Aliphatic polyketone obtained by cationic polymerization of ethylketene
Najib
Hayki
,
Nicolas
Desilles
* and
Fabrice
Burel
INSA de Rouen, Laboratoire Polymères Biopolymères Surfaces, CNRS UMR 6270 & FR 3038, avenue de l'Université BP8, 76801, Saint-Etienne du Rouvray Cedex, France. E-mail: nicolas.desilles@insa-rouen.fr; Fax: +33 2 32 95 66 44; Tel: +33 2 32 95 66 45
First published on 29th July 2011
Abstract
For the first time, ethylketene, synthesized by thermolysis of butyric anhydride, was polymerized cationically in order to obtain a novel aliphatic polyketone. Initiated by Lewis acid initiator AlBr3, this polymerization was undertaken in different solvents and at −78 °C (top polymerization yield of 57% in toluene). 1H-13C HSQC NMR experiments clearly demonstrated the polyketonic microstructure. The thermal properties of this new polymer were analyzed by DSC, TGA and X-ray diffraction: with a glass transition temperature around 70 °C and an average crystallinity of 0.34, this polymer showed a starting degradation temperature near 210 °C.
Introduction
Aliphatic polyketones are a family of polymers generally prepared by the perfectly alternating polymerization of olefins and carbon monoxide, by means of palladium-based catalysts.1,2 In order to produce polymers with varying melting points and therefore tailor-made properties, a second olefin (propylene for example) is introduced in the polymerization medium substituting randomly for ethylene. Actually, aliphatic polyketones are terpolymers of ethylene and propylene with carbon monoxide (a different route, not extensively described so far, is the oxidation of poly(vinyl alcohol)3).Aliphatic polyketones were first prepared and studied in the 1950s,4,5 but it is only in the 1990s, with the development of high-yielding catalysts, that these polymers seriously attracted the scientific community.1 Indeed, they exhibit a unique combination of mechanical (toughness, stiffness, impact resistance), high temperature (heat stability), chemical resistance, and barrier properties (comparable to poly(ethylene terephtalate)). Furthermore, they are processed like polyolefins2 and are also readily compoundable with typical packaging film materials, such as polyolefins and poly(ethylene-co-vinyl alcohol).6
Aliphatic polyketones therefore have significant potential in a broad range of engineering, barrier packaging, fiber and blend applications.7 Finally, polyketones can act as excellent precursors for the preparation of functional polymers by chemical modifications, since the recurring carbonyl functionality can readily be converted into a great variety of functionalized polymers.8–11
Herein, we report another way, not based on the alternating copolymerization of olefins and carbon monoxide, but through the selective polymerization of ketene monomers, to access to new aliphatic polyketone structures. Ketenes are derivatives of carboxylic acids which contain two consecutive double bonds (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CO). First isolated by Staudinger in 1905 with the synthesis of diphenylketene,12 theses substances, containing two double bonds in an adjacent position, show a particularly high reactivity,13,14 so that most of them are unstable products that cannot be isolated and must be handled very carefully. The chemistry of ketenes is dominated by addition reactions with reagents having labile hydrogen atoms, nucleophiles or electrophiles13,15–17 and they characteristically undergo cycloaddition.18,19 One of the most interesting properties of these compounds is their ability to be used as monomers. Most studies of the polymerization of ketenes were carried out with ketene itself20,21 and dimethyl ketene.22–25 These results showed that a selective opening of the double bond was possible with a suitable choice of both initiator and solvent. In the case of dimethyl ketene, it was possible to isolate crystalline polymers consisting of acetal (1), ketone (2) and ester units (3) (Scheme 1) but polyketone is favored only with Lewis acid initiators like AlBr3 and AlCl3.24–26
CO). First isolated by Staudinger in 1905 with the synthesis of diphenylketene,12 theses substances, containing two double bonds in an adjacent position, show a particularly high reactivity,13,14 so that most of them are unstable products that cannot be isolated and must be handled very carefully. The chemistry of ketenes is dominated by addition reactions with reagents having labile hydrogen atoms, nucleophiles or electrophiles13,15–17 and they characteristically undergo cycloaddition.18,19 One of the most interesting properties of these compounds is their ability to be used as monomers. Most studies of the polymerization of ketenes were carried out with ketene itself20,21 and dimethyl ketene.22–25 These results showed that a selective opening of the double bond was possible with a suitable choice of both initiator and solvent. In the case of dimethyl ketene, it was possible to isolate crystalline polymers consisting of acetal (1), ketone (2) and ester units (3) (Scheme 1) but polyketone is favored only with Lewis acid initiators like AlBr3 and AlCl3.24–26
 | ||
| Scheme 1 Dimethyl ketene polymers main-chain structure. | ||
The polymerization of dialkyl ketene acetals and cyclic ketene acetals, initiated by Lewis acid or protonic inorganic acids, and followed by hydrolysis, was extensively studied.27–31 But the obtained polymers were not pure polyketones and were constituted of keto-enolic structures along the chain with, in general, ring-retained structures (in the case of cyclic acetals), chain scisson, cyclization and/or crosslinking.
In this paper, we illustrate the cationic polymerization of ethylketene initiated by Lewis acid initiator AlBr3, in different solvents, in order to obtain a new aliphatic polyketone. The obtained polymers were characterized by FTIR and NMR, their molecular weights were determined by SEC and their thermal properties were analyzed by TGA, DSC and X-ray diffraction.
Experimental section
Materials and methods
n-Decane (Acros Organics; 99+%) and heptane (Acros Organics; 99%) were distilled over sodium under reduced pressure, and packaged in sealed bottles on molecular sieves (4 Å) before use.Dichloromethane (Acros Organics; 99.8%) and toluene (Acros Organics; 98%) were distilled over sodium and packaged in sealed bottles on molecular sieves (4 Å) before use.
Ethyl acetate (Acros Organics; 99+%) was distilled over calcium hydride and packaged in sealed bottles on molecular sieves (4 Å) before use.
Tetrahydrofuran (THF) (Acros Organics; 99.5%) was dried over potassium hydroxide, distilled over sodium benzophenone under atmospheric pressure, and then packaged in sealed bottles on molecular sieves (4 Å) before use.
Methanol (99.5%), ethanol (99.8%), butyric anhydride (98%) and 1,1,1,3,3,3-hexafluoro-2-propanol (99.5+%) were purchased from Acros Organics and used as received.
Aluminum tribromide (Acros Organics; 98+%) was purified by distillation under reduced pressure. AlBr3 solutions were realized under nitrogen atmosphere in a glove box, and then stored in sealed bottles.
For polymers, all measurements were performed after purification by Soxhlet extraction in THF followed by drying under vacuum.
Gas chromatography was carried out on a 1020 GC Plus chromatograph (Perkin Elmer) equipped with a SGE wide-bore 30 QC 5/BP 1/1.0 column.
Molecular weights were determined by size exclusion chromatography (SEC). Samples were solubilized in 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) during 24 hours at room temperature, filtered (0.45 μm) and analyzed at 40 °C using an Alliance 2695 instrument (Waters) equipped with two columns (PSS PFG 1000 Å and 100 Å). The mobile phase was HFIP and poly(methylmethacrylate) standards were used for calibration.
Fourier Transform InfraRed (FTIR) spectra were recorded on a Perkin Elmer Spectrum 2000 FTIR, equipped with a diamond ATR (Attenuated Total Reflection) device.
1H and 13C NMR spectra were recorded on a Bruker AVIII-600 apparatus, respectively at 600 and 150 MHz. Chemical shifts are expressed in ppm relative to CDCl3 which was used as internal reference and solvent. Polymers were studied in a mixture CDCl3/1,1,1,3,3,3-hexafluoro-2-propanol (HFIP).
Ultraviolet spectroscopy (UV) was performed on a Varian Cary 100 spectrophotometer equipped with a Hellma quartz immersion probe (optical pathway: 1 mm) connected with fiber optics, measuring the “in situ” absorbance of the reactive medium at 357 nm. The normalized absorbance was defined as the ratio A(t)/A(t = 0).
Thermogravimetric analysis (TGA) measurements were performed on a TGA Q500 apparatus (TA Instruments) under nitrogen atmosphere at a heating rate of 10 °C min−1. Degradation temperatures were determined at 1% weight loss.
Differential scanning calorimetry (DSC) was performed on a DSC Q2000 apparatus (TA Instruments), under nitrogen with a heating rate of 10 °C min−1. Tg were measured at the midpoint, on the second heating thermogram.
X-Ray diffractograms were performed on a Bruker D8 Discover diffractometer (λCu Kα1 = 1.540598 Å, λCu Kα2 = 1.544426 Å). The crystallinity was computed using the method of Murthy32 and the software PeakFit™ from SPSS Science.
Ethylketene synthesis
All these steps were carried out under Alphagaz™ 2 (Air Liquide) nitrogen atmosphere, which certifies extremely low impurity levels, to prevent the reaction of ethylketene with water and the formation of highly explosive peroxides in the presence of oxygen. In addition, solvents were purified before use to eliminate water, a known inhibitor for cationic polymerizations.Ethylketene (4) (EK) was synthesized by pyrolysis of butyric anhydride (5) (BAN) according to Scheme 2. The apparatus is inspired by those already described elsewhere33–35 and is made of 3 different steps: EK synthesis by BAN pyrolysis, EK purification, and EK polymerization (Scheme 3). Butyric anhydride (200 g, 1260 mmol) was introduced, with a flow rate of 220 g h−1 (thanks to a dosing pump) and under nitrogen atmosphere, into the pyrolysis oven A brought to 575 °C under a reduced pressure of 40 mbar. The resulting gaseous mixture passed through two condensers B and C respectively at 60 and −24 °C: unreacted butyric anhydride and formed by-products were mainly condensed and recovered at this step, and aliquots were analyzed by gas chromatography to estimate the pyrolysis yield (21%). The more volatile gaseous fraction passed through another condenser D at −30 °C, and then bubbled through 19.9 mL of n-decane at −15 °C in flask E, in order to eliminate the last traces of unreacted butyric anhydride and other by-products. Ethylketene was then trapped in liquid nitrogen (−180 °C) in polymerization reactor F. When the monomer synthesis and purification steps were over, ethylketene was allowed to warm gently to −78 °C and brought to atmospheric pressure, giving a yellow solution of pure ethylketene (14.2 g, 203 mmol). The overall synthesis yield (16%) was calculated by mass balance.
 | ||
| Scheme 2 Pyrolysis of butyric anhydride. | ||
 | ||
| Scheme 3 Ethylketene synthesis, purification and polymerization apparatus. | ||
Ethylketene polymerization
To freshly synthesized ethylketene collected in reactor F (14.2 g, 203 mmol) were added slowly, to minimize the variation of temperature, 67.5 mL of toluene, so that [EK] = 3 mol L−1. When the temperature of the mixture was stabilized, 2.0 mL of a solution of AlBr3 in toluene (1.0 mol L−1) were added, to obtain [EK]/[AlBr3] = 100. The polymer, which is insoluble in toluene, precipitated and caused the heterogeneity of the medium as soon as the polymerization started, in parallel with the discoloration of the medium. After complete discoloration, the mixture was allowed to reach room temperature, and then 20 mL of ethanol were added to react with and neutralize residual ethylketene and initiator. The crude polymer was then filtered, washed with methanol, dried under vacuum, and purified by Soxhlet extraction with THF. 8.1 g of polymer were obtained (yield 57%).Results and discussion
As described in the Experimental section, ethylketene was prepared by pyrolysis of butyric anhydride, purified to eliminate unreacted butyric anhydride, formed butyric acid, and other by-products, and finally trapped into liquid nitrogen. Indeed, aldoketenes are well known to be unstable products: while ketoketenes are relatively stable compounds (especially diarylketenes and, to a lower extent arylalkylketenes then dialkylketenes13), aldoketenes can dimerize and trimerize easily,36–38 which may be the reason why their polymerization remains little studied. To our knowledge, the only aldoketene polymerization reported so far in the literature was conducted recently in our research team, where ethylketene was polymerized anionically with lithiated initiators in THF to form a polyester.39First of all, the stability of ethylketeneversus temperature was studied (Fig. 1), by following the UV absorbance at 357 nm of a solution of ethylketene in tetrahydrofuran brought at different temperatures. The decay of ethylketene UV absorbance observed in only several minutes at temperatures as low as −40 °C clearly highlighted the instability of this aldoketene. Thus, great care was taken to maintain ethylketene at low temperatures along its whole synthesis, leading to overall synthesis yields, calculated by mass balance, ranging from 12 to 18%.
 | ||
| Fig. 1 Evolution of the normalized absorbance at 357 nm of a solution of ethylketene in THF, at various temperatures. | ||
In order to minimize the thermal self-reaction of ethylketene, its cationic polymerization was then undertaken at low temperature (−78 °C) with AlBr3 initiator, in several solvents, in order to afford the expected aliphatic polyketone.
The various experimental conditions are presented in Table 1. It is important to notice that every run, starting from the yellow solution of ethylketene, led to the spontaneous discoloration of the medium in parallel with the precipitation of the formed polymer. This heterogeneous polymerization was systematically accompanied with an exothermy, depending on the obtained yield, which could raise the medium temperature by 10 °C. The obtained polymers were found insoluble in all common organic solvents.
| Run | Solvent a | Dielectric constant | Polymerization yieldb (%) |
|---|---|---|---|
| a Each initiator solution was realized with the polymerization solvent, except for Run 2 where toluene was used instead of heptane for solubility concerns. b Defined as the weight ratio of precipitated polymer/ethylketene used, after Soxhlet purification. | |||
| 1 | Dichloromethane | 8.9 | 38 |
| 2 | Heptane | 1.9 | 47 |
| 3 | Toluene | 2.4 | 57 |
| 4 | Ethyl acetate | 6.0 | 26 |
The 57% top yield was obtained in toluene, whereas the polymerization yield dropped down to 26% in ethyl acetate. Heptane and dichloromethane gave rather good yields respectively at 47 and 38%. Globally, better yields were obtained with low dielectric constant solvents, such as heptane and toluene, but the reasons of this variation are not clearly identified. However, this behavior can be linked with the interactions between the involved species and active centers which are very dependent on the dielectric constant of the solvent. Indeed, in high dielectric constant solvents, the propagating chain-end is mainly solvated by the solvent. Whereas in low dielectric constant solvents, since ethylketene is the more polar species in the medium, the propagating chain-end is preferentially solvated by the monomer, creating a local increase in the monomer concentration around the active centers and leading to better yields. The non-precipitating fraction of the reaction medium was analyzed as a very complex mixture of dimer, trimer, and other unidentified by-products.
The obtained polymers were then characterized. Their FTIR-ATR spectra showed a strong absorption band at 1713 cm−1 (Fig. 2), very close to the absorption measured near 1718 cm−1 for the ketene-based polyketone polymer40 and oligomer.41
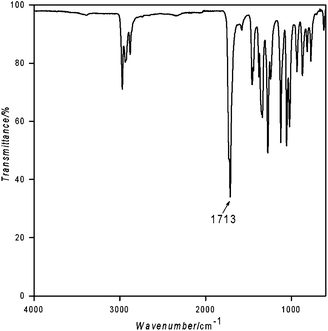 | ||
| Fig. 2 FTIR spectrum of ethylketene-based polyketone. | ||
However, Bunel et al. clearly demonstrated that it was hazardous to attest the structure of a polyketene only on the basis of its FTIR spectrum, and that NMR spectroscopy was definitely more reliable.26 Before going any further, we must specify that, unfortunately, none of the obtained polymers were soluble in common organic solvents. To elucidate their structure, samples were first solubilized in 1,1,1,3,3,3-hexafluoro-2-propanol with subsequent dilution with dichloromethane. The polymers remained soluble in the solvent mixture, which enabled full 1H and 13C NMR characterization. As expected, the study of only 1H NMR spectrum was worthless since the chemical shifts of the different units (polyketone, polyester and polyacetal) are supposed to be very close, as already stated for dimethylketene polymers.26 However, the 13C NMR spectrum was very instructive. Indeed, Fig. 3 revealed the presence of one particular peak at δ = 204.5 ppm, which is characteristic of the ketone structure in the repeating unit. Furthermore, no peak proving the existence of acetal (around 100 ppm) or ester structures (near 180 ppm) could be observed. The attribution of the other signals presented in this figure (δ = 12.1 ppm, 21.7 ppm and 66.0 ppm) correlated with the 1H signals (δ = 0.97 ppm, t, 3H), (δ = 1.84 ppm, m, 2H) and (δ = 4.37 ppm, t, 1H) was made possible, thanks to two-dimensional 1H-13C HSQC NMR. The complete assignment of the NMR signals provided in Fig. 3 clearly demonstrated that these syntheses only led to repeating units consistent with a polyketonic structure. The lack of broad signals ranging from 127 to 210 ppm in 13C NMR, as already observed in polyketene,40 and no signal around 15 ppm in 1H NMR (enol theoretical value), showed the absence of a keto-enolic equilibrium (that could occur between the carbonyl groups and their alpha proton), witness of a pure polyketone structure.
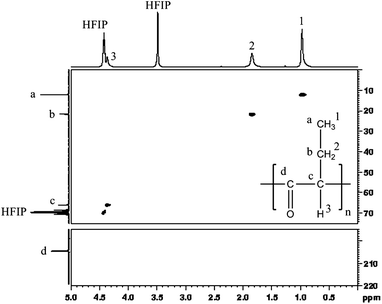 | ||
| Fig. 3 (1H-13C) HSQC NMR spectrum of ethylketene-based polyketone. | ||
Molecular weights (Mn) of the obtained polymers were then studied by steric exclusion chromatography in HFIP (Table 2). Mn ranged from 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300 to 28200 g mol−1. No relation with the polymerization yield could be found, but Mn increased when the dielectric constant of the solvent used decreased. Once again, we could suggest a preferential solvation of the active center and/or counter-ion by the monomer in low dielectric constant solvents, thus supporting the propagation mechanism and the formation of long polymer chains. The polydispersity index was very high, varying between 4.2 and 8.4. These values could be explained either by the heterogeneity of the polymerization medium,26 or by the existence of clusters even in a high dissociating solvent such as HFIP.
300 to 28200 g mol−1. No relation with the polymerization yield could be found, but Mn increased when the dielectric constant of the solvent used decreased. Once again, we could suggest a preferential solvation of the active center and/or counter-ion by the monomer in low dielectric constant solvents, thus supporting the propagation mechanism and the formation of long polymer chains. The polydispersity index was very high, varying between 4.2 and 8.4. These values could be explained either by the heterogeneity of the polymerization medium,26 or by the existence of clusters even in a high dissociating solvent such as HFIP.
| Run | Solvent | M n/g mol−1 | M w/Mn |
|---|---|---|---|
| 1 | Dichloromethane | 14300 |
8.4 |
| 2 | Heptane | 28200 |
4.8 |
| 3 | Toluene | 20600 |
7.8 |
| 4 | Ethyl acetate | 16300 |
4.2 |
The thermal characterization of these polymers was then performed by means of differential scanning calorimetry, thermogravimetric analysis and X-ray diffraction. Main results were reported in Table 3. These polymers were found stable until temperatures ranging from 206 to 214 °C, before they degraded quickly and totally. Tg ranging from 64 to 75 °C was observed, and no endothermic peak corresponding to a melting point could be detected. However, the X-ray diffraction study revealed that these polymers were crystalline, with a main peak at 10.8° and smaller peaks at 15.2, 18.8 and 21.6° corresponding to a 0.34 average crystallinity. It seemed that the fusion temperature was too close to the degradation temperature, so that DSC could not detect the fusion before these polymers degraded.
| Run | Glass transition temperature/°C | Degradation temperature/°C | Cristallinity |
|---|---|---|---|
| 1 | 72 | 209 | 0.40 |
| 2 | 64 | 214 | 0.35 |
| 3 | 72 | 210 | 0.33 |
| 4 | 75 | 206 | 0.28 |
Conclusions
In summary, we have described the synthesis of ethylketene by pyrolysis of butyric anhydride at 575 °C under 40 mbar, followed by its polymerization initiated by aluminium tribromide, in different solvents (dichloromethane, heptane, toluene and ethyl acetate). The best yield was 57% in toluene. The synthesized polymer was fully characterized and revealed to be only constituted of a polyketone structure. Its molecular weight ranged from 14300 to 28200 g mol−1, a solvent with a low dielectric constant favoring the propagation and giving a polymer with a higher Mn. With a glass transition temperature around 70 °C and an average crystallinity of 0.34, this polymer showed a starting degradation temperature around 210 °C. To conclude, the cationic polymerization of ketenes, obtained by pyrolysis of anhydrides, was found to be an effective way to access to new polyketones, unreachable by the classical terpolymerization of ethylene, propylene and carbon monoxide.
Acknowledgements
The authors acknowledge Arkema (SEC analysis in HFIP) and Dr Morgan Sanselme UPRES EA 3233 IMR (X-ray diffractometry) for their technical support. This work was financially supported by French Government (MESR fellowship).Notes and references
- E. Drent and P. H. M. Budzelaar, Chem. Rev., 1996, 96, 663 CrossRef CAS.
- A. Sommazzi and F. Garbassi, Prog. Polym. Sci., 1997, 22, 1547 CrossRef CAS.
- M. I. Abdel-Hamid, G. A.-W. Ahmed and R. M. Hassan, Eur. Polym. J., 2001, 37, 2201 CrossRef CAS.
- M. Brubaker, D. Coffman and H. Hoehn, J. Am. Chem. Soc., 1952, 74, 1509 CrossRef CAS.
- W. Reppe, A. Magin, US Pat., 2 577 208, 1951.
- E. Marklund, U. W. Gedde, M. S. Hedenqvist and G. Wiberg, Polymer, 2001, 42, 3153 CrossRef CAS.
- J. G. Bonner and A. K. Powell, Proceedings of the 213th ACS National Meeting, San Francisco, 1997.
- Z. Jiang, S. Sanganeria and A. Sen, J. Polym. Sci., Part A: Polym. Chem., 1994, 32, 841 CrossRef CAS.
- J. Chen, Y. Yeh and A. Sen, J. Chem. Soc., Chem. Commun., 1989, 14, 965 RSC.
- A. Sen, Z. Jiang and J. Chen, Macromolecules, 1989, 22, 2012 CrossRef CAS.
- Y. Zhang, A. A. Broekhuis, M. C. A. Stuart and F. Picchioni, J. Appl. Polym. Sci., 2008, 107, 262 CrossRef CAS.
- H. Staudinger, Chem. Ber., 1905, 38, 1735 CrossRef.
- T. T. Tidwell, in Ketenes, Wiley Interscience Publication, New York, 1995 Search PubMed.
- T. T. Tidwell, Eur. J. Org. Chem., 2006, 563 CrossRef CAS.
- R. Haener, T. Laube and D. Seebach, J. Am. Chem. Soc., 1985, 107, 5396 CrossRef CAS.
- Y. Ogata and K. Adachi, J. Org. Chem., 1982, 47, 1182 CrossRef CAS.
- D. N. Harpp, L. Q. Bao, C. J. Black, J. G. Gleason and R. A. Smith, J. Org. Chem., 1975, 40, 3420 CrossRef CAS.
- R. J. Clemens, Chem. Rev., 1986, 86, 241 CrossRef CAS.
- W. T. Brady, Tetrahedron, 1981, 37, 2949 CrossRef CAS.
- R. Oda, S. Munemiya and M. Okano, Makromol. Chem., 1961, 43, 149 CrossRef CAS.
- G. Pregaglia and G. Pozzi, La Chimica e l'Industria, 1963, 45, 160 CAS.
- P. Zarras and O. Vogl, Prog. Polym. Sci., 1991, 16, 173 CrossRef CAS.
- H. Staudinger, F. Felix, H. Harder, E. Geiger and P. Meyer, Helv. Chim. Acta, 1925, 8, 306 CrossRef CAS.
- Montecatini, Societa Generale Per L'Industria Mineraria E Chimica, GB Pat., 893 308, 1962.
- G. F. Pregaglia, G. Mazzanti and M. Binaghi, Makromol. Chem., 1961, 48, 234 CrossRef CAS.
- H. Egret, J. P. Couvercelle, J. Belleney and C. Bunel, Eur. Polym. J., 2002, 38, 1953 CrossRef CAS.
- P. R. Johnson, H. M. Barnes and S. M. McElvain, J. Am. Chem. Soc., 1940, 62, 964 CrossRef CAS.
- K. C. Khemani and F. Wudl, J. Am. Chem. Soc., 1989, 111, 9124 CrossRef CAS.
- K. Khemani, S. Askari and F. Wudl, Macromolecules, 1991, 24, 2156 CrossRef CAS.
- Y. Liu, C. E. Keller and C. U. Pittman, Jr, J. Polym. Sci., Part A: Polym. Chem., 1997, 35, 3707 CrossRef CAS.
- P. C. Zhu, Z. Wu and C. U. Pittman, Jr, J. Polym. Sci., Part A: Polym. Chem., 1997, 35, 485 CrossRef CAS.
- N. S. Murthy and H. Minor, Polymer, 1990, 31, 996 CrossRef CAS.
- R. H. Hasek, E. U. Elam, US Pat., 3 201 474, 1965.
- D. C. Hull, W. R. Saunders, US Pat., 2 541 471, 1951.
- T. E. Stanin, V. L. Brown, Jr, US Pat., 255 958, 1963.
- C. C. McCarney and R. S. Ward, J. Chem. Soc., Perkin Trans. 1, 1975, 1600 RSC.
- J. C. Sauer, J. Am. Chem. Soc., 1947, 69, 2444 CrossRef CAS.
- W. E. Hanpord and J. C. Sauer, Org. React., 1946, 3, 108 Search PubMed.
- N. Hayki, N. Desilles and F. Burel, Macromol. Chem. Phys., 2011, 112, 375 Search PubMed.
- G. A. Olah, E. Zadok, R. Edler, D. H. Adamson, W. Kasha and G. K. Surya Prakash, J. Am. Chem. Soc., 1989, 111, 9123 CrossRef CAS.
- S. Swarnalatha and G. Sekaran, J. Mol. Struct., 2007, 840, 90 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |