Controlled synthesis of branched poly(vinyl acetate)s by xanthate-mediated RAFT self-condensing vinyl (co)polymerization†
Julien
Schmitt
ab,
Nicolas
Blanchard
b and
Julien
Poly
*ac
aLaboratoire de Photochimie et d'Ingénierie Macromoléculaires, EA 4567, ENSCMu–UHA, 3 rue Alfred Werner, 68093, Mulhouse, France
bLaboratoire de Chimie Organique et Bioorganique, EA 4566, ENSCMu–UHA, 3 rue Alfred Werner, 68093, Mulhouse, France
cInstitut de Science des Matériaux de Mulhouse, LRC 7228, CNRS–UHA, 15 rue Jean Starcky, 68057, Mulhouse, France. E-mail: julien.poly@uha.fr
First published on 27th July 2011
Abstract
An original xanthate possessing a vinyl ester polymerizable function, namely vinyl 2-[(ethoxycarbonothioyl)sulfanyl]propanoate (Xa2), was synthesized. It was implemented as a chain transfer agent (CTA) to design branched polymers based on vinyl acetate (VAc) by self-condensing vinyl copolymerization (SCVC) by reversible addition–fragmentation chain transfer (RAFT). The branching density as well as the length of the branches were efficiently tuned by adjusting the total initial concentration of polymerizable functions C0 = [VAc]0 + [Xa2]0 and the ratio C0/[Xa2]0. Additionally, Xa2 was also homopolymerized to provide hyperbranched oligomers. These precursors were used as multifunctional CTAs to control a subsequent polymerization of VAc, affording starlike poly(vinyl acetate)s (PVAcs). All the products were characterized by 1H NMR spectroscopy, quadruple detection size exclusion chromatography and differential scanning calorimetry. Reference samples consisted of linear PVAcs which were synthesized using a homologue non-polymerizable xanthate. As expected, the intrinsic viscosity and the glass transition temperature increased when either the number of branches or their length increased.
Introduction
Due to its interesting rheological properties and as it is the main precursor of poly(vinyl alcohol), poly(vinyl acetate) is a widely used polymer. Its major applications concern water-based vinyl paints, adhesives and paper coatings.1 As for PVA, its chief uses are in paper and textile coatings, in adhesives or as a viscosifying agent.2 Partially hydrolyzed PVAc are also commonly used as stabilizing agents, for instance for vinyl chloride suspension polymerization.3 The physico-chemical properties of PVAcs and derived polymers, involved in the aforementioned fields of applications, could be finely tuned by structural modifications. Hence the interest in synthesizing well-defined branched PVAcs.Among the recent attempts to synthesize branched poly(vinyl acetate)s, one can cite the contributions of Rimmer et al. and Baudry and Sherrington, who resorted to a transfer to an allylic carbonate used as a comonomer4 and to crosslinking copolymerization in the presence of a thiol,5 respectively. However, the main disadvantages of these approaches are that they imply a free radical polymerization mechanism, causing irreversible terminations, and additional irreversible transfer reactions, hindering thus a fine control of the structures.
Keeping in mind that vinyl acetate can be polymerized only by chain-growth radical addition, the development of new mechanisms to reach a controlled polymerization of this monomer has been a very challenging area of investigations over the last decade. Several mechanisms have been developed to enable such a control: a reversible addition–fragmentation chain transfer (RAFT)6,7 using either a xanthate8–11 or a dithiocarbamate12,13 as the chain transfer agent (CTA), another degenerative transfer based on alkyl iodides14–16 as CTAs, and reversible terminations mediated by a cobalt complex,17–19 an organostibine20,21 or an iron complex.22 Among them, the xanthate-mediated RAFT polymerization, called also MADIX (for Macromolecular Design via Interchange of Xanthate) by its discoverers,23 has been the most studied.24,25 Thanks to the excellent control afforded, it has been successfully applied to the synthesis of various architectures in terms of composition26–32 or in terms of structure.36–39,44–46
Block copolymers were obtained for instance by sequential copolymerization of VAc with other vinyl esters26–28 or with N-vinylpyrrolidone.29 The functionalization of polyethyleneglycol30 or dextran31 by a xanthate afforded a macromolecular chain transfer agent which then mediated the polymerization of the VAc block. Diblocks based on VAc and styrene or methyl (meth)acrylate were also obtained using an iniferter bearing both xanthate and bromine functionalities32 and enabling thus sequential RAFT polymerization and atom transfer radical polymerization (ATRP).33–35
This xanthate-mediated RAFT polymerization was successfully applied to the synthesis of various branched architectures too. For instance, Stenzel et al. reported the use of multifunctional xanthates to synthesize star polymers.36,37 Whether the chain transfer agent bore several R or Z fragments, the formation of the star proceeded either by divergent or convergent growth of the arms, respectively. The same group described the preparation of poly(vinyl acetate) combs.38 A well-defined poly(vinyl alcohol), obtained by hydrolysis of a PVAc precursor synthesized by xanthate-mediated RAFT, was used as the backbone and decorated with xanthate functions for a subsequent growth. Z- and R-approach were both investigated, but well-defined combs were obtained only using the latter, as the convergent reversible transfer of chains onto the backbone was probably hampered by steric hindrance. The divergent approach was recently improved by Matyjaszewski et al., replacing the initial poly(vinyl alcohol) backbone by a poly(2-hydroxyethylmethacrylate) chain synthesized by ATRP, making thus the R fragments more accessible when generated, since they were further from the vicinity of the backbone.39
Another contribution to the design of branched PVAc by RAFT concerned the synthesis of nanogels by the so-called controlled radical crosslinking copolymerization (CRCC) methodology.40–43VAc was copolymerized with divinyl adipate, a crosslinker having a similar structure.44 The subsequent cleavage of the crosslinks put in evidence an efficient control of the primary chain length. It was possible to modulate the respective contributions of intermolecular or intramolecular crosslinking, which afforded products with various possible crosslink densities for a targeted molar mass.45 These polymers were used to prepare porous films, with a particular focus on the influence of their internal structure on the surface topography.46
In addition to the use of multifunctional CTA or CRCC, RAFT self-condensing vinyl (co)polymerization (SCVP/SCVC) constitutes another powerful strategy enabling the synthesis of well-defined branched architectures involving a RAFT mechanism, which has received surprisingly little coverage. The concept of SCVP was introduced in 1995 by Fréchet et al.47 It consists in the homopolymerization of a so-called inimer (contraction of initiator and monomer), a molecule bearing both an initiating and a polymerizable function, and enabling therefore the formation of hyperbranched polymers.48 Formation of branched polymers by copolymerization of an inimer with a comonomer is referred to as SCVC. In the case of RAFT SCVP/SCVC, the chain transfer agent possesses a polymerizable function. As for the aforementioned RAFT synthesis of star or comb polymers, two strategies can be implemented whether the polymerizable function belongs to the activating group Z or to the leaving group R. The first example of RAFT SCVC described the synthesis of branched polystyrenes, mediated by a dithiobenzoate bearing a pendant vinyl function on the Z group.49 The two main limitations of this Z-approach were the steric hindrance to access the CTA functionalities, inherent to this convergent growth, and the potential weakness of the branch points, ensured by these functions. These restrictions were later overcome by Heidenreich and Puskas, who used a similar CTA with the polymerizable function on the R group.50 Meanwhile, this R-approach was successfully applied by Rimmer et al. and Vogt and Sumerlin to synthesize branched polymers based on N-isopropylacrylamide using a dithioester with a vinylbenzyl R-group51,52 and a trithiocarbonate with an acryloyl R-group,53 respectively. More recently, another polymerizable trithiocarbonate bearing a styryl R-group was synthesised by Zhao et al. and investigated to synthesize branched polymers based on styrene, methyl (meth)acrylate and tert-butyl acrylate, or hyperbranched structures by RAFT SCVP followed by a subsequent chain-extension so as to afford starlike polymers.54
In this contribution, we report the synthesis of a xanthate designed for the RAFT SCVP/SCVC of VAc, namely vinyl 2-[(ethoxycarbonothioyl)sulfanyl]propanoate (Xa2). Its structure is very close to the one of methyl 2-[(ethoxycarbonothioyl)-sulfanyl]propanoate (Xa1), with in particular a similar R group structure, expected to permit an efficient re-initiation (Fig. 1). The Z group is unchanged. Xa2 differs from Xa1 only by the nature of the ester function of the R group, which is a vinyl ester. Therefore, Xa2 exhibits also a monomer behavior, with an excepted reactivity similar to the one of VAc.
![Synthesis of linear and branched poly(vinyl acetate)s by xanthate-mediated reversible addition–fragmentation chain transfer, using methyl 2-[(ethoxycarbonothioyl)sulfanyl]propanoate (Xa1) and vinyl 2-[(ethoxycarbonothioyl)sulfanyl]propanoate (Xa2) as chain-transfer agents, respectively. Re-initiating fragments in red. Xanthate functions including an ethoxy activating group in yellow. Polymerizable functions in white. AcOEt: ethyl acetate. AIBN: azobisisobutyronitrile.](/image/article/2011/PY/c1py00231g/c1py00231g-f1.gif) | ||
| Fig. 1 Synthesis of linear and branched poly(vinyl acetate)s by xanthate-mediated reversible addition–fragmentation chain transfer, using methyl 2-[(ethoxycarbonothioyl)sulfanyl]propanoate (Xa1) and vinyl 2-[(ethoxycarbonothioyl)sulfanyl]propanoate (Xa2) as chain-transfer agents, respectively. Re-initiating fragments in red. Xanthate functions including an ethoxy activating group in yellow. Polymerizable functions in white. AcOEt: ethyl acetate. AIBN: azobisisobutyronitrile. | ||
The synthesis of a xanthate bearing a polymerizable function was already reported.55–59 However, this xanthate was used as a photoiniferter, a way which offers a poor control compared to the RAFT mechanism, and bore a methacryloyl group. The monomers investigated were styrene and methyl methacrylate. Such a xanthate could not be used for the RAFT SCVP/SCVC of VAc, since the reactivity ratios of the methacrylate function with vinyl acetate would not allow a statistical copolymerization and thus a homogeneous distribution of the branch points. Moreover, xanthates are not convenient CTAs for the RAFT polymerization of methacrylic monomers.6,7
Therefore and to the best of our knowledge, this is the first report on a polymerizable xanthate especially designed for the one-step synthesis of well-defined branched PVAc by controlled radical branching polymerization.
Experimental section
Materials
Vinyl acetate (≥99%) and ethyl acetate (>99%) were purchased from Aldrich and Fisher Scientific, respectively, and distilled under reduced pressure over CaH2 prior to use. Azobisisobutyronitrile (AIBN, 98%) was purchased from Aldrich and recrystallized twice from ethanol prior to use. All the other reactants were used as received. 2-Bromopropionic acid (98%) was purchased from Alfa Aesar. Palladium(II) acetate (≥99.9%) and potassium ethyl xanthogenate (96%) were purchased from Aldrich. Methyl 2-[(ethoxycarbonothioyl)sulfanyl]propanoate (Xa1) was provided by Rhodia.Synthesis of vinyl 2-bromopropanoate
Palladium(II) acetate (0.588 g, 0.04 eq.), vinyl acetate (60.4 mL, 10.0 eq.) and 2-bromopropionic acid (10.0 g, 65.4 mmol) were successively introduced in a one-necked round-bottom flask, which was heated at 90 °C for 24 hours. The palladium catalyst was removed by filtration over a short silica chromatography column (eluent: pentane/diethyl ether 50/50 v/v). The organic solution was washed with an aqueous saturated potassium carbonate solution before being dried over anhydrous sodium sulfate. After filtration, it was then concentrated under reduced pressure. The crude product was finally purified by chromatography over a silica column (eluent: pentane/diethyl ether 95/5 v/v) and solvents were evaporated under reduced pressure to give vinyl 2-bromopropanoate (1.424 g, 12%). 1H NMR (CDCl3, 300 MHz), δ(ppm): 7.26 (1H, dd, J = 6.3 Hz, J = 13.8 Hz, CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2), 5.00 (1H, dd, J = 1.8 Hz, J = 13.8 Hz, CHCH2 (trans)), 4.69 (1H, dd, J = 1.8 Hz, J = 6.3 Hz, CHCH2 (cis)), 4.41 (1H, q, J = 6.9 Hz, CH(CH3)Br), 1.86 (3H, d, J = 6.9 Hz, CH3CHBr); 13C NMR (CDCl3), δ(ppm): 167.3 (CO2CHCH2), 141.1 (CH2CH), 99.2 (CH2CH), 39.0 (CH(CH3)Br), 21.3 (CH3CHBr).
CH2), 5.00 (1H, dd, J = 1.8 Hz, J = 13.8 Hz, CHCH2 (trans)), 4.69 (1H, dd, J = 1.8 Hz, J = 6.3 Hz, CHCH2 (cis)), 4.41 (1H, q, J = 6.9 Hz, CH(CH3)Br), 1.86 (3H, d, J = 6.9 Hz, CH3CHBr); 13C NMR (CDCl3), δ(ppm): 167.3 (CO2CHCH2), 141.1 (CH2CH), 99.2 (CH2CH), 39.0 (CH(CH3)Br), 21.3 (CH3CHBr).
Synthesis of vinyl 2-[(ethoxycarbonothioyl)sulfanyl]propanoate
Potassium ethyl xanthogenate (0.986 g, 1.1 eq.), acetonitrile (10 mL) and vinyl 2-bromopropanoate (1.00 g, 5.59 mmol) were successively introduced in a one-necked round-bottom flask under stirring. The reaction was allowed to proceed for 4 h at 0 °C and then for 12 h at ambient temperature. After filtration and evaporation under reduced pressure, the product was dissolved in dichloromethane. The organic phase was washed three times with water and then dried over anhydrous sodium sulfate. Vinyl 2-[(ethoxycarbonothioyl)sulfanyl]propanoate (0.598 g, 49%) was obtained after filtration and evaporation of the solvent under reduced pressure. 1H NMR (CDCl3), δ(ppm): 7.25 (1H, dd, J = 6.2 Hz, J = 13.9 Hz, CHCH2), 4.96 (1H, dd, J = 1.8 Hz, J = 13.9 Hz, CHCH2 (trans)), 4.66 (3H, m, CHCH2 (cis) and OCH2CH3), 4.42 (1H, q, J = 7.4 Hz, C(O)CH(CH3)S), 1.59 (3H, d, J7.4 Hz, C(O)CH(CH3)S), 1.39 (3H, t, J = 7,1 Hz, OCH2CH3); 13C NMR (CDCl3), δ(ppm): 211.0 (CH3CH2OC(S)S), 168.4 (CO2CHCH2), 141.0 (CH2CH), 98.5 (CH2CH), 70.2 (CH3CH2OC(S)S), 46.4 (CH3CHS), 16.2 (CH3CHS), 13.4 (CH3CH2OC(S)S).
Synthesis of linear poly(vinyl acetate)s
The molar concentration of vinyl functions was kept constant in all experiments: C0 = [VAc]0 = 2.50 mol kg−1. The xanthate used as chain transfer agent was Xa1, with a concentration [Xa1]0 equal to C0/50 or C0/100, depending on the targeted degree of polymerization (DP). The initiator concentration [AIBN]0 was equal to 50% × [Xa1]0. In a typical experiment (Table 1, entry 1), corresponding to [Xa1]0 = C0/50, 18.9 mg of AIBN (1.13 × 10−1 mmol), 48.1 mg of Xa1 (2.26 × 10−1 mmol), 970.7 mg of VAc (11.28 mmol) and ethyl acetate (5.00 mL) were added in this order into a flame-dried Schlenk flask. The solution was thoroughly deoxygenated by five freeze–pump–thaw cycles. The flask was filled with nitrogen gas and placed in an oil bath maintained at 80 °C for 24 h under magnetic stirring. Solvent was then evaporated under vacuum and the conversion α of vinyl functions was determined by gravimetry during this step: α = 98.3%. The polymer was then dissolved in a small volume of acetone, precipitated in a large excess of cyclohexane, collected after removal of the supernatant and dried under reduced pressure. The 1H NMR spectrum of this sample is given in the ESI†. SEC and DSC results for this sample are given in Table 1, entry 1.| Entry | CTA | C 0/mol kg−1 | [CTA]0/mol kg−1 | DP | α (%) | M n/g mol−1 | M w/g mol−1 | M w/Mn | N br | R h/nm | [η]/mL g−1 | T g/°C |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a CTA (chain transfer agent): xanthate used; Xa1: methyl 2-[(ethoxycarbonothioyl)sulfanyl]propanoate; Xa2: vinyl 2-[(ethoxycarbonothioyl)sulfanyl]-propanoate. C0: total initial concentration of vinyl functions. [CTA]0: initial CTA concentration. DP: theoretical degree of polymerization of the chains. DP is the degree of polymerization of the branches in the case of branched polymers. Mn: absolute number average molar mass. Mw: absolute mass average molar mass. Mw/Mn: polydispersity index. Rh: hydrodynamic radius. [η]: intrinsic viscosity. Mn, Mw, Rh and [η] determined by quadruple detection size exclusion chromatography. Nbr: average number of branch points per macromolecule. Nbr = Mw (branched polymer)/Mw (linear reference) − 1. Entries 1 and 2 are linear references for odd and even entries, respectively. Tg: glass transition temperature, determined by differential scanning calorimetry. b Conversion of the vinyl functions, determined by gravimetry for VAc. c Conversion of the vinyl functions, determined by 1H NMR for Xa2. | ||||||||||||
| 1 | Xa1 | 2.50 | 5.00 × 10−2 | 50 | 98.3b | 6.18 × 103 | 7.12 × 103 | 1.15 | 0 | 1.7 | 4.7 | 20.2 |
| 2 | Xa1 | 2.50 | 2.50 × 10−2 | 100 | 98.6b | 1.02 × 104 | 1.28 × 104 | 1.25 | 0 | 2.3 | 6.7 | 22.0 |
| 3 | Xa2 | 2.50 | 5.00 × 10−2 | 50 | 93.7b | 1.27 × 104 | 2.27 × 104 | 1.78 | 2.2 | 2.9 | 7.8 | 29.9 |
| 4 | Xa2 | 2.50 | 2.50 × 10−2 | 100 | 90.5b | 1.64 × 104 | 2.80 × 104 | 1.71 | 1.2 | 3.4 | 10.0 | 30.9 |
| 5 | Xa2 | 5.00 | 1.00 × 10−1 | 50 | 97.4b | 1.14 × 104 | 3.73 × 104 | 3.27 | 4.2 | 3.7 | 11.1 | 31.9 |
| 6 | Xa2 | 5.00 | 5.00 × 10−2 | 100 | 99.5b | 2.18 × 104 | 5.75 × 104 | 2.64 | 3.5 | 4.6 | 13.8 | 33.0 |
| 7 | Xa2 | 7.50 | 1.50 × 10−1 | 50 | 99.7b | 1.29 × 104 | 5.91 × 104 | 4.59 | 7.3 | 4.3 | 11.6 | 32.1 |
| 8 | Xa2 | 7.50 | 7.50 × 10−2 | 100 | 99.3b | 2.52 × 104 | 8.24 × 104 | 3.27 | 5.4 | 5.4 | 15.9 | 35.0 |
| 9 | Xa2 | 2.50 | 2.50 | 1 | 84.5c | 8.4 × 102 | 1.5 × 103 | 1.8 | — | 0.7 | 2.0 | — |
| 10 | Xa2 | 2.50 | 5.00 × 10−1 | 5 | 61.1b/100c | 9.0 × 102 | 1.7 × 103 | 1.9 | — | 0.8 | 2.3 | — |
| 11 | 9 | 2.50 | 5.00 × 10−2 | 50 | 99.8b | 1.12 × 104 | 2.00 × 104 | 1.78 | 2.8 | 3.1 | 10.3 | 34.8 |
| 12 | 10 | 2.50 | 5.00 × 10−2 | 50 | 99.8b | 1.12 × 104 | 1.91 × 104 | 1.70 | 2.7 | 3.1 | 10.8 | 33.5 |
Synthesis of branched poly(vinyl acetate)s
The xanthate used as the chain transfer agent was Xa2. The vinyl functions belonging to Xa2 were taken into account in the total molar concentration of vinyl functions: C0 = [VAc]0 + [Xa2]0. Three values of C0 were considered: 2.50 mol kg−1, 5.00 mol kg−1 and 7.50 mol kg−1. [Xa2]0 was equal to C0/50 or C0/100, depending on the targeted degree of polymerization for the branches. The initiator concentration [AIBN]0 was equal to 50% × [Xa2]0 for C0 = 2.50 mol kg−1 and to 25% × [Xa2]0 for C0 = 5.00 mol kg−1 and C0 = 7.50 mol kg−1. The experimental procedure was the same as the one described for linear polymers. In a typical experiment (Table 1, entry 5), corresponding to C0 = 5.00 mol kg−1 and [Xa2]0 = C0/50, 11.1 mg of AIBN (1.13 × 10−1 mmol), 59.6 mg of Xa2 (2.71 × 10−1 mmol), 1.144 g of VAc (13.26 mmol) and ethyl acetate (3.00 mL) were added in this order into the Schlenk flask. The conversion of VAc vinyl functions determined by gravimetry was: α = 97.4%. The 1H NMR spectrum of this sample is given in the ESI†. SEC and DSC results for this sample are given in Table 1, entry 5.Homopolymerization of Xa2
The experimental procedure is the same as the one described for branched polymers. In a typical synthesis (Table 1, entry 9), the experimental parameters were C0 = [Xa2]0 = 2.50 mol kg−1 and [AIBN]0 = 25% × [Xa2]0. 92.6 mg of AIBN (5.64 × 10−1 mmol), 497 mg of Xa2 (2.26 mmol) and ethyl acetate (1.00 mL) were added in this order into the Schlenk flask. After evaporation of the solvent under reduced pressure, a syrupy yellow product was obtained. The conversion of vinyl functions was determined by 1H NMR: α = 84.5%.Chain extension experiment
The hyperbranched PVAc obtained by homopolymerization of Xa2 was considered as a multifunctional xanthate for a subsequent chain extension experiment, leading to starlike VAc-based macromolecular polymers. In a typical experiment (Table 1, entry 11), 18.5 mg of AIBN (1.13 × 10−1 mmol), 47.0 mg of the multifunctional hyperbranched xanthate precursor (corresponding to 2.26 × 10−1 mmol of xanthate functions), 951 mg of VAc (11.0 mmol) and ethyl acetate (5.00 mL) were added in this order into the Schlenk flask. The conversion of vinyl functions determined by gravimetry was: α = 99.8%.Characterization
Compositions and conversions were determined by means of 1H NMR spectroscopy in deuterated chloroform on a Bruker AC-300 spectrometer. After purification by precipitation in cyclohexane, the polymers were dissolved in THF (concentrations of 10.0 mg mL−1; except for samples corresponding to entries 1, 9 and 10 in Table 1: 20.0 mg mL−1) and filtered with a PTFE membrane (0.45 μm). The samples were subsequently analyzed by means of size exclusion chromatography (SEC) in THF at a flow rate of 1.0 mL min−1 using the following setup: a Shimadzu LC-20AD pump, a Shimadzu SIL-20A autosampler and a set of Polymer Laboratories columns in a Shimadzu CTO-20A oven at 30 °C. The columns were composed of a PLgel 10 μm Guard (50 × 7.5 mm) column and either two PLgel 5 μm MIXED-C (300 × 7.5 mm) columns (particle size: 5 μm) or a PLgel 10 μm 100A (300 × 7.5 mm) column (particle size: 10 μm; pore type: 10 nm) with a PLgel 10 μm MIXED-B LS (300 × 7.5 mm) column (particle size: 10 μm) for samples corresponding to entries 1 to 8 or for entries 9 to 12 in Table 1, respectively. Four detectors were used in the present study: a Shimadzu RID-10A refractive index detector, a Shimadzu SPD-20A UV/visible detector at 290 nm and a Viscotek 270 Dual Detector (light scattering and viscosimetry). Molar masses, intrinsic viscosity and hydrodynamic radii were determined by the multi-detection method using OmniSEC Viscotek software (version 4.6.1).Differential scanning calorimetry (DSC) studies were performed on a DSC Q200 TA instrument. For the samples, the temperature range was −20 °C to 80 °C. The heating and cooling rates were 10 °C min−1. The glass transition temperatures were determined during the second heating run, at the tipping point of the heat flow curve.
Results and discussion
Two linear PVAc samples were synthesized with Xa1 as references, following a previously described procedure.44 This route was then adapted for the synthesis of branched polymers using Xa2, which was considered both as a chain transfer agent and as a comonomer of VAc. The efficiency of branching was evidenced by the determination of absolute molar masses and glass transition temperatures (Tg) by SEC and DSC, respectively, and their comparison with the ones of linear references (Table 1). Additionally, Xa2 was also homopolymerized to afford hyperbranched structures, the xanthate functionalities of which were brought into play to synthesize starlike polymers by a subsequent chain-extension with VAc.Investigation of the effect of the nature of the xanthate
When Xa1 was replaced by Xa2 using comparable experimental conditions, higher molar masses and larger polydispersities were observed (Fig. 2a and Table 1, entries 1 to 4). For instance Mw increased in a ratio of 3.2 and 2.2 for targeted chain length of 50 and 100, respectively. While narrow polydispersities were obtained for linear samples prepared from Xa1 (1.15 and 1.25 for DP = 50 and 100, respectively), Xa2 led to broader molar mass distributions. This observation is consistent with the formation of branch points, as in the case of branched polymers the dispersity resulting from chain lengths is increased due to the possible variation of the number of branch points per molecule and their position along the chains. The Tg values measured by DSC support these structural differences too, with an increase around 9–10 °C when the PVAc synthesized using Xa2 is compared with its linear homologue. This higher Tg can be ascribed to the relative motion of chains which is not only limited by entanglements but also by supplementary branch points. Additionally, the presence of xanthate functions in the polymer is evidenced by the good overlap observed by SEC between the RI trace and the UV trace at 290 nm, a wavelength characteristic of these functions (Fig. 2b). Xa2 is therefore efficiently incorporated in the polymer. This consumption is assumed to occur mainly at the early stage of the reaction, during the transfer reaction of primary radicals or oligoradicals to the CTA.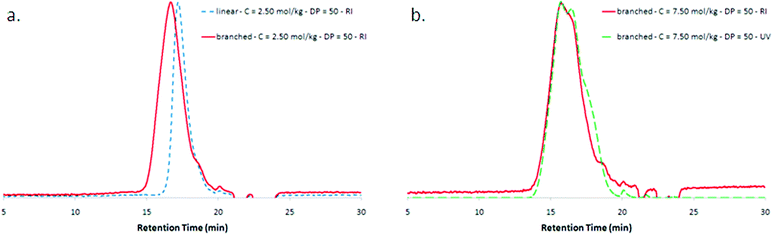 | ||
| Fig. 2 Size exclusion chromatograms of poly(vinyl acetate)s in tetrahydrofuran. (a) Overlap of the RI signals for a branched PVAc and a linear homologue (Table 1, entries 1 and 3). (b) Evidence of the presence of xanthate chain end functionalities for a branched PVAc (Table 1, entry 7): overlap between RI chromatogram and UV chromatogram at 290 nm. | ||
Investigation of the effect of the ratio C0/[Xa2]0
As most of the chains are initiated by the leaving R fragment of the CTA in a RAFT polymerization, their length is directly governed by the ratio C0/[CTA]0; for linear polymers at total conversion, the degree of polymerization is equal to:with [I]0 and f being the initial concentration of the initiator and its efficiency, respectively. f[I]0 can be neglected compared to [CTA]0 either because [I]0 ≪ [CTA]0 or because of a low efficiency. A decrease of [CTA]0 for a fixed value of C0 is hence expected to result in an increase of the degree of polymerization and therefore of molar masses. This effect is observed in the present study for linear as well as for branched polymers (Table 1 and Fig. 3). The glass transition temperature (Tg) is also accordingly impacted by the variation of this ratio, due to more entanglements between chains when DP increases (Table 1).

![Size exclusion chromatograms of poly(vinyl acetate)s in tetrahydrofuran: effect of the ratio C0/[CTA]0.(a) For linear polymers (Table 1, entries 1 and 2). (b) For branched polymers (Table 1, entries 5 and 6).](/image/article/2011/PY/c1py00231g/c1py00231g-f3.gif) | ||
| Fig. 3 Size exclusion chromatograms of poly(vinyl acetate)s in tetrahydrofuran: effect of the ratio C0/[CTA]0.(a) For linear polymers (Table 1, entries 1 and 2). (b) For branched polymers (Table 1, entries 5 and 6). | ||
However, in the particular case of branched PVAc, it is important to note that the ratio C0/[Xa2]0 does not correspond to the overall targeted degree of polymerization, but to the one of the chains constitutive of the structure (the “branches”). Indeed, the total molar mass of a branched polymer depends both on the molar mass and the number of its constitutive branches.
It is therefore possible to make the molar mass of a branched polymer increase either by adding supplementary branches or by lengthening them. In the present study, the polymerizable xanthate is responsible both for the creation of branch points and for the control of the length of the branches. For a fixed value of C0, adjusting [Xa2]0 can thus result in two antagonistic effects. While reducing [Xa2]0 causes the ratio C0/[Xa2]0 and hence the length of the branches to increase, it also reduces the fraction of Xa2 as the branching comonomer and hence the number of branches. It is therefore necessary to adjust independently another parameter, such as C0, in order to enhance both the branching density and the degree of polymerization of the PVAc branched polymers derived from Xa2.
Investigation of the effect of C0
In a RAFT polymerization, most of the chains are initiated by the leaving fragment R of the CTA. In the peculiar case of Xa2, this fragment possesses a vinyl group, the consumption of which results in the formation of branch points. Consequently, a typical branched PVAc synthesized in this work will carry on its main chain such a function, which constitutes the focal point of the structure. For a fixed C0/[Xa2]0 ratio, an increase of C0 is thus expected to improve the consumption of these focal vinyl groups, thanks to their higher concentration conjugated with more concentrated active centers. This is experimentally confirmed with a value of Mw being multiplied by a factor up to 3 when C0 is increased from 2.50 mol kg−1 to 7.50 mol kg−1 for DP = 50 or DP = 100 (Table 1, entries 3 to 8 and Fig. 4). For each series, this Mw increase resulting from a higher number of branches per macromolecule goes with a broader polydispersity, which is consistent with a branched structure. The increase of the number of branches per macromolecule when C0 is increased is also supported by DSC measurements, putting in evidence higher Tg values, consistent with more intermolecular entanglements.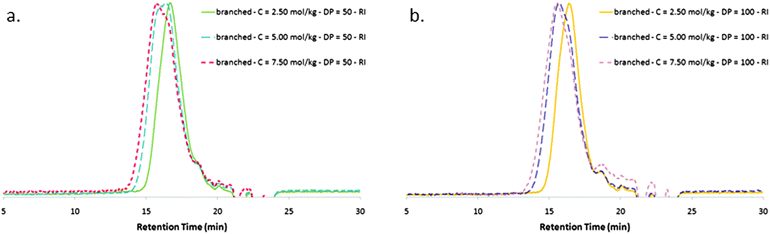 | ||
| Fig. 4 Size exclusion chromatograms of poly(vinyl acetate)s in tetrahydrofuran: effect of C0 for branched polymers. (a) Branched PVAcs with branches having a targeted degree of polymerization of 50 (Table 1, entries 3, 5 and 7). (b) Branched PVAcs with branches having a targeted degree of polymerization of 100 (Table 1, entries 4, 6 and 8). | ||
Investigation of Xa2 branching homopolymerization
Xa2 was also homopolymerized so as to synthesize a hyperbranched oligomer (Fig. 5a and Table 1, entry 9). The overlap of RI and UV at 290 nm SEC traces confirms the presence of xanthate functions in the product, whatever the molar mass fraction (Fig. 6a). However, the presence of a neat peak corresponding to a low molar mass fraction suggests that some vinyl functions of Xa2 are not consumed. Their conversion was indeed estimated to be 84.5% by 1H NMR spectroscopy. This incomplete conversion may be due to the vicinity of xanthate functions in the hyperbranched structure, prone to favor more irreversible termination reactions between active chain ends and thus to lower the concentration of active species, leading to lower conversions. This assumption was supported by a complementary synthesis, in which Xa2 was copolymerized with VAc (Xa2/VAc: 1/4; Table 1, entry 10) to afford a highly branched PVAc. In this case, the lower local concentration of xanthate functions should limit the frequency of termination reactions and thus improve the consumption of vinyl functions. Almost no residual Xa2 peak was observed by SEC (Fig. 6b) and no signal from unreacted vinyl groups was detected by 1H NMR, after evaporation of residual VAc. This hyperbranched poly(Xa2) and this highly branched poly(Xa2-co-VAc) were then used as multifunctional CTAs for the synthesis of starlike VAc-based polymers (Fig. 5b). In both cases, PVAcs with relatively narrow molar mass distributions were obtained (Table 1, entries 11 and 12), with SEC traces shifting significantly towards higher molar masses (Fig. 6c), supporting the chain extension of the precursor. Xanthate chain-end functions are still present in the resulting starlike polymers, as evidenced by the overlap between RI and UV signals (Fig. 6d). The low molar mass shoulder, which is richer in xanthate functions (UV signal more intense for this fraction), is ascribable to residual multifunctional CTA precursor. Interestingly, the hyperbranched and highly branched precursors afforded starlike PVAcs having the same characteristics. As the quantities of branched CTA precursors brought into play were equivalent in terms of xanthate functions, this means that all the xanthate functions are available for a subsequent chain-extension experiment, whatever the density of the branched precursor. It can be suggested that this observation, consistent with a divergent star synthesis (multifunctional R-core), would have been different in the case of a convergent approach (multifunctional Z-core), due to an access to xanthate functions all the more restricted by steric hindrance as the core is dense. Finally, we can also remark that the overall composition of the starlike polymer obtained from the hyperbranched poly(Xa2) precursor is the same as the one of an aforementioned branched PVAc (Table 1, entries 11 and 3, respectively). The only variation is the procedure: batch or sequential for the branched or the starlike PVAcs, respectively. The characterizations support the expected structural differences between them, as the higher viscosity and Tg were measured for the starlike polymer, consistent with more intermolecular entanglements.![(a) Synthesis of a hyperbranched oligomer from vinyl 2-[(ethoxycarbonothioyl)sulfanyl]propanoate (Xa2). (b) Subsequent use as a multifunctional xanthate for the synthesis of a starlike polymer. Re-initiating fragments in red. Xanthate functions including an ethoxy activating group in yellow. Polymerizable functions in white. AcOEt: ethyl acetate. AIBN: azobisisobutyronitrile.](/image/article/2011/PY/c1py00231g/c1py00231g-f5.gif) | ||
| Fig. 5 (a) Synthesis of a hyperbranched oligomer from vinyl 2-[(ethoxycarbonothioyl)sulfanyl]propanoate (Xa2). (b) Subsequent use as a multifunctional xanthate for the synthesis of a starlike polymer. Re-initiating fragments in red. Xanthate functions including an ethoxy activating group in yellow. Polymerizable functions in white. AcOEt: ethyl acetate. AIBN: azobisisobutyronitrile. | ||
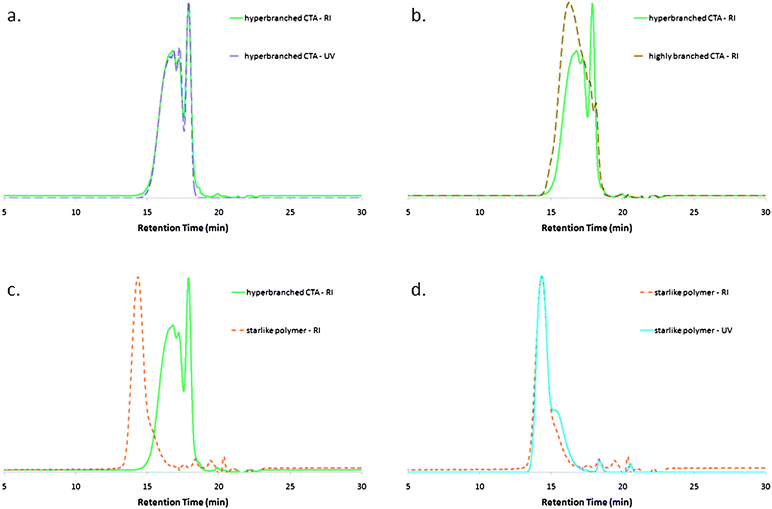 | ||
| Fig. 6 Size exclusion chromatograms in tetrahydrofuran. (a) Hyperbranched chain-transfer agent obtained from Xa2 homopolymerization (Table 1, entry 9): overlap between RI chromatogram and UV chromatogram at 290 nm. (b) Hyperbranched and highly branched chain-transfer agents obtained from Xa2 homopolymerization and copolymerization with VAc (Table 1, entries 9 and 10): RI chromatograms. (c) Starlike polymer resulting from a chain-extension experiment (Table 1, entries 9 and 11): RI chromatograms. (d) Starlike polymer resulting from a chain-extension experiment (Table 1, entry 11): overlap between RI chromatogram and UV chromatogram at 290 nm. | ||
It would have been interesting to compare experimental results with theoretical predictions. Several statistical or kinetic theories of SCVP were indeed developed.60 They are not strictly suitable yet to RAFT SCVP/SCVC because for this mechanism, the branching agent is not an inimer but a “CTAmer”, involving in particular the use of an additional initiator. The development of a refined modelling taking into account these specificities would have gone beyond the scope of this contribution.
Conclusions
Branched PVAcs were successfully synthesized by controlled radical branching polymerization based on a RAFT mechanism, using a polymerizable xanthate. These macromolecular architectures can be described by the number of their branch points and the length of their branches. It was possible to control these structural features by adjusting either the concentration of polymerizable functions C0 or the ratio C0/[Xa2]0. The latter impacts both the length of the branches and the number of branch points per macromolecule: an increase of C0/[Xa2]0 for a fixed value of C0 results on one hand in longer branches, favoring the increase of molar masses, but reduces on the other hand the number of branch points, formed by polymerized Xa2 units, which tends to limit the number of branches. Due to these antagonistic effects, it is necessary to resort to another parameter, such as C0, so as to be able to fix separately the number of branches and their length. The physico-chemical properties of the branched PVAcs varied consistently with the targeted macromolecular architecture, with in particular higher [η] or Tg values for structures with higher numbers of branch points or longer branches. Additionally, Xa2 was also homopolymerized to afford hyperbranched oligomers, which were subsequently used as multifunctional CTA for the divergent synthesis of starlike PVAcs following a chain-extension procedure. It could be interesting to study in detail in a future contribution the rheological behavior of these branched polymers, in comparison with other PVAc-based structures synthesized by xanthate-mediated RAFT polymerization, such as combs or nanogels. We will also extend this methodology to the synthesis of branched polymers of interest, based for instance on acrylamide or on N-vinylpyrrolidone.Notes and references
- W. E. Daniels, in Encyclopedia of Polymer Science and Engineering, ed. H. F. Mark, N. M. Bikales, C. G. Overberger and G. Menges, Wiley, 2nd edn, 1989, vol. 17, p. 393 Search PubMed.
- Polyvinyl Alcohol—Developments, ed. C. A. Finch, Wiley, 2nd edn, 1992 Search PubMed.
- Y. Saeki and T. Emura, Prog. Polym. Sci., 2002, 27, 2055–2131 CrossRef CAS.
- S. Rimmer, S. Collins and P. Sarker, Chem. Commun., 2005, 6029–6031 RSC.
- R. Baudry and D. C. Sherrington, Macromolecules, 2006, 39, 5230–5237 CrossRef CAS.
- J. Chiefari, Y. K. B. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- E. Rizzardo, G. Moad and S. H. Thang, in Handbook of RAFT Polymerization, ed. C. Barner-Kowollik, Wiley, 2008, p. 189 Search PubMed.
- M. H. Stenzel, L. Cummins, G. E. Roberts, T. P. Davis, P. Vana and C. Barner-Kowollik, Macromol. Chem. Phys., 2003, 204, 1160–1168 CrossRef CAS.
- A. Favier, C. Barner-Kowollik and T. P. Davis, Macromol. Chem. Phys., 2004, 205, 925–936 CrossRef CAS.
- R. W. Simms, T. P. Davis and M. F. Cunnigham, Macromol. Rapid Commun., 2005, 26, 592–596 CrossRef CAS.
- A. Theis, T. P. Davis, M. H. Stenzel and C. Barner-Kowollik, Polymer, 2006, 47, 999–1010 CrossRef CAS.
- M. Destarac, D. Charmot, X. Franck and S. Z. Zard, Macromol. Rapid Commun., 2000, 21, 1035–1039 CrossRef CAS.
- E. Rizzardo, J. Chiefari, R. T. A. Mayadunne, G. Moad and S. H. Thang, in Controlled/Living Radical Polymerization, ed. K. Matyjaszewski, American Chemical Society, 2000, vol. 768, p. 278 Search PubMed.
- M. C. Iovu and K. Matyjaszewski, Macromolecules, 2003, 36, 9346–9354 CrossRef CAS.
- K. Koumura, K. Satoh, M. Kamigaito and Y. Okamoto, Macromolecules, 2006, 39, 4054–4061 CrossRef CAS.
- J. Tonnar, E. Pouget, P. Lacroix-Desmazes and B. Boutevin, Eur. Polym. J., 2008, 44, 318–328 CrossRef CAS.
- A. Debuigne, J.-R. Caille and R. Jerome, Angew. Chem., Int. Ed., 2005, 1101–1104 CrossRef CAS.
- R. Bryaskova, C. Detrembleur, A. Debuigne and R. Jerome, Macromolecules, 2006, 39, 8263–8268 CrossRef CAS.
- A. Debuigne, N. Willet, R. Jerome and C. Detrembleur, Macromolecules, 2007, 40, 7111–7118 CrossRef CAS.
- S. Yamago, B. Ray, K. Iida, J.-I. Yoshida, T. Tada, K. Yoshizawa, Y. Kwak, A. Goto and T. Fukuda, J. Am. Chem. Soc., 2004, 126, 13908–13909 CrossRef CAS.
- S. Yamago, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 1–12 CrossRef CAS.
- M. Wakioka, K.-Y. Baek, T. Ando, M. Kamigaito and M. Sawamoto, Macromolecules, 2001, 35, 330–333 CrossRef.
- D. Charmot, P. Corpart, H. Adam, S. Z. Zard, T. Biadatti and G. Bouhadir, Macromol. Symp., 2000, 150, 23–32 CrossRef CAS.
- M. Destarac, D. Taton, S. Z. Zard, T. Saleh and Y. Six, in Advances in Controlled/Living radical polymerization, ed. K. Matyjaszewski, American Chemical Society, 2003, vol. 854, p. 536 Search PubMed.
- D. Taton, M. Destarac and S. Z. Zard, in Handbook of RAFT Polymerization, ed. C. Barner-Kowollik, Wiley, 2008, p. 373 Search PubMed.
- C. E. Lipscomb and M. K. Mahanthappa, Macromolecules, 2009, 42, 4571–4579 CrossRef CAS.
- Y. Gu, J. He, C. Li, C. Zhou, S. Song and Y. Yang, Macromolecules, 2010, 43, 4500–4510 CrossRef CAS.
- M. H. Repollet-Pedrosa, R. L. Weber, A. L. Schmitt and M. K. Mahanthappa, Macromolecules, 2010, 43, 7900–7902 CrossRef CAS.
- T. L. U. Nguyen, K. Eagles, T. P. Davis, C. Barner-Kowollik and M. H. Stenzel, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 4372–4383 CrossRef CAS.
- G. Pound, F. Aguesse, J. B. McLeary, R. F. M. Lange and B. Klumperman, Macromolecules, 2007, 40, 8861–8871 CrossRef CAS.
- J. Bernard, M. Save, B. Arathoon and B. Charleux, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 2845–2857 CrossRef CAS.
- R. Nicolay, Y. Kwak and K. Matyjaszewski, Chem. Commun., 2008, 5336–5338 RSC.
- M. Kato, M. Kamigaito, M. Sawamoto and T. Higashimura, Macromolecules, 1995, 28, 1721–1723 CrossRef CAS.
- J.-S. Wang and K. Matyjaszewski, J. Am. Chem. Soc., 1995, 117, 5614–5615 CrossRef CAS.
- N. V. Tsarevsky, W. A. Braunecker, W. Tang and K. Matyjaszewski, in Controlled/Living radical polymerization: Progress in ATRP, ed. K. Matyjaszewski, American Chemical Society, 2009, vol. 1023, p. 85 Search PubMed.
- M. H. Stenzel, T. P. Davis and C. Barner-Kowollik, Chem. Commun., 2004, 1546–1547 RSC.
- J. Bernard, A. Favier, L. Zhang, A. Nilasaroya, T. P. Davis, C. Barner-Kowollik and M. H. Stenzel, Macromolecules, 2005, 38, 5475–5484 CrossRef CAS.
- J. Bernard, A. Favier, T. P. Davis, C. Barner-Kowollik and M. H. Stenzel, Polymer, 2006, 47, 1073–1080 CrossRef CAS.
- A. Nese, Y. Kwak, R. Nicolay, M. Barrett, S. S. Sheiko and K. Matyjaszewski, Macromolecules, 2010, 43, 4016–4019 CrossRef CAS.
- D. Taton, in Macromolecular Engineering, ed. K. Matyjaszewski, L. Leibler and Y. Gnanou, Wiley, 2007, vol. 2, p. 1007 Search PubMed.
- H. Gao and K. Matyjaszewski, Prog. Polym. Sci., 2009, 34, 317–350 CrossRef CAS.
- N. Sanson and J. Rieger, Polym. Chem., 2010, 1, 965–977 RSC.
- J. Poly, E. Ibarboure, J. Rodriguez-Hernandez, D. Taton and E. Papon, Macromolecules, 2010, 43, 1299–1308 CrossRef CAS.
- J. Poly, D. J. Wilson, M. Destarac and D. Taton, Macromol. Rapid Commun., 2008, 29, 1965–1972 CrossRef CAS.
- J. Poly, D. J. Wilson, M. Destarac and D. Taton, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 5313–5327 CrossRef CAS.
- J. Poly, E. Ibarboure, J.-F. Le Meins, J. Rodriguez-Hernandez, D. Taton and E. Papon, Langmuir, 2011, 27, 4290–4295 CrossRef CAS.
- J. M. J. Fréchet, M. Henmi, I. Gitsov, S. Aoshima, M. R. Leduc and R. B. Grubbs, Science, 1995, 269, 1080–1083 Search PubMed.
- H. Mori, A. H. E. Müller and P. F. W. Simon, in Macromolecular Engineering, ed. K. Matyjaszewski, L. Leibler and Y. Gnanou, Wiley, 2007, vol. 2, p. 973 Search PubMed.
- Z. Wang, J. He, Y. Tao, L. Yang, H. Jiang and Y. Yang, Macromolecules, 2003, 36, 7446–7452 CrossRef CAS.
- A. J. Heidenreich and J. E. Puskas, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7621–7627 CrossRef CAS.
- S. Carter, B. Hunt and S. Rimmer, Macromolecules, 2005, 38, 4595–4603 CrossRef CAS.
- S. Rimmer, S. Carter, R. Rutkaite, J. W. Haycock and L. Swanson, Soft Matter, 2007, 3, 971–973 RSC.
- A. P. Vogt and B. S. Sumerlin, Macromolecules, 2008, 41, 7368–7373 CrossRef CAS.
- C. Zhang, Y. Zhou, Q. Liu, S. Li, S. Perrier and Y. Zhao, Macromolecules, 2011, 44, 2034–2049 CrossRef CAS.
- R. Francis and A. Ajayaghosh, Polymer, 1995, 36, 1091–1096 CrossRef CAS.
- A. Ajayaghosh, Polymer, 1995, 36, 2049–2053 CrossRef CAS.
- A. Ajayaghosh and R. Francis, Macromolecules, 1998, 31, 1436–1438 CrossRef CAS.
- A. Ajayaghosh and R. Francis, J. Am. Chem. Soc., 1999, 121, 6599–6606 CrossRef CAS.
- R. Francis and A. Ajayaghosh, Macromolecules, 2000, 33, 4699–4704 CrossRef CAS.
- Z.-F. Zhao, H.-J. Wang and X.-W. Ba, J. Chem. Phys., 2009, 131, 074101 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Typical 1H NMR spectra with peak assignments. See DOI: 10.1039/c1py00231g |
| This journal is © The Royal Society of Chemistry 2011 |