Unusual activation by solvent of the ethylene free radical polymerization†
Etienne
Grau
,
Jean-Pierre
Broyer
,
Christophe
Boisson
,
Roger
Spitz
and
Vincent
Monteil
*
Université de Lyon, Univ. Lyon 1, CPE Lyon, CNRS, UMR 5265 Laboratoire de Chimie Catalyse Polymères et Procédés (C2P2), LCPP team Bat 308F, 43 Bd du 11 novembre 1918, F-69616, Villeurbanne, France. E-mail: monteil@lcpp.cpe.fr; Fax: +33 4 72 43 17 68
First published on 28th July 2011
Abstract
Ethylene polymerization is performed industrially either by radical polymerization under severe conditions (1000–4000 bar, 200–300 °C) or by a catalytic mechanism at lower temperatures (usually less than 100 °C) and pressures (below 50 bar). Standard radical polymerization conditions are too severe to permit a fine control of the macromolecular architecture. Under milder conditions (100 bar, 70 °C), radical ethylene polymerization is assumed ineffective which has been confirmed in bulk (supercritical ethylene). However, we have shown that the efficiency of this polymerization is strongly dependent on the solvent. This unusual activation by solvent has been rationalized using theoretical considerations. A second effect investigated is the influence of solvent on PE molecular weight. Indeed PE with either low molecular weight and high chain-end functionality or higher molecular weight can be synthesized according to the solvent used.
Introduction
Polyethylene is one of the most important polymers in everyday life. Although it has been seven decades since polyethylene's first commercialization, polyolefins remain highly technology driven. The three major classes of polyethylene are described by acronyms HDPE, LDPE, and LLDPE. High-density polyethylene (HDPE) is a linear, semi-crystalline ethylene homopolymer prepared by the Phillips or Ziegler–Natta polymerization process.1,2 Linear low-density polyethylene (LLDPE) is a random copolymer of ethylene and α-olefins produced commercially using Phillips, Ziegler–Natta, and metallocene catalysts. These catalytic processes were developed generally under mild experimental conditions (T < 100 °C and P < 100 bar). Low-density polyethylene (LDPE) is a branched homopolymer prepared under high-temperature and high-pressure via a free radical polymerization process.3,4Free radical polymerization of ethylene is industrially conducted under high pressures (1000–4000 bar) and high temperatures (200–300 °C) in bulk. LDPE melts between 100 and 120 °C and exhibits a crystallinity in the range of 30–60%. We have investigated the efficiency of this reaction under milder slurry conditions: pressure up to 250 bar, T = 70 °C.5,6 These conditions are less efficient than the industrial process but easier to handle. The PE produced possesses higher crystallinities (∼70%) than the traditional industrial LDPE but molecular weights remain too low for applications (Mn < 5000 g mol−1). Moreover the influence of the solvent appears to be crucial for the free radical ethylene polymerization. For instance we managed to synthesize polyethylene down to 10 bar in a polar solvent (THF) by a free radical process with activity 6 times higher than in a nonpolar solvent (toluene).5 This unusual activation by solvent will be further studied in the present work. Indeed free radical polymerization of ethylene will be investigated in a wide range of solvents in order to improve the medium pressure process.
The influence of solvent on free radical polymerization of vinyl compounds was previously reported by Kamachi.7 For almost all monomers it is a tiny effect except for vinyl acetate8 and ethylene.9–12 For these two monomers the kinetics of radical polymerization could vary by a factor up to 10 depending on the solvent. The early studies for ethylene remain partial due to the experimental facilities available at this time (before 1980s). Machi13 suggested that the solubility of the growing polyethylene chain could induce the activation effect by solvent through a Trommsdorff–Norrish effect but his interpretation is still controversial.14 Myshkin8 assumed it was a fully different mechanism based on the dielectric constant ε of the solvent. For other monomers several explanations for this influence have been proposed but none of them is consistent with all sets of data. The diverse influences of solvent on the free radical polymerization are due to variations of kinetic constants according to solvent parameters. The termination rate15–17 was partially related to the viscosity of the solvent due to diffusion mechanisms. For the propagation rate,7,18,19 different origins have been proposed as polarity, interactions between polymer and solvent, interactions between monomer and solvent, and complexation between the propagating macroradical and the solvent. Other authors20–22 suggested that local monomer concentration could play a major role. In the present paper, we performed the radical polymerization of ethylene in a wide range of solvents and studied their influences on yield of polymerization and molecular weight of produced polyethylene. Thanks to theoretical studies, we suggest a new relation which links the yield of polymerization to a combination of key solvent parameters.
Experimental section
All chemicals were purified using standard Schlenk procedures and handled under argon atmosphere. Solvents were distilled from drying agents and degassed under argon. Ethylene (purity 99.95%) was purchased from Air Liquide and AIBN from Acros and used without any further purification.Polymer characterizations
Molecular weights of polyethylenes were determined by size exclusion chromatography (SEC) using a Waters Alliance GPCV 2000 instrument (columns: PLgel Olexis); two detectors (viscosimeter and refractometer) in trichlorobenzene (flow rate: 1 mL min−1) at 150 °C. The system was calibrated with polystyrene standards using universal calibration. Differential scanning calorimetry (DSC) was performed on a Mettler Toledo DSC1 at a heating rate of 5 K min−1. Two successive heating and cooling steps of the samples were performed. We have considered data (Tm values, crystallinity) obtained during the second heats.Standard polymerization procedure of ethylene
Caution : all polymerizations involve high pressure and explosive gas.Ethylene polymerizations were done in a 160 mL stainless steel autoclave (equipped with safety valves, stirrer, oven) from Parr Instrument Co. The azobisisobutyronitrile (AIBN) was dissolved in 50 mL of desired solvent in a Schlenk tube under argon. The mixture was introduced through cannula into the reactor. Ethylene was introduced and the mixture was heated at the desired temperature under stirring (300 rpm). To manage safely polymerization over 50 bar of ethylene we use a 1.5 L intermediate tank. The tank was cooled down to −20 °C to liquefy ethylene at 35 bar. When thermodynamic equilibrium was reached, the intermediate tank was isolated and heated to reach up to 300 bar of ethylene pressure. This tank was used to charge the reactor and maintain the pressure of ethylene constant in the reactor by successive manual ethylene addition. After 4 hours of polymerization the reactor was slowly cooled down and degassed. The precipitated polymer was then dried under vacuum at 70 °C.
Results and discussion
Ethylene free radical polymerization in various organic solvents
Free radical polymerizations of ethylene under identical conditions (100 bar, 70 °C, 50 mL of solvent) were performed in a wide range of solvents. Polymerization results are presented in Table 1. As it can be seen from Table 1, polymerization yield is highly dependent on the solvent of polymerization (from 0.1 g to 4 g). If we consider that solubility of ethylene is almost the same for all solvents (470 g L−1 under 100 bar at 70 °C)23 then conversions in the presence of solvent are between 3% and 17%.| Runa | Solvent | Dielectric constant (ε) (at 20 °C)b | Dipole momentum (μ) (10−30 C m)b | Yield/g | Melting pointc/°C [crystallinity (%)] | M n d/g mol−1 | PDId |
|---|---|---|---|---|---|---|---|
| a The reactions were carried out using 50 mg of AIBN in 50 mL of purified solvent at 70 °C under 100 bar of ethylene pressure during 4 hours. b Obtained from ref. 24. c Determined by DSC. d Determined by HTSEC, nd = not determined. | |||||||
| 1 | Supercritical ethylene | — | 0 | 0.1 | 105.3 [46] | 3010 | 1.3 |
| 2 | Cyclohexane | 2.0 | 0 | 0.6 | 115.5 [58] | 4800 | 2.2 |
| 3 | Heptane | 1.9 | 0 | 0.65 | 116.7 [55] | 4700 | 2.1 |
| 4 | Toluene | 2.4 | 1 | 0.7 | 115.9 [63] | 2340 | 1.9 |
| 5 | Dimethylsulfoxide | 46.4 | 13.5 | 1 | 112.7 [43] | 1910 | 3.5 |
| 6 | Acetonitrile | 35.9 | 11.8 | 1.1 | 115.5 [59] | 1370 | 2.2 |
| 7 | Diethylcarbonate | 2.8 | 3.7 | 1.2 | 117.8 [62] | 7150 | 2.5 |
| 8 | N,N-Dimethylformamide | 36.7 | 10.8 | 1.3 | 108.5 [47] | 530 | 2.9 |
| 9 | Dibutylether | 3.1 | 3.9 | 1.3 | 109.0 [52] | 1370 | 1.4 |
| 10 | Ethanol | 24.5 | 5.8 | 1.4 | 117.6 [63] | 2130 | 2.4 |
| 11 | Acetone | 20.6 | 9 | 1.5 | 115.2 [62] | 1710 | 2.0 |
| 12 | Dimethylcarbonate | 3.2 | 3.7 | 1.6 | 117.9 [57] | 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 720 720 |
2.5 |
| 13 | Butanone | 18.5 | 9.2 | 1.8 | 61 [nd] | 370 | 1.2 |
| 14 | Butyrolactone | 39.0 | 14.2 | 1.8 | nd [nd] | 570 | 1.4 |
| 15 | Butan-2-ol | 16.6 | 5.5 | 1.9 | 116.4 [68] | 2070 | 2.8 |
| 16 | Cyclohexanone | 16.0 | 10.2 | 2.1 | nd [nd] | 1760 | 1.5 |
| 17 | Butan-1-ol | 17.5 | 5.8 | 2.2 | 117.8 [58] | 4130 | 2.4 |
| 18 | Ethyl acetate | 6.0 | 6.1 | 2.3 | 115.2 [54] | 3760 | 3.3 |
| 19 | Dichloromethane | 8.9 | 5.2 | 2.7 | 105.1 [46] | 1050 | 1.6 |
| 20 | 1,4-Dioxane | 2.2 | 1.5 | 3.2 | 118.9 [65] | 1300 | 2.2 |
| 21 | Tetrahydrofuran | 7.6 | 5.8 | 3.9 | 115.2 [58] | 1190 | 1.9 |
In pure ethylene, without any solvent, the polymerization of ethylene is almost inefficient (run 1, 0.1 g corresponding to a conversion of 0.3%). The resulting polyethylene exhibits a low melting point, 105 °C, close to LDPE thermal properties, and a low molecular weight (3000 g mol−1). In all cases, a higher activity is measured in the presence of solvent. Therefore solvents seem to play a major role in the activation of the radical polymerization of ethylene. However, all solvents did not lead to the same activation (yield ranges from 0.6 g in cyclohexane to 3.9 g in THF) and in first approximation, it appears that nonpolar solvents are less efficient than polar ones.
Effect of solvent on the PE molecular weights
PE molecular weights are strongly related to the solvent (see Table 1) due to transfer reaction to the solvent. The highest molecular weights are reached in dimethylcarbonate (Mn = 11700 g mol−1—run 12), and the lowest in butanone (Mn = 370 g mol−1—run 13). The transfer ability of the solvent can be related to the calculated number of chains per initiator.25Cyclohexane (Mn = 4800 g mol−1—run 2) is the less transferring solvent, while butanone is found to be the highest. Toluene (run 4) is the nonpolar solvent with the highest transfer ability, 2.4 times more than cyclohexane. Finally dimethylcarbonate is the less transferring polar solvent (only 1.1 times more than cyclohexane) and consequently leads to the highest molecular weights.
Solvent with high transfer ability can be used to obtain a high content of PE chain-end functionality. One of the most transferring solvents is THF, 26 times higher than cyclohexane. Transfer to solvent provided THF-ended polyethylenes which were fully identified by 13C NMR (see ESI†, Fig. S1).5 The 13C NMR spectrum exhibits standard chemical shift26 of a branched polyethylene (9 branches/1000C). Additionally two different structures, 1- and 2-polyethylenyl-THF, were identified. The chain-end functionalized PE obtained from transfer to solvent may be used further as macro-monomers. Chain-end labelling from transfer to solvent was also determined by 13C NMR for other solvents such as toluene, dioxane (run 20), dichloromethane (run 19, see ESI†, Fig. S2–S4).
An interesting solvent for further use of chain-end functionalized PE is butyrolactone (run 14). The resulting macromonomer could be used in copolymerization with lactonesviaring-opening polymerization in order to obtain polyester with PE branches. Otherwise, chloro-terminated PE from transfer to dichloromethane could also be used as starting reactant for nucleophilic substitution.
On the other hand, solvents with low transfer ability and high activity will provide non-functional polyethylenes. Usually, high transfer ability is related to high activities, except for ethyl acetate (run 18) which is a particularly poor transferring solvent (but still 4.9 times more transferring than cyclohexane), for butan-1-ol (run 17), and for carbonates (runs 7 and 12). These solvents provide PE with relatively higher molecular weights (over 10000 g mol−1).
Consequently, free radical polymerization of ethylene in solvent can provide either, functional/low-molecular weight polyethylenes or non-functional/higher-molecular weight polyethylenes.
Rationalization of the activation by solvent during ethylene radical polymerization
We have evidenced a strong solvent influence on yield in the free radical polymerization of ethylene. This high solvent effect could not be related directly to solvent parameters such as solvent viscosity, dipole momentum, dielectric constant or solubility parameters. THF activates the polymerization 2.8 times more than ethanol (run 12) despite similar dipole momenta. Toluene is 4.6 times less efficient than 1,4-dioxane while they possess the same dielectric constants. No simple relations between the yield and solvent properties such as dielectric constant (ε) or dipole momentum (μ) or other physical constants seemed to exist.To quantify the solvent effect we used the theory of the activated complex27 (eqn (1)) which links a kinetic constant in a solvent to a kinetic constant without a solvent. In this theory, the solvent effect is due to the preferential interactions between the solvent and the activated complex or the reactants. In the case of the free radical polymerization of ethylene, this stabilization is only due to van der Waals interactions, that is, Keesom (dipole–dipole), Debye (dipole–induced dipole) and London (instantaneous dipole–induced dipole) interactions28 (eqn (2)–(4)). Keesom interactions are assumed to be the principal interactions which stabilize the macroradical (EKeesom > EDebye and ELondon). Consequently, each kinetic constant of the polymerization (kd, kp, kt or ktot) exhibits a relation (eqn (5)) with different solvent properties (ε, μ). Thus, according to the free radical kinetic law (eqn (6)), ktot and therefore yield can be related to (μ/ε)2 (eqn (7)). Since only ktot is accessible in this experiment we cannot determine the relative dependency of kp, kt, kd with (μ/ε)2. However it is expected that kt is only slightly influenced by solvent properties as it is mostly controlled by diffusion processes.16,17
It should be noted that the transfer to solvent usually does not impact the kinetic of the polymerization. Indeed the transfer reaction does not modify the radical concentration (Steady States Approximation). However in specific cases the resulting radical cannot efficiently react with monomer and consequently slows down the polymerization. It appears not to be the case for ethylene polymerization as for example THF and dibutylether lead to similar radical (O–CH–CH2) but different activities (runs 9 and 21).
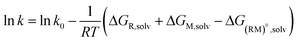 | (1) |
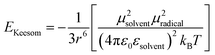 | (2) |
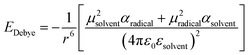 | (3) |
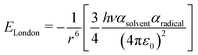 | (4) |
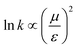 | (5) |
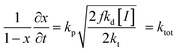 | (6) |
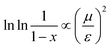 | (7) |
We plotted the conversion versus (μ/ε)2 (Fig. 1), in order to confirm this relation.
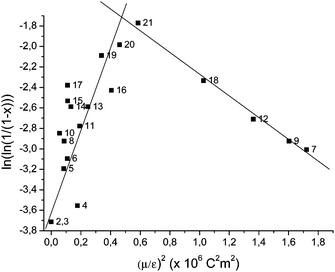 | ||
| Fig. 1 Solvent influence due to Keesom interactions on radical polymerization of ethylene (labels correspond to run numbers in Table 1). ■: 50 mg AIBN, 50 mL of solvent 4 h at 70 °C under 100 bar of ethylene pressure. | ||
The curve obtained is unexpectedly Λ-shaped. A change of behavior is observed for Keesom interactions higher than the ones for THF ((μ/ε)2optimum ≈ 0.58 × 10−60 C2 m2). At lower value of (μ/ε)2, yield increases with this parameter, while it decreases over this value. Most of the solvents showed a good correlation between polymerization yield and (μ/ε)2.
For alcohols (runs 10, 15 and 17) such as ethanol an “over yield” is observed. This can be due to H-bond interactions which have been neglected in the theory. Indeed stabilization by solvent can take place not only by van der Waals interactions but also by H-bond interactions in this case.
Case of solvent mixtures
In order to validate the interpretation of this solvent effect we performed polymerizations using different binary and ternary mixtures of toluene, THF, and diethylcarbonate (DEC) as solvents (Fig. 2). By this way, we artificially change the (μ/ε)2 of the solvent by mixing solvents.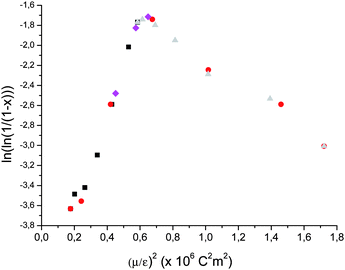 | ||
Fig. 2
Solvent mixture composition influence related to Keesom interactions on radical polymerization of ethylene. 50 mg AIBN 4 h at 70 °C under 100 bar of ethylene pressure in 50 mL of ■: THF–toluene;  : THF–DEC; : THF–DEC;  : toluene–DEC; : toluene–DEC;  : THF–toluene–DEC mixture. : THF–toluene–DEC mixture. | ||
Standard mixing rules29 respectively for relative permittivity εmixture = Σxiεi, with xi the volume fraction of solventi and εi the solventi relative permittivity and for dipole μmixture = ΣΣxixj(μiμj)1/2, with μi the dipole momentum of the solventi are used.
In all cases, whatever the mixture composition, the Λ-shaped curve is observed once again between conversion and values of (μ/ε)2. The maximum of activity (yield 4.1 g) was reached for (μ/ε)2optimum ≈ 0.65 × 10−60 C2 m2. Polymerizations in toluene–DEC mixture follow the same curve than toluene–THF and THF–DEC mixtures. So by tuning properly the proportion of toluene–DEC mixture we are able to provide the same activity than the ethylene polymerization in THF. This evidences that the solvent interaction with the alkyl radical is an exact average of the solvent composition and is not due to the nature of the solvent itself. In other words the solvation shell of the alkyl radical presents the same composition as that of the solvent composition, so there are not preferred interactions with one of the solvents.
Using solvent mixtures, conversion higher than in THF can be reached. Moreover the activation by the solvent appears to be uncoupled from the control of molecular weight by transfer to solvent as evidenced by the molecular weights of synthesized polyethylenes (see ESI†, Table S1). For example, a toluene/DEC 50/50 v/v mixture provides about the same polymerization activity as THF but does not lead to the same molecular weight: respectively 3200 g mol−1 and 1200 g mol−1. Therefore as the solvent activation effect is a global solvent effect only related to μ and ε, and molecular weight mostly controlled by the nature of the solvent (or solvent mixture) used, yield and average molecular weight can be tuned easily by choosing a suitable mixture of solvents.
Arrhenius parameters of free radical polymerization of ethylene in various solvents
To go further we calculated the global activation energy and global pre-exponential factor of the ethylene free radical polymerization. For this purpose, we performed polymerizations at various temperatures (50 °C, 70 °C and 90 °C) and ethylene pressures (at 50, 100, 150, 200 and 250 bar) in all three solvents used previously in the mixture. One can remark that ethylene conversion seemed not to be linked to ethylene pressure (see ESI†, Table S3). From these experiments, we plotted ln ktot (global kinetic constant of the radical polymerization) versus 1/T to determine Arrhenius parameters. Corresponding Etot and ln (Atot) are summarized in Table 2.| Solvent | (μ/ε)2/10−60 C2 m2 | Global activation energy (Etot)b/kJ mol−1 | Global preexponential factor (ln (Atot))b |
|---|---|---|---|
| a Assuming the validity of Arrhenius law. b Determined by a linear regression from 15 experiments at 50, 70 and 90 °C and for each temperature at 50, 100, 150, 200, 250 bars. | |||
| Toluene | 0.18 | 27.7 | 7.6 |
| THF | 0.58 | 32.8 | 10.3 |
| DEC | 1.72 | 40.0 | 12.2 |
Ideally, the determination of the Arrhenius parameters for each polymerization step should be performed, but this kind of study is currently incompatible with our experimental conditions of pressure (stopped flow or pulsed laser polymerization techniques cannot be easily used).
Both global activation energy and pre-exponential factor increase with (μ/ε)2. On one hand, lower global activation energy is usually linked to a more favorable reaction. In all solvents, the polymerization mechanism is considered to be the same, so the change in the global activation energy is only due to the relative stabilization of intermediate and activated states, which differ from one solvent to the other.30 Solvation by toluene provides a lower energy barrier than in THF and DEC. On the other hand, the global pre-exponential factor is proportional to the frequency of efficient collisions. With a higher pre-exponential factor the probability of the mechanism involved is supposed to increase. Differences in geometry of activated states26 in toluene, in THF and in DEC could explain the difference of pre-exponential factors. Toluene is less electron donor than THF, more toluene molecules are necessary to stabilize the radical. Thus the radical should have a denser solvation shell in toluene than in THF. This could explain why preexponential factor is higher in THF than in toluene. The same reason could be applied for DEC. For these three solvents, a linear relationship seems to exist between Etot and (μ/ε)2, in the same way ln (Atot) vs. (ε/μ)2 is linear. These two relations allow to calculate the Arrhenius parameters for every (μ/ε)2 and to predict the optimum of solvent activation. The optimum depends on the temperature (in Kelvin the relation is (μ/ε)2optimum ≈ 0.03 T1/2 × 10−60 C2 m2).31 Therefore at 70 °C the predict optimum is (μ/ε)2optimum ≈ 0.56 × 10−60 C2 m2.
Interpretation of the solvent optimum
The optimum of solvent properties was calculated by three different techniques (μ/ε)2optimum ≈ 0.56–0.65 × 10−60 C2 m2 at 70 °C (different solvents, mixture of solvents and Arrhenius parameters). This optimum solvent property is close to the (μ/ε)2 ≈ 0.62 × 10−60 C2 m2 of an alkyl macroradical (μ ≈ 1.5 × 10−30 C m and ε ≈ 1.9). The punctual dipole momentum of an alkyl macroradical has been determined on the basis of a 1-hexyl radical. It has been determined by Gaussian 03 (ref. 32) calculation (see ESI†, Fig. S5 and Table S3) of partial charge and geometry of the radical, μradical = Σqiri with qi the partial charge of the i atom, and ri a vector from some reference to the atom i. The relative permittivity ε corresponds to the permittivity of the growing chain-end, and therefore it could be approximated to a molecule similar to the saturated chain end (for macroradical of the free radical polymerization of ethylene we have chosen heptane). Consequently optimal solvent is reached when its (μ/ε)2 is the closest to the (μ/ε)2 of the propagating radical.Possible interpretation of the activation of ethylene free radical polymerization by solvent
The solvent activation effect on the free radical polymerization of ethylene is correlated to Keesom interactions between the radical and the solvent. This interaction is not punctual but is due to the average composition of the solvation shell of the macroradical. The decrease of the Keesom interaction lowers the global activation energy (due to a decrease of the stabilization), as the global pre-exponential factor (due to a thickening of the macroradical's solvation shell). The intensity of the solvent effect remains an open question. This optimum (μ/ε)2 is close to the corresponding same parameters of the macroradical (μ/ε)2.Otherwise the optimum corresponds to the radical stemming from monomer, not to the radical fragment issued from AIBN. Therefore the initiation (first addition of the monomer) is not the determining step in the solvent activation effect. Indeed if it was the case the optimum (μ/ε)2 would correspond to the (μ/ε)2 ≈ 0.02 × 10−60 C2 m2 of the radical resulting from AIBN dissociation (μ ≈ 1.6 × 10−30 C m and ε ≈ 25—see ESI†, Fig. S6 and Table S4). Consequently it must be the propagation and/or termination steps which are influenced by the solvent.
It should be noted that for standard monomers (MMA, Sty, BuA), the solvent effect remains tiny.7,10 These monomers possess higher propagation rate and lower termination rate than the monomers (ethylene, vinyl acetate) which exhibit a solvent activation effect. So only monomers, which possess low propagation rate or high termination rate, seem to express a high solvent effect.
Conclusion
In conclusion, we have reported the radical polymerization of ethylene under mild conditions (P = 100 bar, T = 70 °C) in a wide range of solvents. Important activation by solvent has been observed.Moreover, the crucial importance of solvent transfer capacities on the nature of synthesized PE was evidenced. Indeed since the alkyl radical possesses a high reactivity, the transfer constants to solvents are high and the molecular weight of the PE synthesized is controlled by transfer to solvent. This transfer to solvent can be used to functionalize PE chain-end. Carbonates are the less transferring solvents and Mn values up to 15000 g mol−1 are reached. This molecular weight far over the entanglement molecular weight should lead to some attractive mechanical properties. Activation by solvent and molecular weight control by transfer appear to be unlinked.
We observe that the major factor to explain the solvent activation effect is the Keesom interaction of the solvent on the macroradical and therefore demonstrated a good correlation between yield of polymerization and solvent properties ((μ/ε)2). This Λ-shaped relation demonstrates that an optimal solvent exists for ethylene radical polymerization and possesses a (μ/ε)2 close to 0.6 × 10−60 C2 m2. The same optimum has been identified using two other techniques: solvent mixtures and Arrhenius parameters. Moreover, this optimum is correlated to the corresponding (μ/ε)2 of the propagating radical. Finally, we have demonstrated that this solvent activation effect is not a punctual effect of the solvent but a global average effect of the solvation shell of the growing radical and is only dependent on the average μ and ε of the solvent mixture.
Acknowledgements
E.G. thanks the “Ministère de la Recherche et de l'Enseignement Supérieur” for fellowship. The authors thank Mettler-Toledo for the thermal analysis.Notes and references
- R. Mulhaupt, Macromol. Chem. Phys., 2003, 204, 289 CrossRef CAS.
- D. L. Beach and Y. V. Kissin, in Encyclopedia of Polymer Science and Engineering, ed. H. F. Mark, N. M. Bikales, C. G. Overberger and G. Menges, WileyInterscience, New York, 2nd edn, 1985, vol. 6, pp. 454–489 Search PubMed.
- K. W. Doak, in Encyclopedia of Polymer Science and Engineering, ed. H. F. Mark, N. M. Bikales, C. G. Overberger and G. Menges, WileyInterscience, New York, 2nd edn, 1985, vol. 6, pp. 386–428 Search PubMed.
- S. L. Aggarwal and O. J. Sweeting, Chem. Rev., 1957, 57, 665 CrossRef CAS.
- E. Grau, J. P. Broyer, C. Boisson, R. Spitz and V. Monteil, Macromolecules, 2009, 42, 7279 CrossRef CAS.
- E. Grau, J. P. Broyer, C. Boisson, R. Spitz and V. Monteil, Phys. Chem. Chem. Phys., 2010, 12, 11665 RSC.
- M. Kamachi, Adv. Polym. Sci., 1981, 38, 55 CrossRef CAS.
- A. G. Shostenko and V. E. Myshkin, Dokl. Akad. Nauk SSSR, 1979, 246, 1429 CAS.
- V. F. Gromov and P. M. Khomikovskii, Russ. Chem. Rev., 1979, 48, 1943 Search PubMed.
- M. Buback, Prog. Polym. Sci., 2002, 27, 191 CrossRef CAS.
- M. Buback and J. Schweer, Z. Phys. Chem. NF, 1989, 161, 153 CAS.
- M. Buback, Macromol. Symp., 2009, 275–276, 90 CrossRef CAS.
- F. Suganuma, S. Machi, H. Mitsui, M. Hagiwara and T. Kagiya, J. Polym. Sci., Part A-1: Polym. Chem., 1968, 6, 2069 CrossRef CAS.
- S. Munari and S. Russo, J. Polym. Sci., 1979, 4, 773 Search PubMed.
- V. F. Gromov and P. M. Khomikovskii, Russ. Chem. Rev., 1979, 48, 1040 Search PubMed.
- C. Barner-Kowollik and G. T. Russell, Prog. Polym. Sci., 2009, 34, 1211 CrossRef CAS.
- J. Barth and M. Buback, Macromol. React. Eng., 2010, 4, 288 Search PubMed.
- O. F. Olaj and E. Scnoll-Bitai, Monatsh. Chem., 1999, 130, 731 CAS.
- S. Beuermann and N. García, Macromolecules, 2004, 37, 3018 CrossRef CAS.
- G. Henrichi-Olivé and S. Olivé, Z. Phys. Chem. N.F, 1965, 47, 286.
- G. Henrichi-Olivé and S. Olivé, Z. Phys. Chem. N.F, 1966, 48, 35.
- L. A. Smirnova, N. A. Kopylova and V. V. Izvolenskii, Eur. Polym. J., 1996, 32, 1213 CrossRef CAS.
- Identical solubilities were measured for toluene, THF, and DEC, thanks to the method described in our previous work.6 Then we assume that the ethylene solubility is mostly independent of the solvent.
- A. Loupy, in Effets de milieu en synthèse organique: Des effets de solvants aux méthodes d'activation non classiques, ed. A. Loupy, Dunod, 2nd edn, 1996, pp. 1–40 Search PubMed.
- Assuming that AIBN dissociation being similar in all solvents, and that the molecular weight is only controlled by the transfer to solvent.
- G. B. Galland, R. F. de Souza, R. S. Mauler and F. F. Nunes, Macromolecules, 1999, 32, 1620 CrossRef CAS.
- C. Reichardt, in Solvents and Solvent Effects in Organic Chemistry, ed. C. Reichardt, VCH: Weinhei, 2nd edn, 1988, pp. 121–205 Search PubMed.
- C. Reichardt, in Solvents and Solvent Effects in Organic Chemistry, ed. C. Reichardt, VCH: Weinhei, 2nd edn, 1988, pp. 5–41 Search PubMed.
- R. C. Reid, J. M. Prausnitz and B. E. Poling, in The Properties of Gases and Liquids, ed. R. C. Reid, J. M. Prausnitz and B. E. Poling, McGraw-Hill Book Company, Singapore, 1988, pp. 74–84 Search PubMed.
- To determine the exact structure of the activated state, theoretical calculations have to be done.
- For details of the calculation see ESI†.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Rob, J. R. Cheeseman, J. A. Montgomery, Jr, T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian 03, Gaussian, Inc., Wallingford, CT, 2003 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Text detailing calculations; NMR spectra of PE synthesized in THF, toluene, dioxane and dichloromethane; tables showing the influence of the mixture composition and the temperature on the free radical polymerization of ethylene, and GAUSSIAN 03 output for 1-hexyl radical and AIBN radical fragment. See DOI: 10.1039/c1py00200g |
| This journal is © The Royal Society of Chemistry 2011 |