DOI:
10.1039/C1PY00238D
(Paper)
Polym. Chem., 2011,
2, 2341-2349
Exfoliation of layered silicates through in situ controlled free radical polymerization mediated by a silicate-anchored initiator
Received
24th May 2011
, Accepted 5th July 2011
First published on 29th July 2011
Abstract
In this study, a new type of controlled free radical polymerization (CFRP) initiators from azetidine-2,4-dione functional groups were synthesized and then embedded within the interlayer spaces of layered silicates to prepare a series of polymer-layered silicate nanohybrids. Kinetics analyses and determination of the molecular weights and distributions of the polymerization products revealed that these CFRP initiators were viable species for 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO)-mediated stable free radical polymerization. Anchoring the CFRP initiator within the galleries of montmorillonite (MMT) enlarged the d-spacing from 15 to 38 Å. The incorporated cationic CFRP initiator (a silicate-anchored TEMPO derivative) provides the initiation functionality for the controlled free radical polymerization within the interlayers. The molecular weights of the resulting polystyrenes varied from 6800 to 85![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 200 g mol−1, depending on the initiator–MMT/styrene weight ratio, with polydispersities of 1.14–1.45 for the lower-molecular-weight samples and 1.96–2.43 for the higher-molecular-weight samples. X-Ray diffraction patterns and transmission electron microscopy revealed that the growing polymer chains separated the layered silicates, thereby leading to the formation of exfoliated individual nanoplatelets. Thus, in situ controlled polymerization within the two-dimensional MMT platform, mediated by silicate-anchored initiators, is an effective dispersion/exfoliation route toward polymer-layered silicate nanohybrids.
200 g mol−1, depending on the initiator–MMT/styrene weight ratio, with polydispersities of 1.14–1.45 for the lower-molecular-weight samples and 1.96–2.43 for the higher-molecular-weight samples. X-Ray diffraction patterns and transmission electron microscopy revealed that the growing polymer chains separated the layered silicates, thereby leading to the formation of exfoliated individual nanoplatelets. Thus, in situ controlled polymerization within the two-dimensional MMT platform, mediated by silicate-anchored initiators, is an effective dispersion/exfoliation route toward polymer-layered silicate nanohybrids.
Introduction
Layered inorganic materials, such as clay minerals and layered double hydroxides, exhibit diverse physicochemical properties because of their two-dimensional platelet structures, nanoscale interlayer spacings, and high specific surface areas; therefore, they are being used for the preparation of organic/inorganic hybrids in efforts to develop new functional materials.1–5 Much effort has been focused on dispersing inorganic layered silicates into a variety of polymer matrices, including polystyrene (PS),6,7poly(methyl methacrylate),7,8polyimide,9,10 polypropylene,11 and epoxy resin.12,13 The most common approaches for the preparation of polymer-layered silicate (PLS) nanohybrids are melt blending, solution blending, and in situpolymerization.14 Exfoliated PLS nanohybrids containing uniformly dispersed inorganic phases often outperform their intercalated counterparts and can exhibit properties superior to those of the pristine polymers. Nevertheless, developing a simple method for distributing randomized clays homogeneously in polymer matrices remains a challenging task.1 In general, enhancing the degree of adhesion between the inorganic and organic phases improves the compatibility of the hybrids and facilitates the transfer of stress to the reinforced phase. When individual nanolayer exfoliation occurs, the properties of the polymer embedded with the nano-layered materials typically include an increase in tensile properties, enhanced gas permeability, decreased solvent uptake, increased dimensional, thermal stability, and flame retardance.15–17
Although high-aspect-ratio, plate-like silicate clays appear to be ideal materials for reinforcement, nanoclays are difficult to disperse in most hydrophobic polymers due to their preference for face-to-face stacking and, thereby, agglomeration and intrinsic incompatibility. To improve their miscibility with polymers, layered silicates must be modified by widening and preoccupying the interlayer spaces with surfactants or so-called modifiers, thereby causing them to become organophilic. The modification generally involves swelling the pristine layered silicate and then exchanging the interlayer ions with organic surfactant salts. Through these modifications, the formation of nanohybrids is facilitated. Nanoscale assemblies of layered inorganic clays, including montmorillonite (MMT), layered double hydroxides (LDH), and mica, have been used recently in various applications: as inorganic/organic nanohybrids, in optoelectronic devices, and in biomedical materials.18–22 In this study, we developed an in situ approach toward organic/inorganic nanohybrids by anchoring controlled free radical polymerization (CFRP) initiators within the galleries of layered silicates and then polymerizing the monomers. As the polymerization within the galleries progresses and the polymer chain density increases, the interlayers are gradually pushed apart, eventually exfoliating into individual nanoplatelets and forming a well-dispersed nanohybrid.
For precise control of the polymerization process, many well-defined homopolymers, block copolymers, grafted and star-like polymers, and hyperbranched polymers have been developed through controlled living polymerization. There are various forms of living polymerization, including group transfer polymerization (GTP),23 atom transfer radical polymerization,24 reversible addition/fragmentation chain transfer (RAFT),25 and nitroxide-mediated controlled radical polymerization (NMCRP).26 All have been studied extensively for the production of polymers with specifically designed structures and narrow polydispersities as a result of the stepwise growth mechanism. Because some of these methods, particularly GTP and ionic polymerizations, are often sensitive to moisture and trace impurities, they would be difficult to perform within inorganic silicate hosts, which typically contain water and ionic impurities. Direct syntheses of dispersed nanohybrids through in situ controlled polymerization using various silicate-anchored initiators and monomers have been reported.27–32 Several polymers, including PS, poly(methyl methacrylate), and poly(styrene-b-butadiene-b-styrene), have been produced with controlled molecular weights and distributions within the inorganic apertures or intergalleries of silicates. The tethered interlayer initiator improves the organophilicity of the pristine hydrophilic layered silicate, thereby increasing the homogeneity for polymerization.
Based on our previous study,33 the azetidine-2,4-dione group undergoing facile ring-opening reactions with selective aliphatic primary amine groups to form malonamide linkages has been known for many years. This is due to the azetidine-2,4-dione group with a strained cyclic structure is subject to be attacked by anionic species under basic conditions. The selective end-group functionalization is useful for preparing specific polymer intermediates. We have synthesized a series of polyurea/malonamide structures, including dendritic polyurethane,34 polymalonamide elastomers,35 thermo-reversible polyurethane supramolecules,36 dendritic intercalating agents for clays,18,19,21,22 multiple hydrogen-bonding epoxy reactive modifiers,37 and hyperbranched polymers for nonlinear optical materials.38 In this study, peroxide intermediates were efficiently prepared with high yields at mild temperature conditions via anionic ring-opening mechanisms. Subsequently, we describe the synthesis of dispersed layered silicate nanohybrids through CFRP of styrene monomers within interlayers featuring silicate-anchored initiator units. Our inserted cationic organic initiators were 2,2,6,6-tetramethyl-1-piperidyloxy (TEMPO)-based derivatives that provided the initiation functionality for controlled free radical polymerization. We also employed mono- and bis-types of initiators to analyze the kinetic constants and the evolution of the molecular weight. A TEMPO-based amine salt derivative containing an initiation group was synthesized and anchored within the intergallery spaces of MMT. We then investigated the characteristics of the resulting polymer (molecular weight, polydispersity) and layered silicate (d-spacing).
Experimental
Materials
Methylenedi-p-phenyl diisocyanate (MDI), isobutyryl chloride, triethylamine (TEA), 4-aminobenzylamine, tert-butyl hydroperoxide (TBHPO), styrene, and 1,8-diazabicyclo[5,4,0]undec-7-ene (DBU) were purchased from Arcos. Phenyl isocyanate, 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO), and lithium bromide were purchased from Lancaster. Styrene was freshly distilled under reduced pressure. Toluene, xylene, and cyclohexane were distilled under reduced pressure over CaH2 and stored over 4 Å molecular sieves. Na+-MMT (Nanocor) was a sodium-type silicate with a cationic exchange capacity (CEC) of 1.2 mequiv g−1 and a surface area of 750 m2 g−1; the naturally occurring clay had a generic structure of 2:1 layered silicate/aluminium oxides with two tetrahedron sheets sandwiching an edge-shared octahedral sheet (averaging 8–10 sheets in a primary stack).
Measurements
1H NMR spectra were recorded using a Varian Gemini-200 FT-NMR spectrometer and CDCl3, acetone-d6 and DMSO-d6 as solvents. IR spectra were recorded using a Perkin–Elmer Spectrum One FT-IR spectrometer. Thermal analysis was performed in air using a TA Instruments DSC2010 operated at a heating rate of 10 °C min−1. Thermogravimetric analysis (TGA) was performed using a Seiko SSC-5200 thermogravimetric analyzer operated at a heating rate of 10 °C min−1 under N2. Gel permeation chromatography (GPC) was performed using a Waters apparatus equipped with Waters Styragel columns and a refractive index detector with PS calibration and THF as the eluent. Mass spectrometry was performed using a JEOL JMS SX/SX 102, an instrument equipped with a fast atom bombardment (FAB) source. X-Ray diffraction (XRD) was performed using a Shimadzu SD-D1 diffractometer equipped with a Cu target (λ = 1.54 Å), operated at a generator voltage of 35 kV, a current of 30 mA, and a scanning rate of 2° min−1. The d-spacings of the initiator–MMT and MMT–PS nanohybrids were analyzed using Bragg's equation (nλ = 2dsin θ). The microstructures of the nanohybrids were studied using bright-field transmission electron microscopy (TEM); sections (50–100 nm thick) were microtomed using a diamond knife. TEM was performed using a Zeiss EM 902A microscope operated at 80 kV. The sections were supported by 100 mesh grids sputter-coated with a 3 nm thick layer of Cu.
Mono(azetidine-2,4-dione) (Scheme 1)
A solution of TEA (15.0 g, 0.150 mol) in xylene (100 mL) was added to a solution of phenyl isocyanate (23.8 g, 0.200 mol) and isobutyryl chloride (13.4 g, 0.100 mol) in xylene (150 mL). The reaction mixture was heated under reflux for 7 h, cooled to room temperature, filtered to remove TEA·HCl, and concentrated to a volume of 50 mL. The product was then crystallized from cyclohexane, yielding a white powder (67%). Mp: 72.5 °C. FT-IR (KBr, cm−1): 1856 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1736 (CO). 1H NMR (DMSO-d6) δ (ppm): 1.40 (6H, CH3), 7.30–7.34 (1H, ArH), 7.48–7.51 (2H, ArH), 7.68–7.71 (2H, ArH).
O), 1736 (CO). 1H NMR (DMSO-d6) δ (ppm): 1.40 (6H, CH3), 7.30–7.34 (1H, ArH), 7.48–7.51 (2H, ArH), 7.68–7.71 (2H, ArH).
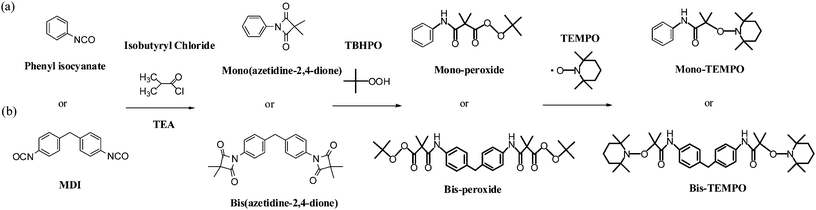 |
| | Scheme 1 Synthesis of CFRP initiators. | |
Bis(azetidine-2,4-dione) (Scheme 1)
Using a similar procedure to that described above, the reaction of MDI (100 g, 0.400 mol) with isobutyryl chloride (95.6 g, 0.880 mol) and TEA (101 g, 1.00 mol) yielded a white powder (82%). Mp: 103.5 °C. FT-IR (KBr, cm−1): 1858 (CO), 1734 (CO). 1H NMR (DMSO-d6) δ (ppm): 1.38 (12H, CH3), 3.98 (2H, ArCH2Ar), 7.29–7.33 (4H, ArH), 7.59–7.63 (4H, ArH).
The purified mono(azetidine-2,4-dione) (1.63 g, 0.860 mmol) was added to a solution of TBHPO (1.3 g, 1.0 mmol) and a catalytic amount of DBU in toluene (30 mL). The solution was stirred at room temperature under N2 for 6 h. After evaporating the solvent, the product was purified chromatographically (SiO2; EtOAc/hexane, 1:4) to yield mono-peroxide (55%). Mp: 94.2 °C. FT-IR (KBr, cm−1): 1767 (O–O–CO), 1688 and 3342 (amide). 1H NMR (CDCl3) δ (ppm): 1.26 (6H, t-Bu), 1.53 (6H, CH3), 7.05–7.09 (1H, ArH), 7.20–7.30 (2H, ArH), 7.43–7.47 (2H, ArH), 8.43 (1H, NH). MS: m/z 279.3 [M+].
The reaction of bis(azetidine-2,4-dione) using the procedure described above yielded bis-peroxide (55%). Mp: 139.6 °C. FT-IR (KBr, cm−1): 1768 (O–O–CO), 1678 and 3348 (amide). 1H NMR (CDCl3) δ (ppm): 1.27 (6H, t-Bu), 1.53 (6H, CH3), 3.84 (2H, CH2), 7.03–7.07 (4H, ArH), 7.34–7.38 (4H, ArH), 7.43–7.47 (2H, ArH), 8.36 (2H, NH). MS: m/z 570.5 [M+].
Bis-one peroxide (Scheme 2)
The reaction of bis(azetidine-2,4-dione) using the procedure described above, but with half an equivalent of the peroxide, yielded bis-one-peroxide (30%). Mp: 137.2 °C. 1H NMR (DMSO-d6) δ (ppm): 1.20 (9H, CH3), 1.39 (6H, CH3), 1.47 (6H, CH3), 3.89 (2H, CH2), 7.11–7.15 (2H, ArH), 7.29–7.33 (2H, ArH), 7.48–7.52 (2H, ArH), 7.59–7.63 (2H, ArH), 9.54 (1H, NH).
 |
| | Scheme 2 Synthesis of an intercalating molecule for MMT. | |
Bis-one
-
amine (Scheme 2)
4-Aminobenzylamine (0.244 g, 2.00 mmol) was added to a solution of bis-one-TEMPO (0.78 g, 1.5 mmol) in toluene (5 mL) and then the mixture was stirred at room temperature under N2 for 4 h. The resulting solution was poured into water; the precipitate was filtered off and dried under vacuum for 6 h to give a white solid (89%). Mp: 174.5 °C. 1H NMR (DMSO-d6) δ (ppm): 0.97 (6H, CH3), 1.14–1.25 (6H, CH3), 1.39 (12H, CH3), 1.41 (6H, CH3), 3.82 (2H, CH2), 4.12 (2H, CH2), 4.89 (2H, NH2), 6.43–6.46 (2H, ArH), 6.86–6.90 (2H, ArH), 7.10–7.14 (4H, ArH), 7.48–7.57 (4H, ArH), 8.02 (1H, NH), 9.26 (1H, ArNH), 9.31 (1H, ArNH).
Intercalation of Na+-MMT with bis-one-amine-salt
Na+-MMT (2.00 g, 2.40 equiv kg−1) and deionized water (150 mL) were stirred vigorously in a 250 mL flask heated at 80 °C for 3–4 h until fully swollen. In a separate vessel, bis-one-amine (1.54 g, 2.40 mmol) in toluene (20 mL) was acidified with hydrochloric acid (37% in water, 0.234 g, 2.40 mmol). The solution of the acidified intercalating agent was poured into the flask containing the Na+-MMT slurry; the ensuing mixture was stirred vigorously at 80 °C for 4 h and then it was cooled to room temperature. The resulting agglomerated precipitate was collected and washed sequentially with water and acetone (twice) to remove any residual ions or free intercalating agents. The pre-intercalated initiator/organoclay was dried under vacuum at 60 °C for 6 h and characterized using X-ray diffraction.
Preparation of PS using silicate-anchored initiators
For bulk polymerization, a 250 mL round-bottom flask containing a magnetic stirring bar was charged with styrene (30 mL) and an initiator [mono-peroxide (0.376 g, 1.17 mmol), mono-TEMPO (0.373 g, 1.17 mmol), or bis-TEMPO (0.379 g, 0.585 mmol)]. The reactant solution was heated at 125 °C under N2 for several hours to obtain the PS samples, which were purified through repeated precipitations from CH2Cl2 into MeOH. Under the same polymerization conditions, the silicate-anchored initiator (MMT intercalated with bis-one-amine-salt) was used to form dispersed polymer/silicate nanohybrids having silicate contents ranging from 0.1 to 7 wt%. The polymerization recipes and results are presented in Table 1.
Table 1 Characteristics of the PS/MMT nanohybrids
| Samplea |
Styrene/g |
Initiator–MMTb/g |
Initiator–MMT dosage (wt %) |
M
n of PSc/g mol−1 |
PDId |
MMT contente (wt%) |
d-Spacingf/Å |
|
MMT–PS: MMT/PS nanohybrid.
Bis-one
-
amine
-salt-intercalated MMT.
Determined by GPC analysis (eluent: THF; calibration: PS standards).
Polydispersity index, Mw/Mn.
Determined by TGA under an atmosphere of air.
Determined from XRD and TEM analyses. Featureless: no diffraction peaks.
|
|
2 h
|
|
MMT
–PS-0.1
|
4 |
0.004 |
0.1 |
50700 |
2.43 |
1.5 |
Featureless |
|
MMT
–PS-1
|
4 |
0.04 |
1 |
24800 |
2.13 |
4.4 |
152–exfoliated |
|
MMT
–PS-3
|
4 |
0.12 |
3 |
11100 |
2.06 |
8.3 |
100–exfoliated |
|
MMT
–PS-5
|
4 |
0.21 |
5 |
8300 |
1.45 |
13.3 |
100–196 |
|
MMT
–PS-7
|
4 |
0.3 |
7 |
6800 |
1.14 |
20.6 |
90–158 |
|
8 h
|
|
MMT
–PS-0.1
|
4 |
0.004 |
0.1 |
85200 |
2.04 |
0.1 |
Featureless |
|
MMT
–PS-1
|
4 |
0.04 |
1 |
58600 |
1.96 |
1.3 |
Featureless |
Results and discussion
Synthesis and characterization of starting materials
We used our previously developed procedures to synthesize the azetidine-2,4-dione-containing starting materials.33,34 The mono-functional mono(azetidine-2,4-dione) and di-functional bis(azetidine-2,4-dione), derived from phenyl isocyanate and MDI, respectively, exhibit selective reactivity toward primary amines. Next, we reacted the azetidine-2,4-dione-containing starting materials with TBHPO through a catalytic ring opening transformation to form their peroxide ester intermediates, which we subsequently combined with the stable nitroxide free radical TEMPO to form the CFRP initiators depicted in Scheme 1. The reactions of the functionalized azetidine-2,4-diones with TBHPO and TEMPO were readily monitored using FT-IR spectroscopy. For example, the starting mono(azetidine-2,4-dione) exhibits characteristic IR absorptions at 1856 and 1736 cm−1 (Fig. 1(a)). Its conversion to the peroxide ester functionality of mono-peroxide was evidenced by a characteristic signal at 1768 cm−1, and additional malonamide absorptions at 1678 and 3348 cm−1 (Fig. 1(b)). The hydroperoxide-containing mono-peroxide reacted with TEMPO to form a propanamide linkage, accompanied by the release of CO2. After incorporating TEMPO into the structure, the absorption peaks of the hydroperoxide unit disappeared completely, whereas the relative intensities of the amide signals were enhanced (Fig. 1(c)).
 |
| | Fig. 1
FTIR spectra of (a) mono(azetidine-2,4-dione), (b) mono-peroxide, and (c) mono-TEMPO. | |
To serve as the silicate-anchored initiator, we synthesized the precursor bis-one-amine according to Scheme 2. By reacting just one end of bis(azetidine-2,4-dione), we prepared bis-one-peroxide and bis-one-TEMPO using procedures similar to those described above. We then coupled bis-one-TEMPO, featuring one stable TEMPO unit and capped with another reactive azetidine-2,4-dione moiety, with 4-aminobenzylamine, which reacted selectively through its aliphatic amino group.34Fig. 2 showed the 1H NMR spectra of mono-peroxide, mono-TEMPO, bis-TEMPO and bis-one-amine. For mono-peroxide (Fig. 2(a)), chemical shifts for tert-butyl group (1.26 ppm) and methyl group (1.53 ppm) were observed. As shown in Fig. 2(b), the emergence of 0.98 and 1.43 ppm absorption peaks indicated the presence of TEMPO. In addition, mass data provided the direct quantitative change of molecular weights that corroborated the covalent attachment of TEMPO. 1H NMR spectrum of bis-TEMPO was also provided in Fig. 2(c) for the sake of comparison. As shown in Fig. 2(d), the signals at 3.82 and 4.12 ppm represent the protons of the CH2 linkages of the MDI and 4-aminobenzylamine units, respectively. The signals for the NH protons of the amide linkages appear at 8.02, 9.26, and 9.31 ppm. The signals appearing in the range from 0.97 to 1.41 ppm correspond to the CH3 groups of the malonamide, propanamide, and TEMPO units. The chemical shifts of the signals for the aromatic protons and aromatic NH2 units appear at 6.43–7.57 and 4.89 ppm, respectively. The integral intensities of the various peaks are consistent with the values expected for the proposed structure. Thus, the spectroscopic evidence supports the successful synthesis of bis-one-amine.
 |
| | Fig. 2
1H NMR spectra of (a) mono-peroxide, (b) mono-TEMPO, (c) bis-TEMPO and (d) bis-one-amine. | |
Polymerizations of styrene using the TEMPO-based initiators
For comparison with the TEMPO-terminated initiators derived from azetidine-2,4-dione-based starting materials, we also used the mono-peroxide as a free radical polymerization initiator for styrene. Under conditions similar to those of TEMPO-mediated CFRP, the mono-peroxide-initiated polymerization exhibited higher reactivity and rapid growth of PS (Fig. 3(a)). An increase in the higher molecular weight resulted in an increased rate of monomer conversion and a broadened molecular weight distribution (increasing from 2.1 to 2.9 upon increasing the reaction time), suggesting inefficient control of the mono-peroxide-initiated free radical polymerization. In contrast, the controlled free radical polymerization initiated by mono-TEMPO (Fig. 3(a)) exhibited lower reactivity and a lower growth molecular weight, but with better molecular weight distributions (<1.3), indicating that the stable TEMPO free radical effectively coupled with the propagating polymer radical to retard propagation, chain transfer, or termination. In general, stable free radical polymerizations involving bimolecular initiators featuring TEMPO moieties, conventional radical initiators (e.g., AIBN, BPO), and vinyl monomers (e.g., styrenic or acrylic monomers) are very effective for styrenic monomers, but their initial rates are quite slow to reach high conversions.39,40
 |
| | Fig. 3 Molecular weights and polydispersities as a function of time for the polymerizations of styrene using initiators (a) mono-peroxide and mono-TEMPO, and (b) bis-TEMPO and mono-TEMPO. | |
At the same concentration of free radical and TEMPO groups, the molecular weight of the bis-TEMPO-initiated PS grew faster than that of the mono-TEMPO-initiated polymer; that is, the initial rate of the reaction mediated by bis-TEMPO was greater than that of mono-TEMPO. As shown in Fig. 3, an additional time of 900 min is needed to kick-start both mono-TEMPO and bis-TEMPO systems. This indicates that these TEMPO-derived initiators were more stable and exhibited an incubation period when compared to CFRP of styrene initiated by BPO in the presence of TEMPO. The starting polydispersity index (PDI) of bis-TEMPO was relatively high (ca. 1.6), but it decreased upon increasing the reaction time to a constant value of 1.45. The mono-TEMPO-initiated polymer still possessed lower molecular weight and polydispersity at twice the reaction time. In general, the relative initiation ability deduced from Fig. 3(a) and (b) followed the order mono-peroxide ≫ bis-TEMPO ≥ mono-TEMPO. At the same conversion, the growths in the molecular weight of PS followed the trend mono-peroxide ≫ bis-TEMPO ≥ mono-TEMPO. The trend in terms of the polydispersity (molecular weight distribution) was mono-TEMPO > bis-TEMPO ≫ mono-peroxide.
Pasquale et al.40 reported a real-time monitoring of the stable free radical polymerization of styrene and the calculation of an apparent first-order rate constant from the slope, which is equal to the rate of propagation times the concentration of growing polymer radicals, assuming no termination or side reactions occur. The polymerization process could be evaluated as follows: Rp = −d[M]/dt = kp[P˙][M] = K[M]; ln (M0/M) = K × t. Therefore, the linear relationship between ln (M0/M) and time implies the living polymerization behavior. The polymerizations of styrene using the mono-TEMPO- and bis-TEMPO-based initiators at 125 °C revealed pseudo-first-order polymerization kinetics, as expected for linear radical polymerizations; this feature is indicative of a constant number of propagating centers. From the first-order plots in Fig. 4, the kinetic constants exhibited good correlations (R > 0.99), with apparent rate constants for the mono-TEMPO- and bis-TEMPO-initiated polymerizations of 4.1 × 10−5 and 4.6 × 10−5s−1, respectively. The mono-TEMPO- and bis-TEMPO-initiated polymers both maintained narrow polydispersity (1.3–1.45). The observed rate constants are higher than literature values for TEMPO-mediated polymerizations of styrene using a similar method and uracil-based initiators (2.69–3.12 × 10−5s−1),39,40 but are similar to those of dinitroxide mediators (5.89 × 10−5s−1).41 In comparison with the well-known reaction between BPO and TEMPO systems (1.15–2.09 × 10−5s−1),40 the rate constants of the TEMPO-derived initiators in this work are twice as large. This initiator derived from azetidine-2,4-dione could be directly used for CFRP without involving BPO. Moreover, the molecular weight of the bis-TEMPO-initiated polymer was approximately double that of the mono-TEMPO-initiated polymer at the same conversion, as anticipated (Fig. 5). This finding corroborates the similar kinetic constants (4.1–4.6 × 10−5s−1) and signifies that our bis-TEMPOinitiator provides two viable sites for linear-growth polymerization at the same free radical content. Besides, the decrease in the polydispersity (from 1.65 to 1.46) of the bis-TEMPO-initiated PS upon increasing the conversion (time) was consistent with its structure.
 |
| | Fig. 4 Apparent first-order kinetics analysis for the polymerizations of styrene using the various CFRP initiators. | |
 |
| | Fig. 5 Evolution of molecular weights and PDIs, plotted with respect to conversion, for the polymerizations of styrene using the various CFRP initiators. | |
Synthesis and characterization of PS/MMT hybrids
Using the approach outlined in Scheme 2, we synthesized bis-one-amine from bis(azetidine-2,4-dione). Treatment of bis-one-amine with an equivalent of hydrochloric acid afforded its ammonium salt, which we used to intercalate Na+-MMT. Fig. 6 displays XRD patterns of the pristine MMT and the MMTs intercalated with the bis-one-amine-salt CFRP initiator. Our XRD analyses revealed the degree of expansion of the basal spacing and, by implication, the dispersion of the layered silicate in the PS matrices. For the pristine Na+-MMT, a strong diffraction peak appeared at a value of 2θ of 6°, corresponding to an interlayer spacing of 15 Å. In contrast, the intercalated initiator–MMT hybrid possessed a d spacing of 38 Å (curve b in Fig. 6), derived from Bragg's equation using values of n of 1, 2, and 3. Moreover, the TGA study indicates the actual organic/inorganic ratio of 52/48 for initiator–MMT. This is consistent with the theoretical organic fractions based on the originally intended CEC ratios. For example, for initiator–MMT at 1.0 CEC equivalent, a theoretical organic/inorganic ratio of 50/50 is derived, which is close to the ratio (52/48) obtained from the TGA study. This finding suggests that the bis-one-amine-saltinitiator could diffuse into the interlayer of MMT through ionic exchange. In this study, an approximate estimation of the extended bis-one-amine-salt size is shown as follows: the phenyl group is 4 Å, the terminated cyclic TEMPO and amine salts are <10 Å and the length of building block is about 16 Å based on our previous studies.18,21,22 After elimination of the thickness of silicate plate (approximately 10 Å), the occupied space of bis-one-amine-salt is about 28 Å, indicating about one repeat unit. Therefore, it is concluded that the conformation of the initiator–MMT hybrid was an ordered intercalation containing bis-one-amine-salt monolayers between layered silicates. Furthermore, we used the silicate-anchored initiator to mediate the in situ CFRP of styrene at 110 °C. If the styrene monomers migrated into the clay galleries and subsequently reacted with the anchored bis-one-amine-saltinitiator, the molecular weight would be controlled and the polydispersity would be narrow. The systems solidified completely after 2–8 h to yield MMT–PS nanohybrids as homogeneous transparent films; Table 1 lists the compositions and abbreviated names of the various MMT–PS samples. The XRD patterns for the nanohybrids MMT–PS-1, MMT–PS-3, MMT–PS-5, and MMT–PS-7 (Fig. 6) were completely featureless (no diffraction peaks), indicating that initiation and polymerization occurred for the hybrid system. To determine the extent to which their physical characteristics were controlled, we detached the grafted polymer chains from the platelet surfaces through reverse ion exchange by refluxing the nanohybrids in THF solutions comprising LiBr and then investigated their molecular weights and molecular weight distributions (GPC). According to Table 1, at a constant styrene weight, the value of Mn of PS increased upon decreasing the initiator–MMT/styrene weight ratio after a polymerization time of 2 h, thus confirming the effective molecular weight control induced by the weight fraction of the bis-one-amine-saltinitiator. When the polymerization time increased to 8 h, we obtained a higher value of Mn (85200 g mol−1) of MMT–PS-0.1. Thus, the isolated polymers exhibited values of Mn varying from 6800 to 85200 g mol−1, with PDIs of 1.14–1.45 for the lower-molecular-weight samples and 1.96–2.43 for the higher-molecular-weight samples. It is noteworthy that the molecular weight increased as the MMT content decreased. Therefore, the molecular weight was controlled by the initiator–MMT/monomer ratio—consistent with previously reported results.27 The real MMT contents of the synthesized nanohybrids (Table 1) ranged from 0.1 to 20.6 wt%, as determined through TGA under an air atmosphere.
 |
| | Fig. 6
XRD patterns of (a) Na+-MMT, (b) initiator–MMT, (c) MMT–PS-1, (d) MMT–PS-3, (e) MMT–PS-5, and (f) MMT–PS-7. | |
For all of the MMT–PS samples prepared at different initiator–MMT/styrene monomer ratios, we observed no diffraction peaks in the range 2–10° (2θ) in their XRD patterns, implying a disordered form or interlayer exfoliation (Fig. 6). To confirm these results, we used TEM to visualize the spatial distributions of the MMT–PS nanohybrids (Fig. 7). The TEM micrograph of initiator–MMT indicates that the well-defined basal spacing around 38 Å was present for the ordered intercalation between the layered silicate structures (Fig. 7(a)). The micrographs of the MMT–PS nanohybrids after 2 h of polymerization exhibited morphologies varying from intercalated to exfoliated, due to in situ controlled polymerization of styrene mediated by the bis-one-amine-saltinitiator-modified MMT. As a result, most of the styrene monomer units reacted with the silicate-anchored initiator to form their polymers within the MMT galleries, which further expanded and eventually exfoliated to form individual nanoplatelets. As indicated in Table 1, the degree of enlargement of the basal spacing depended mainly on the molecular weight of the polymer chains. Unlike a conventional process for solution exfoliation, melt intercalation, or in situpolymerization methods, the in situ CFRP system in this work is capable of controlling the overall polymer architecture. Intergallery initiation of CFRP will provide an opportunity to control not only such crucial characteristics as molecular weights, polydispersity, block copolymer formation, and functionality directly inside the clays, but also the overall architecture of the nanohybrids. This would open up enormous opportunities to use the nano-materials for diverse applications via molecular assembling and nanostructure manipulation. Scheme 3 presents a possible conceptual model of the transformation. The spatially expanded interlayers resulting from the presence of the bis-one-amine-saltinitiator allow the styrene monomers to enter into the layered galleries, where they subsequently undergo in situ controlled polymerization. The growing PS chains then induce the separation of the layered silicates, ultimately forming the exfoliated individual nanoplatelet morphology.
 |
| | Scheme 3 Preparation of PLS nanohybrids through in situ CFRP mediated by the silicate-anchored initiator. | |
 |
| | Fig. 7
TEM micrographs of (a) initiator–MMT, (b) MMT–PS-1, (c) MMT–PS-3, (d) MMT–PS-5, and (e) MMT–PS-7. | |
Conclusion
Novel azetidine-2,4-dione-based precursors were used for the synthesis of stable mediators of the bulk free radical polymerization of styrene. Subsequently, PS polymers with lower radical initiation ability and lower growth molecular weights, but with narrow molecular weight distributions were obtained, when initiated with the TEMPO-terminated initiators derived from the azetidine-2,4-dione-based starting materials. The resulting apparent rate constants were similar for the mono- and di-functional initiators (4.1–4.6 × 10−5s−1). Furthermore, we found that interlayer in situpolymerization using the silicate-anchored initiators is a viable approach for the direct intergallery expansion of well-dispersed MMT–PS nanohybrids. The resulting MMT–PS nanohybrids possessed exfoliated or intercalated structures, depending on the initiator–MMT/styrene monomer weight ratios. Therefore, embedding CFRP initiators into the silicate gallery is a promising method for obtaining clay-exfoliated nanohybrids featuring polymers of high molecular weight and low polydispersity.
Acknowledgements
We thank the National Science Council of Taiwan for financial support.
Notes and references
- J. N. Coleman, M. Lotya, A. O'Neill, S. D. Bergin, P. J. King, U. Khan, K. Young, A. Gaucher, S. De, R. J. Smith, I. V. Shvets, S. K. Arora, G. Stanton, H. Y. Kim, K. Lee, G. T. Kim, G. S. Duesberg, T. Hallam, J. J. Boland, J. J. Wang, J. F. Donegan, J. C. Grunlan, G. Moriarty, A. Shmeliov, R. J. Nicholls, J. M. Perkins, E. M. Grieveson, K. Theuwissen, D. W. McComb, P. D. Nellist and V. Nicolosi, Science, 2011, 331, 568 CrossRef CAS.
- P. Podsiadlo, A. K. Kaushik, E. M. Arruda, A. M. Waas, B. S. Shim, J. Xu, H. Nandivada, B. G. Pumplin, J. Lahann, A. Ramamoorthy and N. A. Kotov, Science, 2007, 318, 80 CrossRef CAS.
- J. J. Lin, C. C. Chu, M. L. Chiang and W. C. Tsai, Adv. Mater., 2006, 18, 3248 CrossRef CAS.
- J. M. Oh, T. T. Biswick and J. H. Choy, J. Mater. Chem., 2009, 19, 2553 RSC.
- R. Simons, G. G. Qiao, S. A. Bateman, X. Zhang and P. A. Lynch, Chem. Mater., 2011, 23, 2303 CrossRef CAS.
- X. Fu and S. Qutubuddin, Polymer, 2001, 42, 807 CrossRef CAS.
- H. Akata, M. A. Tasdelen, F. D. Prez and Y. Yagci, Eur. Polym. J., 2008, 44, 1949 CrossRef.
- M. C. Costache, D. Wang, M. J. Heidecker, E. Manias and C. A. Wilkie, Polym. Adv. Technol., 2006, 17, 272 CrossRef CAS.
- T. Y. Chao, H. L. Chang, W. C. Su, J. Y. Wu and R. J. Jeng, Dyes Pigm., 2008, 77, 515 CrossRef CAS.
- A. B. Morgan, J. W. Gilman and C. L. Jackson, Macromolecules, 2001, 34, 2735 CrossRef CAS.
- J. Y. Wu, T. M. Wu, W. Y. Chen, S. J. Tsai, W. F. Kuo and G. Y. Chang, J. Polym. Sci., Part B: Polym. Phys., 2005, 43, 3242 CrossRef CAS.
- J. Ma, Z. Z. Yu, Q. X. Zhang, X. X. Xie, Y. W. Mai and I. Luck, Chem. Mater., 2004, 16, 757 CrossRef CAS.
- Y. N. Chan, T. Y. Juang, Y. L. Liao, S. A. Dai and J. J. Lin, Polymer, 2008, 49, 4796 CrossRef CAS.
-
Y. C. Ke and P. Stroeve, Polymer-Layered Silicate and Silica Nanocomposites, Elsevier B.V., Netherlands, 2005 Search PubMed.
- C. S. Triantafillidis, P. C. LeBaron and T. J. Pinnavaia, Chem. Mater., 2002, 14, 4088 CrossRef CAS.
- J. W. Gilman, C. L. Jackson, A. B. Morgan, R. Harris, Jr, E. Manias, E. P. Giannelis, M. Wuthenow, D. Hilton and S. H. Philips, Chem. Mater., 2000, 12, 1866 CrossRef CAS.
- J. M. Brown, J. M. Curliss and R. A. Vaia, Chem. Mater., 2000, 12, 3376 CrossRef CAS.
- T. Y. Juang, C. C. Tsai, T. M. Wu, S. A. Dai, C. P. Chen, J. J. Lin, Y. L. Liu and R. J. Jeng, Nanotechnology, 2007, 18, 205606 CrossRef.
- Y. C. Chen, T. Y. Juang, S. A. Dai, T. M. Wu, J. J. Lin and R. J. Jeng, Macromol. Rapid Commun., 2008, 29, 587 CrossRef CAS.
- J. J. Lin, J. C. Wei, T. Y. Juang and W. C. Tsai, Langmuir, 2007, 23, 1995 CrossRef CAS.
- C. C. Tsai, T. Y. Juang, S. A. Dai, T. M. Wu, W. C. Su, Y. L. Liu and R. J. Jeng, J. Mater. Chem., 2006, 16, 2056 RSC.
- T. Y. Juang, Y. C. Chen, C. C. Tsai, S. A. Dai, T. M. Wu and R. J. Jeng, Appl. Clay Sci., 2010, 48, 103 CrossRef CAS.
- O. W. Webster, W. R. Hertler, D. Y. Sogah, W. B. Fahnham and T. V. RajanBabu, J. Am. Chem. Soc., 1983, 105, 5706 CrossRef CAS.
- J. S. Wang and K. Matyjaszewski, J. Am. Chem. Soc., 1995, 117, 5614 CrossRef CAS.
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina and J. Jefery, Macromolecules, 1998, 31, 5559 CrossRef CAS.
- W. H. Ting, S. A. Dai, Y. F. Shih, I. K. Yang, W. C. Su and R. J. Jeng, Eur. Polym. J., 2008, 49, 1497 CAS.
- M. W. Weimer, H. Chen, E. P. Giannelis and D. Y. Sogah, J. Am. Chem. Soc., 1999, 121, 1615 CrossRef CAS.
- J. Di and D. Y. Sogah, Macromolecules, 2006, 39, 5052 CrossRef CAS.
- J. Di and D. Y. Sogah, Macromolecules, 2006, 39, 1020 CrossRef CAS.
- P. Ding, M. Zhang, J. Gai and B. Qu, J. Mater. Chem., 2007, 17, 1117 RSC.
- H. Böttcher, M. L. Hallensleben, S. Nuß, H. Wurm, J. Bauer and P. Behrens, J. Mater. Chem., 2002, 12, 1351 RSC.
- J. Pyun and K. Matyjaszewski, Chem. Mater., 2001, 13, 3436 CrossRef CAS.
- S. A. Dai, T. Y. Juang, C. P. Chen, H. Y. Chang, W. J. Kuo, W. C. Su and R. J. Jeng, J. Appl. Polym. Sci., 2007, 103, 3591 CrossRef CAS.
- C. P. Chen, S. A. Dai, H. L. Chang, W. C. Su, T. M. Wu and R. J. Jeng, Polymer, 2005, 46, 11849 CrossRef CAS.
- M. C. Kuo, Y. C. Tung, C. L. Yeh, H. Y. Chang, R. J. Jeng and S. A. Dai, Macromolecules, 2008, 41, 9637 CrossRef CAS.
- M. C. Kuo, R. J. Jeng, W. C. Su and S. A. Dai, Macromolecules, 2008, 41, 682 CrossRef CAS.
- J. K. Liu, S. M. Shau, T. Y. Juang, C. C. Chang, S. A. Dai, W. C. Su, C. H. Lin and R. J. Jeng, J. Appl. Polym. Sci., 2011, 120, 2411 CrossRef CAS.
- H. L. Chang, T. Y. Chao, C. C. Yang, S. A. Dai and R. J. Jeng, Eur. Polym. J., 2007, 43, 3988 CrossRef CAS.
- B. D. Mather, J. R. Lizotte and T. E. Long, Macromolecules, 2004, 37, 9331 CrossRef CAS.
- A. J. Pasquale and T. E Long, Macromolecules, 1999, 32, 7954 CrossRef CAS.
- J. R. Lizotte, S. G. Anderson and T. E. Long, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 1547 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here.