf.ref. 39).
Acknowledgements
Grateful thanks are accorded to CSIR, New Delhi, for the award of a fellowship to GKM (No. 31/28(87)/2008-EMR-I). One of the authors (SK) gratefully acknowledges the Ministry of Earth Sciences, New Delhi, for a fellowship (MoES/9-DS/6/2007-PC-IV).
References
-
(a)
F. L. Buchholz and A. T. Graham, Modern Superabsorbent Polymer Technology, Wiley, New York, 1997 Search PubMed;
(b)
L. B. Peppas and R. S. Harland, Absorbent Polymer Technology, Elsevier, Amsterdam, 1990 Search PubMed.
- H. Yang, H. Liu, H. Kang and W. Tan, J. Am. Chem. Soc., 2008, 130, 6320–6321 CrossRef CAS.
- X. Zhang, S. Wang, M. Hu and Y. Xiao, Biosens. Bioelectron., 2006, 21, 2180–2183 CAS.
-
(a) S. G. Lévesque and M. S. Shoichet, Biomaterials, 2006, 27, 5277–5285 CrossRef CAS;
(b) M. S. Shoichet, Macromolecules, 2010, 43, 581–591 CrossRef CAS.
- R. Meena, K. Prasad and A. K. Siddhanta, Food Hydrocolloids, 2009, 23, 497–509 CrossRef CAS.
-
(a) K. Li and B. Liu, Polym. Chem., 2010, 1, 251–259 Search PubMed;
(b) H. Tan and K. G. Marra, Materials, 2010, 3, 1746–1767 Search PubMed.
- K. Pal, A. K. Banthia and D. K. Majumdar, Des. Monomers Polym., 2009, 12, 197–220 Search PubMed.
-
Hydrogels in Medicine and Pharmacy, ed. N. A. Peppas, CRC, Boca Raton, FL, 1987, vol. I–III Search PubMed.
-
L. Abad, C. San Diedo, L. Relleve, C. Aranilla, A. M. Dela Rosa, I. Janik and J. Rosiak, Proceedings of the 15th Philippine Chemistry Congress, Cebu City, Philippines, 1999, p. 466 Search PubMed.
-
J. P. Kennedy, G. Fenyvesi, B. Keszler and K. S. Rosenthal, Polymer Gels Fundamentals and Applications, ACS Symposium Series 833: ACS, Washington, DC, 2003; pp. 290–299 Search PubMed.
- Y. M. Lee, S. H. Kim and S. J. Kim, Polymer, 1996, 37, 5897–5905 CrossRef CAS.
- D. Hwang and S. Damodaran, J. Appl. Polym. Sci., 1996, 62, 1285–1293 CrossRef CAS.
- A. Paperella and K. Park, ACS Symp. Ser., 1994, 620, 180.
- B. Ramaraj and G. Radhakrishnan, J. Appl. Polym. Sci., 1994, 52, 837–846 CrossRef CAS.
- R. Meena, K. Prasad and A. K. Siddhanta, J. Appl. Polym. Sci., 2007, 104, 290–296 CrossRef CAS.
- R. Meena, K. Prasad and A. K. Siddhanta, Int. J. Biol. Macromol., 2007, 41, 94–101 CrossRef CAS.
-
A. K. Siddhanta, R. Meena, G. Prasad, M. U. Chhatbar, G. K. Mehta, M. D. Oza, S. Kumar and K. Prasad, Development of Carbohydrate Polymer Based New Hydrogel Materials Derived from Seaweed Polysaccharides, in Handbook of Carbohydrate Polymers: Development, Properties and Applications, ed. I. Ryouichi and M. Youta, NOVA Science Publishers Inc., New York, 2010, pp 555–582 Search PubMed.
- R. Meena, M. Chhatbar, K. Prasad and A. K. Siddhanta, Polym. Int., 2008, 57, 329–336 CrossRef CAS.
- C. T. Aranilla, F. Yoshii, A. M. Dela Rosa and K. Makuuchi, Radiat. Phys. Chem., 1999, 55, 127–131 CrossRef.
- L. Relleve, F. Yoshii, A. M. Dela Rosa and T. Kume, Radiat. Phys. Chem., 1999, 273, 63–68 CAS.
- C. Araki, Proc.–Int. Seaweed Symp., 1966, 5, 3–19 Search PubMed.
-
(a) A. Biswas, R. L. Shogren, S. Kim and J. L. Willett, Carbohydr. Polym., 2006, 64, 484–487 CrossRef CAS;
(b) A. Biswas, B. K. Sharma, J. L. Willet, K. Vermillion, S. Z. Erhan and H. N. Cheng, Green Chem., 2007, 9, 85–89 RSC;
(c) A. Biswas, R. L. Shogren, G. Selling, J. Salch, J. L. Willett and C. M. Buchanan, Carbohydr. Polym., 2008, 74, 137–141 CrossRef CAS.
-
(a) D. M. Kalaskar, R. V. Ulijn, J. E. Gough, M. R. Alexander, D. J. Scurr, W. W. Sampson and S. J. Eichhorn, Chemistry and Materials Science, 2010, 17, 747–756 Search PubMed;
(b) J. Kapus'niak, W. Ciesielski, J. J. Kozioł and P. Tomasik, Eur. Food Res. Technol., 1999, 209, 325–329 CrossRef CAS.
-
T. Heinze, T. Liebert and A. Koschella, Esterification of Polysaccharides, Springer Berlin Heidelberg, New York, [ISBN: 3-540-32103-9], 2006 Search PubMed.
- D. M. Vriezema, A. Kros, R. de Gelder, J. Cornelis-sen, A. E. Rowan and R. J. M. Nolte, Macromolecules, 2004, 37, 4736–4739 CrossRef CAS.
- T. W. Baughman and K. B. Wangener, Adv. Polym. Sci., 2005, 176, 1–42 CAS.
- K. Okoshi, K. Sakajiri, J. Kumaki and E. Yashima, Macromolecules, 2005, 38, 4061–4064 CrossRef CAS.
- C. Djerassi, J. D. Gray and F. A. Kincl, J. Org. Chem., 1960, 25, 2174–2177 CrossRef CAS.
- T. Akao, K. Kobashi and M. Aburada, Biol. Pharm. Bull., 1994, 17, 1573–1576 CAS.
- R. Touyama, Y. Takeda, K. Inoue, I. Kawamura, M. Yatsuzuka, T. Ikumoto, T. Shingu, T. Yokoi and H. Inouye, Chem. Pharm. Bull., 1994, 42, 668–673 CAS.
- J. E. Park, J. Y. Lee, H. G. Kim, T. R. Hahn and Y. S. Paik, J. Agric. Food Chem., 2002, 50, 6511–6514 CrossRef CAS.
- M. D. Oza, R. Meena, K. Prasad, P. Paul and A. K. Siddhanta, Carbohydr. Polym., 2010, 81, 878–884 CrossRef CAS.
- M. U. Chhatbar, R. Meena, K. Prasad, D. R. Chejara and A. K. Siddhanta, Carbohydr. Res., 2011, 346(5), 527–533 CrossRef CAS.
- R. Meena, A. K. Siddhanta, K. Prasad, B. K. Ramavat, K. Eswaran, S. Thiruppathi, M. Ganesan, V. A. Mantri and P. V. Subbarao, Carbohydr. Polym., 2007, 69, 179–188 CrossRef CAS.
-
(a) D. A. Rees, Adv. Carbohydr. Chem. Biochem., 1969, 24, 267–332 CAS;
(b) S. K. De, N. R. Aluru, B. Johnson, W. C. Crone, D. J. Beebe and J. Moore, J. Microelectromech. Syst., 2002, 11(5), 544–555 CrossRef CAS.
- G. K. Mehta, R. Meena, K. Prasad, M. Ganesan and A. K. Siddhanta, J. Appl. Phycol., 2010, 22, 623–627 Search PubMed.
- D. Christiaen and M. Bodard, Bot. Mar., 1983, 26, 425–427 Search PubMed.
- R. Mahalakshmi, S. X. Jesuraja and D. S. Jerome, Cryst. Res. Technol., 2006, 8, 780–783 CrossRef.
-
(a) M. Casolaro, S. Bottari and Y. Ito, Biomacromolecules, 2006, 7(5), 1439–1448 CrossRef CAS;
(b) L. Jiang, J. Chen and L. Chen, Int. J. Biol., 2010, 2(1), 184–189 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1py00250c |
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here.