Influence of macromolecular characteristics of RAFT/MADIX poly(vinyl acetate)-based (co)polymers on their solubility in supercritical carbon dioxide†
Etienne
Girard
ab,
Thierry
Tassaing
c,
Jean-Daniel
Marty
*b and
Mathias
Destarac
*a
aLHFA, UMR 5069, Université de Toulouse, 118, route de Narbonne, F-31062, Toulouse, Cedex 9, France. E-mail: destarac@chimie.ups-tlse.fr
bIMRCP, UMR 5623, Université de Toulouse, 118, route de Narbonne, F-31062, Toulouse, Cedex 9, France. E-mail: marty@chimie.ups-tlse.fr
cISM, UMR 5255 CNRS- Université Bordeaux 1, 351 cours de la Libération, 33405, Talence Cedex, France
First published on 20th July 2011
Abstract
We investigated the structure–property relationships between poly(vinyl acetate)-based (co)polymers and their solubility in supercritical carbon dioxide. Building on RAFT/MADIX polymerization, key macromolecular characteristics—chain length, chain-end group and composition—of these (co)polymers were studied. Their solubility in sc-CO2 was determined by high-pressure infrared spectroscopy, thereby providing guidelines for their design.
Introduction
With the enforcement of new environmental protocols and regulations such as the Montreal Protocol and the Registration, Evaluation Authorization and restriction of Chemicals (REACH), the substitution of hazardous chemicals, intensification of chemical processes and reduction in volatile organic compounds (VOCs) have been promoted. Thus, the replacement of hazardous organic solvents by “greener” solvents exhibiting less toxicity and inflammability—like water, ionic liquids and supercritical fluids (SCFs)—is of notable interest.1 In particular, carbon dioxide is a very promising alternative. Comparatively to other SCFs, its supercritical state (sc-CO2) is easily accessible since its critical point is located at 31.1 °C and 7.38 MPa. Using sc-CO2 as a reaction medium in chemical processes offers many advantages such as complete elimination of the solvent traces via a simple depressurization. The latter particularity has been successfully exploited in carbon-based technologies dedicated to the extractions of aromas in the food industry and the purification of pharmaceutical actives.However, carbon dioxide presents certain physical limitations which have hampered the expansion of CO2-based processes in chemical synthesis. It is a weak solvent compared to usual organic solvents, being non-polar with both low cohesion energy and weak dielectric constant. Thus, solubilization in liquid or sc-CO2 has been mostly limited to low molar mass molecules or specific polymers bearing fluorinated and siloxane moieties. Moreover, low solubility of most hydrophilic compounds and polymers in sc-CO2 rules out many potential applications. In the past decade, several approaches, like addition of polar co-solvents such as alcohols or acetone, have been explored to enhance the solubility of polar substances in sc-CO2. The combination of the two most abundant and inexpensive solvents on Earth, CO2 and water as environmentally benign, non-toxic and non-flammable fluids, offers new possibilities in waste minimization for the replacement of organic solvents. However, water and carbon dioxide exhibit a weak mutual solubility (depending on pressure and temperature): the solubility of water in CO2 at 15 °C and 45 MPa is 0.1 wt%.2 Water-in-CO2 (W/C) microemulsions and emulsions, formed by the addition of appropriate surfactants, have consequently the ability to function as a “universal” solvent medium by solubilizing high concentrations of polar, ionic and non-polar molecules within the dispersed and continuous phases.
A peculiar safety reason to sc-CO2-based processes is the necessity to work under practicable conditions with pressures and temperatures down to 40 MPa and 100 °C. This consequently requires polymeric surfactants soluble in such conditions. In order to develop efficient families of surfactants, extensive studies were dedicated to determine the “CO2-philicity” of various polymers in sc-CO2.3,4 Due to strong Lewis acid–Lewis base interactions and low cohesive energies, perfluoroacrylates were shown to be the most “CO2-philic” polymers exhibiting low cloud points with a Mn of 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1.5 Yet, their toxicological profile6 as well as their high price7 constitute a severe limitation for industrial applications. To a lesser extent, siloxanes also exhibited significant solubilities, though with a Mn of 10000 g mol−1 lower than their fluorinated counterparts.8,9 However, their chemical stability in specific applications and their price7 limit their industrial use. There is consequently a substantial need for developing cheap, stable and environmental-friendly polymeric surfactants for water/sc-CO2 emulsions.
000 g mol−1.5 Yet, their toxicological profile6 as well as their high price7 constitute a severe limitation for industrial applications. To a lesser extent, siloxanes also exhibited significant solubilities, though with a Mn of 10000 g mol−1 lower than their fluorinated counterparts.8,9 However, their chemical stability in specific applications and their price7 limit their industrial use. There is consequently a substantial need for developing cheap, stable and environmental-friendly polymeric surfactants for water/sc-CO2 emulsions.
Thus, a large body of research was devoted to the synthesis of novel hydrocarbon-based polymers exhibiting enhanced solubilities in sc-CO2. Firstly, Sarbu et al. synthesized poly(ether carbonates) from propylene oxide and carbon dioxide which showed a greater solubility than perfluoroacrylates at 22 °C and pressures down to 16 MPa.10 Comparative solubility studies with commodity polymers in sc-CO2 by Shen et al. also demonstrated that poly(vinyl acetate) (PVAc) exhibited a higher solubility than poly(methyl methacrylate), poly(lactide) and poly(propylene oxide).4 Their pioneering work emphasized both the crucial role of the molecular weight and the presence of an accessible pendant acetate group for the Lewis acid–Lewis base interactions between the electron-rich carbonyl oxygen and the electron-deficient carbon of the CO2 molecule. This notable difference coming from the acetate groups confirmed the findings reported by Kazarian et al. using IR spectroscopy.11 Computational studies also pointed out the existence of cooperative H-bond interactions between the methyl of the acetate group and the oxygen atom of the CO2 molecule.12 Then, further works on acetylated sugars13 and siloxanes with acetate-functional side-chains14 were built on these observations to suggest new CO2-philic entities. Finally, the statistical copolymerization of vinyl acetate with bulky monomers such as vinyl butyrate15 and dibutyl maleate (DBM)16 led to hydrocarbon-based polymers with improved “CO2-philicity”. A P(VAc-alt-DBM) copolymer with a Mn equal to 3800 g mol−1 was shown to be soluble at pressures and temperatures close to the cloud point coordinates of a siloxane polymer with a Mn equal to 10000 g mol−1.16
However, since PVAc is a non-conjugated monomer whose polymerization has longstandingly been difficult to control, few examples of amphiphilic diblock copolymers based on a PVAc block have been published in the field of sc-CO2 polymeric surfactants. For a long time, synthetic strategies to produce PVAc-based amphiphilic copolymers relied on telomerizations, i.e. irreversible chain transfer reactions. For instance, Tan et al. synthesized PVAc-b-PEG and PVAc-b-PEG-b-PVAc copolymers according to a tedious three-step process, including radical polymerization of VAc in the presence of 2-isopropoxyethanol to produce a OH-terminated PVAc, transformation of the OH group into an imidazole ester and finally coupling of the resulting PVAc with hydroxy-terminated PEGs.17 These block polymers were reported as efficient templates for the synthesis of acrylamide-based porous materials in sc-CO2. The developments in reversible-deactivation radical polymerization18 (RDRP) constitute the most promising advances to produce well-defined VAc oligomers and combine them with hydrophilic blocks or specific functionalities. In particular, the reversible addition–fragmentation chain transfer polymerization/macromolecular design by interchange of xanthates (RAFT19,20/MADIX)21 with the specific use of xanthate transfer agents is a very easy-to-handle technique to control the polymerization of vinyl esters,22 allowing the access to PVAcs of controlled Mn and low dispersities.23 Hence, xanthates offer great potential to produce new PVAc-based copolymers for sc-CO2 applications. Tan et al. synthesized a xanthate-functionalized PEG to produce PVAc–PEG–PVAc triblock co-oligomers for C/W emulsion templating.24 However, this strategy offers little versatility to tune the length of the hydrophilic segment. Actually, large potentialities of RAFT/MADIX polymerization still remain unexploited in this field of research.
In this study, we thus aimed at establishing the structure–property relationships between RAFT/MADIX PVAc-based polymers and their solubility in sc-CO2. PVAc blocks were used as building materials to understand the influence of key macromolecular characteristics—such as chain length, chain end group and hydrophilic/CO2-philic balance—on their behaviour in sc-CO2. We also took advantage of the chemistry of xanthates to introduce a CO2-philic fluorinated moiety in the Z-group. We firstly focused on PVAc and then extended the study to novel poly(N,N-dimethylacrylamide)–poly(vinyl acetate) amphiphilic diblock copolymers which could be potentially active at the W/C interface. Following an original approach developed by Martinez et al., their solubility was determined from high-pressure infrared spectroscopy.25
Experimental section
Materials
Vinyl acetate (VAc, Acros Organics, 99+%), N,N-dimethylacrylamide (DMA, Aldrich, 99%) and ethyl acetate (Fischer Scientific, Laboratory reagent grade) were distilled on CaH2. 2,2′-Azobisisobutyronitrile (AIBN, Acros Organics, 98%) was recrystallized twice from methanol and dried in vacuo. Carbon disulfide (Sigma-Aldrich, anhydrous, >99%) was distilled on P2O5. The O-ethyl-S-(1-methoxycarbonyl) ethyldithiocarbonate RAFT/MADIX agent (Rhodixan A1 (Xa), see Scheme 1) was obtained from Rhodia and used as received. Carbon dioxide (CO2 N45 TP, Air Liquide), N,N-dimethylformamide (DMF, Sigma-Aldrich, Chromasolv Plus for HPLC, >99.9%), tetrahydrofuran (THF for preparative HPLC, stabilized with BHT, SDS), lithium bromide (Aldrich, 99+%), 1H,1H,2H,2H-perfluoro-1-octanol (Aldrich, 97%), sodium hydride (Aldrich, 60% dispersion in mineral oil), methyl 2-bromopropionate (Aldrich, 98%) were used as received.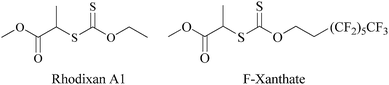 | ||
| Scheme 1 Chemical structure of the RAFT/MADIX agents used in this study. | ||
Synthesis of methyl 2-((3,3,4,4,5,5,6,6,7,7,8,8,8-tridecafluorooctyloxy)carbonothioylthio)propanoate (F-xanthate)
Carbon disulfide (1.217 g, 15.98 mmol) was added to a solution of 1H,1H,2H,2H-perfluoro-1-octanol (3 g, 7.99 mmol) in N,N-dimethylformamide (40 mL). The mixture was cooled down to 0 °C in an ice bath and sodium hydride (319.7 mg, 7.99 mmol) was added. The mixture was stirred at 0 °C for 1 h and methyl 2-bromopropionate (1.334 g, 7.99 mmol) was then added dropwise. The mixture was stirred at 0 °C for 1 h and then at RT for 2 h. After concentration under vacuum, the mixture was diluted with diethyl ether, washed twice with water and finally with brine. The organic phase was collected and dried overnight on MgSO4. After concentration, the product was purified by chromatography on silica gel using heptane/ethyl acetate (9:1) to yield 2.73 g (64%) of pure F-xanthate (FXa, see Scheme 1) as a yellow viscous oil. 1H NMR (300 MHz, CDCl3): δ (ppm) = 4.81 (t, 2H), 4.40 (q, 1H), 3.69 (q, 2H), 3.64 (s, 3H), 2.79 (m, 2H), 1.47 (d, 3H). 13C NMR (300 MHz, CDCl3): δ (ppm) = 211.2 (s, SC(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S)), 170.7 (s, C-C(O)-O), 100–125 (m, fluorinated carbon atoms), 65.6 (s, O-CH2-C), 52.3 (s, CH3-O), 46.3 (s, C-CH3), 29.0 (t, CH2-CH2-CF2), 16.3 (s, CH3-C). 19F NMR (300 MHz, CDCl3): δ (ppm) = −81.7 (t, 3F, CF3), −113.5 (m, 2F, CF2-CH2), −122.4 (m, 2F, CF2-CF2-CH2), −123.5 (m, 2F, C3F7-CF2), −123.9 (m, 2F, C2F5-CF2), −126.9 (m, 2F, CF3-CF2). GC-MS: accurate mass: 527.0 amu (theoretical mass of C13H11S2O3F13 = 526.3 amu).
S)), 170.7 (s, C-C(O)-O), 100–125 (m, fluorinated carbon atoms), 65.6 (s, O-CH2-C), 52.3 (s, CH3-O), 46.3 (s, C-CH3), 29.0 (t, CH2-CH2-CF2), 16.3 (s, CH3-C). 19F NMR (300 MHz, CDCl3): δ (ppm) = −81.7 (t, 3F, CF3), −113.5 (m, 2F, CF2-CH2), −122.4 (m, 2F, CF2-CF2-CH2), −123.5 (m, 2F, C3F7-CF2), −123.9 (m, 2F, C2F5-CF2), −126.9 (m, 2F, CF3-CF2). GC-MS: accurate mass: 527.0 amu (theoretical mass of C13H11S2O3F13 = 526.3 amu).
RAFT/MADIX synthesis of PVAc
In a typical experiment, a mixture of vinyl acetate (2.8 g, 3.2 × 10−2 mol), AIBN (8 mg, 0.049 mmol) and Rhodixan A1 (97 mg, 0.47 mmol) in ethyl acetate (2.9 g) was degassed by four freeze–pump–thaw cycles, backfilled with argon, sealed and placed in an oil bath at 60 °C. After 15 h, the reaction was stopped by cooling in liquid nitrogen. Conversion was determined by 1H NMR in CDCl3. The polymer was dried in vacuo. (Mn,theo = 6000 g mol−1, Mn,SEC = 6050 g mol−1, Đ = 1.61) The same protocol was applied to produce a PVAc3.8k–FXa sample replacing Rhodixan A1 with F-xanthate.RAFT/MADIX synthesis of PDMA macro-chain transfer agents
In a typical experiment, a mixture of N,N-dimethylacrylamide (1.62 g, 1.63 mmol), AIBN (4 mg, 0.024 mmol) and Rhodixan A1 (166 mg, 0.8 mmol) in ethyl acetate (7.2 g) was degassed by four freeze–pump–thaw cycles, backfilled with argon, sealed and placed in an oil bath at 60 °C. After 15 h, the reaction was stopped by cooling in liquid nitrogen. Conversion was determined by 1H NMR in CDCl3. The polymer was dried in vacuo. (Mn,theo = 2200 g mol−1, Mn,SEC = 2050 g mol−1, Đ = 1.46).RAFT/MADIX synthesis of PDMA–PVAc–Xa block copolymers
In a typical experiment, AIBN (7.2 mg, 0.04 mmol) and vinyl acetate (1.52 g, 17.6 mmol) were added to a solution of PDMA macro-chain transfer agent (0.5 g, 0.42 mmol) in ethyl acetate (2.5 g), then degassed by four freeze–pump–thaw cycles, backfilled with argon, sealed and placed in an oil bath at 60 °C. After 15 h, the reaction was stopped by cooling in liquid nitrogen. Conversion was determined by 1H NMR in CDCl3. The polymer was dried in vacuo. (Mn,theo = 3900 g mol−1, Mn,SEC = 5100 g mol−1, Đ = 1.24) (Scheme 2).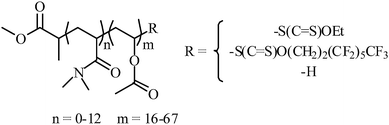 | ||
| Scheme 2 Chemical structure of the copolymers synthesized in this study. | ||
Characterization techniques
000 g mol−1. Flow rate was 1 mL min−1 and toluene was used as a flow marker. Prior to injections, samples were diluted at a concentration of 10 g L−1 overnight and filtered through a 0.45 μm PTFE filter. Data were handled with the Wyatt Astra 5.3.4.14 software.
SEC in DMF containing 10 mM of LiBr was performed using a 100 μL injection loop, a set of three Shodex columns (KD-G, K-805L and KD-802) thermostated at 55 °C and a Waters 410 Differential Refractometer thermostated at 40 °C. A calibration was performed using the same poly(methyl methacrylate) standards that described above. Flow rate was 1 mL min−1 and toluene was used as a flow marker. Prior to injections, samples were diluted at a concentration of 10 g L−1, stirred overnight and filtered through a 0.45 μm PTFE filter.
Results and discussion
RAFT/MADIX polymerization of vinyl acetate
As of today, almost all RDRP strategies were proved to mediate the polymerization of VAc at the notable exception of nitroxide-mediated polymerization. Atom-transfer radical polymerization (ATRP) with iron26 or copper complexes27 offers moderate control over the polymerization of VAc with dispersities of 1.5–1.8. Alternatively, iodine transfer polymerization (ITP) with alkyl iodides produced narrow PVAcs with dispersities inferior to 1.5.28 However, a gradual decomposition of the terminal iodide leading to an aldehyde group was observed. Up to date, cobalt-mediated radical polymerization (CoMRP) allowed to control VAc polymerization29 but the possibilities of combining various blocks are scarce. Organoheteroatom-mediated polymerization (OMRP) based on tellurium,30 and antimony31 derivatives has also been successful methods to produce well-defined PVAcs with Mn less than 5000 g mol−1. Due to the toxicity, cost and air-sensitivity of these controlling agents, their scope remains rather limited. Ultimately, RAFT/MADIX gives excellent control of VAc polymerization in a more straightforward way than the previously cited methods.32 With an appropriate design of the RAFT/MADIX agent,33 dispersities as low as 1.2 can be achieved at moderate conversions.34,35 For this purpose, Rhodixan A1 was thus designed with a methyl propionate R-leaving group and an O-ethylZ-group and showed a good control for VAc polymerization with no reported retardation. In addition, the chemistry of xanthate agents used in RAFT/MADIX polymerization opens up the possibility of varying polymeric architectures and modifying the chain-end groups.36,37Following the principles of RAFT/MADIX polymerization, we consequently synthesized PVAcs with Mn ranging from 2k (standing for 2000 g mol−1) to 6k (see entries 1 to 3 in Table 1) to probe the effect of chain length on the solubility in sc-CO2. This aimed at constituting reference materials for the following solubility studies of block copolymers. The polymerization of VAc was carried out at 60 °C in ethyl acetate with Rhodixan A1 (Scheme 1). While Mn values determined by SEC were in very good agreement with theoretically predicted ones up to high conversion, dispersities were found to increase with targeted Mn (see Table 1). As suggested by the excellent matching between the theoretical and experimental Mn, transfer to solvent was negligible over the course of the polymerization. This was not surprising given that a low transfer constant to ethyl acetate (Ctr,s = 3 × 10−4)38 at 60 °C was reported for VAc. Therefore, the noticeable increase in dispersity may be due to combined effects of chain transfer monomer and polymer39 together with head-to-head defects30 during VAc polymerization. Chain transfer to polymer is known to be responsible for the formation of a substantial amount of chain branches.39 The corresponding transfer constant (Ctr,pol) value ranges from 2.4 to 47 × 10−4 depending on the polymerization conditions.38 In parallel to this, the polymerization of VAc also suffers from a non-negligible proportion of regioirregularities.38 Few head-to-head additions lead to the formation of reactive primary radicals which, once in the dormant state, presumably have a much lower interchain transfer constant (Cex = kex/kp) than secondary radicals generated by head-to-tail additions.30 In the case of organotellurium-mediated polymerization of VAc, this resulted in a significant increase in Đ with the targeted chain length.30 Such a behaviour was also reported with the iodine-transfer polymerization of vinylidene fluoride.40 It is worth mentioning that both chain transfer to polymer and head-to-head additions are expected not to impact the values of Mn but only Đ values. The ω-functionalization of PVAc with Rhodixan end groups was confirmed by MALDI-TOF mass spectrometry analysis irrespective of the chain length and dispersity (see Fig. S1 in the ESI†).
| Sample | Conv.(%)a | M n,theo b/g mol−1 | M n,SEC/g mol−1(Đ)c | T g/°C |
|---|---|---|---|---|
| a Determined by 1H NMR in CDCl3. b Theoretical Mn = [VAc]0/[Rhodixan A1]0 × Mw(VAc) × conv./100 + Mw(xanthate). c Determined by SEC in THF with PMMA standards. | ||||
| PVAc1.8k–Xa | 95 | 2000 | 2000 (1.20) | 13.5 |
| PVAc3.8k–Xa | 92 | 4050 | 4000 (1.30) | 21.2 |
| PVAc5.8k–Xa | 97 | 6000 | 6050 (1.61) | 29.7 |
| PVAc4.2k–H | — | 4200 | 4200 (1.30) | — |
| PVAc3.8k–FXa | 87 | 4300 | 3850 (1.26) | 15.6 |
In addition to the chain length effect, chain-end groups also play an important role in the solubility of low Mn polymers in sc-CO2.16,41 To probe this effect, PVAc4.2k–H, PVAc3.8k–Xa and PVAc3.8k–FXa (entries 2, 4 and 5 in Table 1)—all exhibiting Mn close to 4k—were synthesized. As the xanthate moiety was suggested to decrease the solubility of polymers in sc-CO2,16 the xanthate end-capping group was removed by radical reduction using dilauroyl peroxide in a H-donor solvent (see ESI†).42 In our case, a mixture of THF and propan-2-ol was used to ensure the complete solubilization of the PVAc starting materials, following the methodology published by Tong et al.43 The removal was confirmed by SEC-UV at a wavelength of 290 nm and MALDI-TOF MS measurements (see Fig. S3(a) and (b) in the ESI†). No high-molecular weight shoulder appeared on the SEC-RI traces suggesting the absence of coupling reactions between PVAc macroradicals (see Fig. S3(a) in the ESI†).
Along with a post-polymerization modification, we also took advantage of the chemistry of xanthates to introduce a CO2-philic fluorinated moiety in the Z-group. Such fluorinated xanthates have been scarcely reported in the litterature.44–47 Recently, the copolymerization of vinylidene fluoride and 3,3,3-trifluoropropene was mediated by a xanthate with a fluorinated R-group to probe the polymerization by 19F NMR.44 Xanthates bearing an O-trifluoromethyl Z group were also designed to investigate the polymerization of styrene in bulk and dispersed media.45–47 In these latter studies, the fluorinated moiety was aimed at increasing the reactivity of the transfer agent towards the propagating styryl radicals. In our case, the fluorinated moiety was incorporated with an ethylene spacer between the perfluoro group and the CS bond in order to preserve a reactivity similar to that of Rhodixan A1. F-Xanthate was subsequently used to mediate VAc polymerization to give PVAc3.8k–FXa (see entry 5 in Table 1). As expected, similar macromolecular characteristics (Mn, Đ) with Rhodixan A1 were found.
RAFT/MADIX synthesis of the PDMA macro-chain transfer agents
The scope of RAFT/MADIX polymerization is broad considering the range of hydrophilic monomers whose polymerization can be controlled. Indeed, the RAFT/MADIX process was successfully implemented for vinyl lactams48–50 and a very large number of water-soluble (meth)acrylic, (meth)acrylamido51 and styrenic monomers52 offering large possibilities to conceive amphiphilic copolymers for the stabilization of W/C emulsions. In particular, DMA was chosen among other non-ionic monomers as it exhibits a good solubility in water and a high reactivity. In addition, its polymerization is efficiently controlled up to full conversion compared to the other hydrophilic monomers (e.g. vinyl lactams) polymerizable by RAFT/MADIX. This allowed us to avoid purification steps which are problematic considering the partial solubilities of oligomers in non-solvents used for precipitation.A large variety of RAFT/MADIX agents including trithiocarbonates and dithioesters successfully mediates the polymerization of DMA.53 However, as these agents tend to inhibit the RAFT/MADIX polymerization of VAc, PDMA–PVAc diblock copolymers are not attainable following this synthetic route. The recent developments of universal RAFT agents could address this limitation. An elegant alternative using switchable N-(4-pyridinyl)-N-methyldithiocarbamate RAFT agents was thus published by Benaglia et al.54 who synthesized well-defined PVAc-based diblock copolymers from polystyrene or poly(methyl methacrylate) macromolecular RAFT agents.55 Fluorodithioformates were also suggested as universal RAFT agents56 but their synthesis remains challenging and their applicability yet to be demonstrated.57 Ultimately, O-ethyl xanthates like Rhodixan A1 offer a much simpler access to PDMA–PVAc diblock copolymers given that they were shown to control the polymerization of acrylamido monomers.51
We applied a golden rule of RAFT/MADIX polymerization by first polymerizing DMA—giving the most stable growing radicals—and then chain extending with VAc. The polymerization of DMA was conducted at 60 °C in ethyl acetate under dilute conditions of 1.66 mol L−1 to minimize exothermy. As shown in Table 2, all polymerizations of DMA were well controlled with number-average molar masses determined by either NMR or SEC in agreement with the theoretical ones at the end of the polymerization. This means that Rhodixan A1 has fully reacted, which was expected with a chain transfer constant to Rhodixan A1 equal to 2.3 measured at 60 °C in another solvent (ethanol:water 4.5:1 wt%).58 Dispersity was in the 1.4–1.6 range, at the end of the polymerization suggesting a relatively slow exchange of the xanthate group between dormant and active chains.59 Ultimately, the functionalization of PDMA chains by the xanthate fragments at both ends was confirmed by MALDI-TOF (see Fig. S2 in the ESI†).
| Sample | PDMA MADIX agent | PDMA-b-PVAc–Xa copolymers | |||||||
|---|---|---|---|---|---|---|---|---|---|
| M n theo a/g mol−1 | M n NMR b/g mol−1 | M n SEC c/g mol−1 (Đ) | T g/°C | Conv.d(%) | M n theo e/g mol−1 | M n,SEC c/g mol−1 (Đ) | DMA/VAc massf (%) | T g/°C | |
| a Theoretical Mn = [DMA]0/[xanthate]0 × Mw(DMA) × conv./100 + Mw(xanthate). b Determined by integration of the signals of the proton located at the α position of the xanthate moieties. c Determined by SEC in DMF + LiBr with PMMA standards. d Determined by 1H NMR in CDCl3. e Theoretical Mn = [VAc]0/[PDMA-MADIX agent]0 × Mw(VAc) × conv./100 + Mn(PDMA MADIX agent). f Based on theoretical masses. | |||||||||
| PDMA0.4k–PVAc1.4k–Xa | 600 | 500 | — | −6.0 | 81.0 | 2050 | 2700 (1.11) | 21 | 26.2 |
| PDMA0.8k–PVAc3k–Xa | 1000 | 1200 | 750 (1.56) | 13.3 | 85.6 | 4050 | 4600 (1.24) | 21 | 35.0 |
| PDMA1.2k–PVAc5k–Xa | 1400 | 1450 | 1250 (1.46) | 57.6 | 91.9 | 6450 | 6900 (1.41) | 19 | 38.0 |
| PDMA0.4k–PVAc3.5k–Xa | 600 | 500 | — | −6.0 | 86.1 | 4050 | 4300 (1.21) | 10 | 29.2 |
| PDMA0.8k–PVAc3k–Xa | 1000 | 1200 | 750 (1.56) | 13.3 | 85.6 | 4050 | 4600 (1.24) | 21 | 35.0 |
| PDMA1k–PVAc2.7k–Xa | 1200 | 1450 | 1050 (1.48) | 42.4 | 86.4 | 3900 | 5100 (1.24) | 27 | 37.5 |
| PDMA2k–PVAc1.9k–Xa | 2200 | 2400 | 2050 (1.46) | 79.5 | 83.9 | 4100 | 5500 (1.20) | 52 | 50.1 |
| PDMA0.8k–PVAc3.2k–FXa | 1300 | 950 | 1800(1.11) | — | 87.0 | 4500 | 4600(1.21) | 20 | — |
RAFT/MADIX polymerization of PDMA–PVAc–Xa block copolymers
The PDMA RAFT/MADIX macromolecular agents were then used to mediate the polymerization of VAc. By using PDMA oligomers with a wide range of Mn, we targeted PDMA-b-PVAc polymers with both various chain lengths and compositions. The polymerizations were conducted at 60 °C with VAc in the presence of AIBN. Chain extensions were proved successful by the displacement of the peaks on the SEC chromatograms (see Fig. 1) and the absence of residual traces of the initial PDMA RAFT/MADIX agent. Furthermore, an increase in the chemical shift of the 1H NMR signals corresponding to the proton located at the α position of the xanthate moieties from 4.6–4.8 (for PDMA) to 6.4–6.6 ppm (for PVAc) confirmed the SEC results. The synthesized PDMA-b-PVAc copolymers showed good agreements between theoretical and experimental Mn too (see in Table 2). The slight discrepancies of these values were attributed to differences in hydrodynamic volumes with the PMMA standards. Dispersities were low (1.11 < Đ < 1.41) and matched the values previously reported for PVAc oligomers in this range of Mn. Hence, the PDMA radicals generated during the chain extension process proved their ability to be a good leaving group and efficiently reinitiate the RAFT/MADIX polymerization of VAc.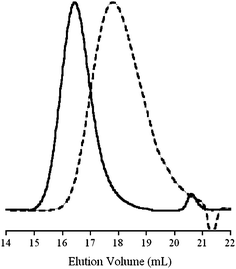 | ||
| Fig. 1 Overlays of SEC-RI chromatograms in DMF (including 10 mM LiBr) with a PDMA2k (dashed line) and a PDMA2k–PVAc1.9k–Xa block copolymer (solid line). | ||
Thermal behaviour in the solid state
Along with the specific favourable Lewis interactions, Beckman et al. suggested that a low cohesive energy density and a high free volume were key features for the design of CO2-philic polymers.60 The measurements of glass transition temperatures (Tg) (which are correlated to the entropy of mixing and high free volumes) can consequently provide qualitative information for perspectives of solubility of our (co)polymers in scCO2.We performed differential scanning calorimetry (DSC) for all homopolymers and block copolymers. The Tg values of PVAc polymers exhibited a slight dependence of Tg towards Mn (see in Table 1). Hence, the experimental Tg value for a PVAc5.8k–Xa was close to the infinite Tg value for PVAc (i.e. 35–40 °C). In contrast, PDMA oligomers showed a strong molar mass dependency of Tg (see in Table 2). This may be due to the combined effects of Mn and xanthate end-groups, the latter being particularly significant for the PDMAs of lowest Mn. To clarify this, further PDMA samples with increasing Mn were synthesized confirming this mass dependence up to 20000 g mol−1, as found by Fuchise et al.61
Surprisingly, the PDMA-b-PVAc copolymers exhibited a single Tg value (see in Table 2). Tg values ranged between 28 °C and 55 °C, i.e. between the Tg of each block taken separately. This excluded the possibility of us having not observed the Tg of one particular block due to low molar mass effects. As indicated by the presence of a single Tg, these copolymers were miscible regardless of both their chain lengths and compositions. This located our copolymers under the order–disorder transition (ODT), indicating a low NχAB product (i.e. a low degree of polymerization (N) and/or a low interaction parameter (χAB)). In order to explain our observations, we thus synthesized a PDMA–PVAc block copolymer with a theoretical Mn of 20000 g mol−1 and a 50 wt% PDMA. This copolymer exhibited a single intermediate Tg too and this clearly suggested a low interaction parameter as a result. Ultimately, a blend of PDMA–Xa (Mn,theo = 40800 g mol−1) and PVAc–Xa (Mn,theo = 38100 g mol−1) polymers revealed two distinct Tg that confirmed the results published by Parada et al.62 More interestingly, the plot of Tgversus the theoretical DMA weight fraction appeared to be linear with an excellent correlation coefficient of 0.98 in the case of PDMA-b-PVAc copolymers whose Mn was close to 4000 g mol−1 (see Fig. 2). According to the Gordon–Taylor equation,63 this linearity suggests an ideal volume additivity of the repeating monomer units in PDMA-b-PVAc copolymers.
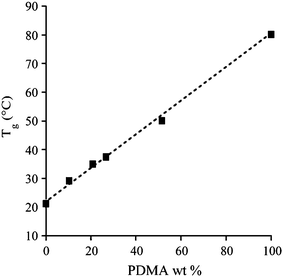 | ||
| Fig. 2 Dependence of Tg on PDMA weight fractions for the PDMA–PVAc–Xa block copolymers (Mn = 4000 g mol−1). | ||
Back to the problematics of sc-CO2 surfactants, increasing the weight fraction of PDMA led to an increase in Tg and a consequent higher entropy of mixing. Yet, the observed miscibility is not expected to impact the solubility of these copolymers in sc-CO2 due to their asymmetrical affinity towards CO2. This will be put into perspective with the following solubility results measured by infrared spectroscopy.
Solubility of PVAc in sc-CO2
Usual measurements of polymer solubility rely on repeated visual observations of (de)mixing transitions in a variable-volume view cell. This allows the determination of cloud point coordinates for a sample at a single polymer/CO2 ratio over a large range of temperature. Based on the work of Martinez et al., our approach used an isochoric cell to measure the solubility of samples versusCO2 density in a more straightforward manner.25 It has been shown by a number of authors that the logarithm of the solubility of solid solutes in sc-CO2 is linearly proportional to the density or to the logarithm of density.64 Hence, solubility data in sc-CO2 are usually presented as a function of CO2 density. High pressure infrared spectroscopy actually offered an alternative way of determining the solubility of polymers in sc-CO2 by measuring the growth of a characteristic vibrational band with increasing pressures (see Fig. 3). Using the CO2 state equation, the solubility of the polymer can be thus plotted as a function of CO2 density. Consequently, Martinez et al. reported the solubility of various hyperbranched polymers in sc-CO2 along with two isotherms at 40 °C and 100 °C.25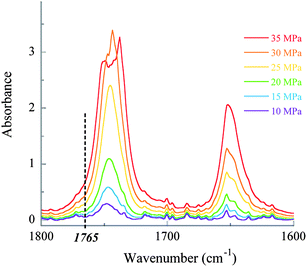 | ||
| Fig. 3 Overlays of infrared spectrograms for a PDMA1k–PVAc2.7k–Xa block copolymer with increasing pressures. | ||
Following these principles, the vibrational band of esters at 1745 cm−1 was selected to measure the solubility of the (co)polymers synthesized in this study. Since the absorbance of the characteristic band at 1745 cm−1 reached values out of the linear range of the Beer–Lambert law, absorbance values for all samples were measured at 1765 cm−1. Interestingly, a red shift of around 10 cm−1 was observed at low pressures, which is characteristic of an electron donor–acceptor complex, i.e. a Lewis acid–base interaction between CO2 (acid) and the acetate group (base). Similar findings were thus observed during the formation of an acetone–CO2 complex and its favoured conformation.65
Yet, we assumed ε was independent of pressure and temperature at higher pressures. The molar extinction coefficient (ε) of the CO vibrational band was consequently determined at 1765 cm−1 from a fully solubilized sample of PVAc2k–Xa at 40 °C and 25 MPa. This coefficient was evaluated to 39.9 L mol−1 cm−1. The same experiment was repeated both in carbon tetrachloride and with other samples in sc-CO2 to confirm this experimental value.
Then, we determined the solubility of PVAc polymers in CO2 as a function of their number-average molecular masses to constitute a comparison platform for the entire study (see Fig. 4). At a CO2 density of 0.88 g cm−3 (i.e. 22 MPa and 40 °C), 1 wt% of PVAc1.8k–Xa was soluble. The longer PVAc polymers were far less soluble: 0.81 wt% of PVAc3.8k–Xa was solubilized at 0.94 g cm−3 whereas only 0.38 wt% of PVAc5.8k–Xa was detected at the same density (see Fig. 4). In comparison, Lee et al. reported slightly lower cloud point coordinates for 0.2 wt% of a PVAc (Mn = 2300 g mol−1) at 18.6 MPa and 40 °C.15 Kilic et al. measured a solubility of 1 wt% for a PVAc polymer (Mn = 3090 g mol−1) at 25 °C and 31 MPa.66
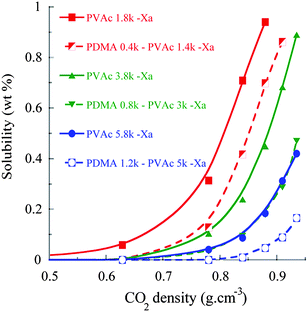 | ||
| Fig. 4 Solubility (wt%) of PVAc–Xa polymers and PDMA–PVAc–Xa block copolymers in sc-CO2 with increasing chain lengths. | ||
Along with the chain length, the nature of the chain end group may play an important role on the solubility of polymers in sc-CO2.67,68 In particular, the xanthate group was suggested to strongly lower the solubility of poly(vinyl acetate)-alt-poly(dibutyl maleate) copolymers as concluded from a comparison between samples synthesized by telomerization and by RAFT/MADIX polymerization.16 In the case of our PVAcs, we attempted to clarify this by comparing a xanthate-capped PVAc (PVAc3.8k–Xa) and a xanthate-free equivalent (PVAc4.2k–H) obtained by a radical reduction of the xanthate group by dilauroyl peroxide at 80 °C in a mixture of THF and propan-2-ol.42,43 No substantial differences of solubility in sc-CO2 were found (see Fig. 5) since they were respectively soluble in proportions of 0.89 wt% and 0.87 wt% at 35 MPa. In other words, the influence of the xanthate group was negligible in our case for this range of Mn. Differences in microstructures for poly(vinyl acetate)–poly(dibutyl maleate) copolymers due to dissimilar types of compositional drifts between conventional and controlled radical polymerizations might explain the differences of solubilities observed by Lee et al.16 Moreover, the xanthate can be turned into a solubility enhancervia the introduction of a fluorinated Z-group. The F-xanthate agent was thus designed to incorporate a C6F13 moiety at the ω-end of the synthesized (co)polymers. As hoped, the incorporation of this latter moiety significantly increased the solubility of PVAc3.8k–FXa. Indeed, 1 wt% of this sample was soluble at conditions down to 30 MPa and 40 °C (see Fig. 5). Although this sample was less CO2-philic than PVAc1.8k–Xa, this clearly demonstrates that the limited intrinsic solubilities of PVAc polymers in sc-CO2 can be counterbalanced and improved via an appropriate RAFT/MADIX synthesis strategy.
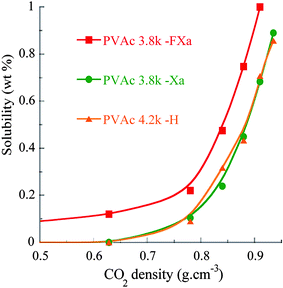 | ||
| Fig. 5 Solubility (wt%) of PVAc polymers (Mn = 4k) with different chain end groups. | ||
Solubility of PDMA–PVAc–Xa block copolymers with different chain lengths in sc-CO2
To the best of our knowledge, the influence of a CO2-phobic polymer block on the solubility of amphiphilic polymer surfactants has never been examined. To clarify this point, the solubility of PDMA-b-PVAc–Xa samples comprising approximately 20 wt% of PDMA in sc-CO2 was first investigated. As in the case of PVAc–Xa polymers, three different chain lengths were investigated from 2k to 6k (entries 1 to 3 in Table 1). While 1 wt% of the shortest copolymer (Mn = 2k) was solubilized at 35 MPa, the solubility dropped to 0.47 wt% for an equivalent copolymer with a Mn of 4k (see Fig. 4). The diblock of highest molar mass (Mn = 6.4k) was poorly soluble (0.17 wt% at 35 MPa). Considering the samples with similar DMA/VAc mass percentages, both the chain length and the presence of the PDMA block consequently appeared to have a dramatic influence on the solubility in sc-CO2. This can be simply explained by a dual increase of “CO2-phobicity” due to both an increase in the length of the CO2-phobic block and the decrease of the CO2-philicity of the longest PVAc blocks.A comparison with their PVAc homopolymer equivalents allows to measure the effect of the replacement of VAc units by DMA units (see Fig. 4). The values of solubility were almost divided by a factor of 2 for both PDMA-b-PVAc–Xa copolymers exhibiting the longest chain lengths. These results appeared logical considering the immiscibility of PDMA in sc-CO2. Indeed, Kilic et al. synthesized PDMA with a Mn of 1.3kvianitroxide-mediated polymerization and 0.7 wt% of this polymer was found to be insoluble even at 25 °C and a high pressure of 45 MPa. This was attributed to the high cohesive energy density and the high Tg of the polymer.69
As a consequence, short chain lengths must be targeted when synthesizing amphiphilic CO2 polymer surfactants based on such CO2-philic PVAc blocks. Yet, preserving steric stabilization proper to polymer surfactants also requires chain lengths long enough to prevent destabilization viaOstwald-ripening, aggregation or coalescence. Thus, PDMA–PVAc–Xa block copolymers with Mn close to 4k appeared as a good balance between solubility and steric stabilization.
Solubility of PDMA–PVAc–Xa diblock copolymers with varying compositions in sc-CO2
Ultimately, we investigated the effect of incorporating a CO2-phobic block through a variation in PDMA weight fraction of PDMA–PVAc block copolymers. The impact of PDMA/PVAc mass ratio on the solubility in CO2 was consequently studied at an approximate number-average molar mass of 4k. Four different ratios from 10:90 to 50:50 were targeted to evaluate their impact on the copolymer solubility in sc-CO2 (see entries 4 to 7 in Table 2). The solubility in sc-CO2 decreased with increasing weight fractions of PDMA: the copolymers exhibited solubilities of 0.71, 0.47, 0.36 and 0.24 wt% for corresponding PDMA weight fractions of 0.10, 0.210, 0.27 and 0.52, respectively (see Fig. 6). This particularly emphasizes the necessity for selecting polymer surfactants with low CO2-phobic/CO2-philic ratio to gain sufficient solubility in CO2 and subsequent stabilization of W/C emulsions.
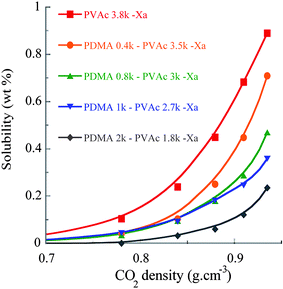 | ||
| Fig. 6 Solubility (wt%) of PDMA–PVAc–Xa block copolymers with comparable chain lengths but varying PDMA weight fractions. | ||
The strategy using a fluorinated xanthate could further help. A PDMA0.8k–PVAc3.2k–FXa diblock copolymer was synthesized with F-xanthate (see entry 8 in Table 2). This copolymer exhibited a solubility of 0.81 wt% in sc-CO2 at 35 MPa, whereas the soluble weight fraction of its non-fluorinated counterpart was only 0.47 wt%. This result confirms the interest of such an approach in the design of amphiphilic polymer surfactants with both improved CO2-philicity and amphiphilicity.
Conclusions
Building on RAFT/MADIX polymerization, a novel family of poly(N,N-dimethylacrylamide)–poly(vinyl acetate) diblock copolymers was synthesized with excellent control over number-average molar masses and dispersities. High pressure infrared spectroscopy measurements allowed to determine the key macromolecular parameters—such as hydrophilic/CO2-philic balance, chain length and chain end group—controlling their solubility in sc-CO2. Both chain length and PDMA weight fraction were proved to be limiting parameters for the solubility in sc-CO2. Moreover, the CO2-philicity of such copolymers can be easily tuned by the appropriate choice of the starting xanthate transfer agent. Further studies investigating the relationships between the structure of these amphiphilic copolymers, their self-assembly in sc-CO2 and the stabilization of water/sc-CO2 interfaces will be reported in forthcoming publications.Acknowledgements
EG thanks PRES of Toulouse for PhD funding.References
- P. T. Anastas and M. M. Kirchhoff, Acc. Chem. Res., 2002, 35, 686 CrossRef CAS.
- Y. Takebayashi, Y. Mashimo, D. Koike, S. Yoda, T. Furuya, M. Sagisaka, K. Otake, H. Sakai and M. Abe, J. Phys. Chem. B, 2008, 112, 8943 CrossRef CAS.
- F. Rindfleisch, T. P. DiNoia and M. A. McHugh, J. Phys. Chem., 1996, 100, 15581 CrossRef CAS.
- Z. Shen, M. A. McHugh, J. Xu, J. Belardi, S. Kilic, A. Mesiano, S. Bane, C. Karnikas, E. J. Beckman and R. Enick, Polymer, 2003, 44, 1491 CrossRef CAS.
- P. Lacroix-Desmazes, P. Andre, J. M. DeSimone, A. V. Ruzette and B. Boutevin, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 3537 CrossRef CAS.
- G. Kostov, F. Boschet and B. Ameduri, J. Fluorine Chem., 2009, 130, 1192 CrossRef CAS.
- E. J. Beckman, J. Supercrit. Fluids, 2004, 28, 121 CrossRef CAS.
- Y. Xiong and E. Kiran, Polymer, 1995, 36, 4817 CrossRef CAS.
- R. Fink and E. J. Beckman, J. Supercrit. Fluids, 2000, 18, 101 CrossRef CAS.
- T. Sarbu, T. Styranec and E. J. Beckman, Nature, 2000, 405, 165 CrossRef CAS.
- S. G. Kazarian, M. F. Vincent, F. V. Bright, C. L. Liotta and C. A. Eckert, J. Am. Chem. Soc., 1996, 118, 1729 CrossRef CAS.
- P. Raveendran and S. L. Wallen, J. Am. Chem. Soc., 2002, 124, 12590 CrossRef.
- P. Raveendran and S. L. Wallen, J. Am. Chem. Soc., 2002, 124, 7274 CrossRef CAS.
- R. Fink, D. Hancu, R. Valentine and E. J. Beckman, J. Phys. Chem. B, 1999, 103, 6441 CrossRef CAS.
- H. Lee, E. Terry, M. Zong, N. Arrowsmith, S. Perrier, K. J. Thurecht and S. M. Howdle, J. Am. Chem. Soc., 2008, 130, 12242 CrossRef CAS.
- H. Lee, J. W. Pack, W. Wang, K. J. Thurecht and S. M. Howdle, Macromolecules, 2010, 43, 2276 CrossRef CAS.
- B. Tan, J. Y. Lee and A. I. Cooper, Macromolecules, 2007, 40, 1945 CrossRef CAS.
- A. D. Jenkins, R. G. Jones and G. Moad, Pure Appl. Chem., 2010, 82, 483 CrossRef CAS.
- C. Barner-Kowollik, Handbook of RAFT Polymerization, Wiley-VCH, Weinheim, 2008 Search PubMed.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2011, 62, 1402.
- D. Taton, M. Destarac and S. Z. Zard, in Handbook of RAFT Polymerization, ed. C. Barner-Kowollik, Wiley-VCH, Weinheim, 2008, ch. 10 Search PubMed.
- M. H. Repollet-Pedrosa, R. L. Weber, A. L. Schmitt and M. K. Mahanthappa, Macromolecules, 2010, 43, 7900 CrossRef CAS.
- R. G. Gilbert, M. Hess, A. D. Jenkins, R. G. Jones, P. Kratochvil and R. F. T. Stepto, Pure Appl. Chem., 2009, 81, 351 CrossRef.
- K. Chen, N. Grant, L. Liang, H. Zhang and B. Tan, Macromolecules, 2010, 43, 9355 CrossRef CAS.
- V. Martinez, S. Mecking, T. Tassaing, M. Besnard, S. Moisan, F. Cansell and C. Aymonier, Macromolecules, 2006, 39, 3978 CrossRef CAS.
- M. Wakioka, K. Y. Baek, T. Ando, M. Kamigaito and M. Sawamoto, Macromolecules, 2001, 35, 330.
- H. Tang, M. Radosz and Y. Shen, AIChE J., 2009, 55, 737 CrossRef CAS.
- M. C. Iovu and K. Matyjaszewski, Macromolecules, 2003, 36, 9346 CrossRef CAS.
- A. Debuigne, J. R. Caille, C. Detrembleur and R. Jerome, Angew. Chem., Int. Ed., 2005, 44, 3439 CrossRef CAS.
- Y. Kwak, A. Goto, T. Fukuda, Y. Kobayashi and S. Yamago, Macromolecules, 2006, 39, 4671 CrossRef CAS.
- S. Yamago, B. Ray, K. Iida, J. Yoshida, T. Tada, K. Yoshizawa, Y. Kwak, A. Goto and T. Fukuda, J. Am. Chem. Soc., 2004, 126, 13908 CrossRef CAS.
- D. Charmot, P. Corpart, H. Adam, S. Z. Zard, T. Biadatti and G. Bouhadir, Macromol. Symp., 2000, 150, 23 CrossRef CAS.
- M. L. Coote and L. Radom, Macromolecules, 2003, 37, 590.
- M. H. Stenzel, L. Cummins, G. E. Roberts, T. P. Davis, P. Vana and C. Barner-Kowollik, Macromol. Chem. Phys., 2003, 204, 1160 CrossRef CAS.
- A. Favier, C. Barner-Kowollik, T. P. Davis and M. H. Stenzel, Macromol. Chem. Phys., 2004, 205, 925 CrossRef CAS.
- J. Bernard, A. Favier, L. Zhang, A. Nilasaroya, T. P. Davis, C. Barner-Kowollik and M. H. Stenzel, Macromolecules, 2005, 38, 5475 CrossRef CAS.
- C. Barner-Kowollik, T. P. Davis and M. H. Stenzel, Aust. J. Chem., 2006, 59, 719 CrossRef CAS.
- G. Moad and D. H. Solomon, in The Chemistry of Free Radical Polymerization, ed. G. Moad and D. H. Solomon, Pergamon Press, Oxford, 1995, ch. 5 Search PubMed.
- D. Britton, F. Heatley and P. A. Lovell, Macromolecules, 1998, 31, 2828 CrossRef CAS.
- C. Boyer, D. Valade, L. Sauguet, B. Ameduri and B. Boutevin, Macromolecules, 2005, 38, 10353 CrossRef CAS.
- B. Tan, C. L. Bray and A. I. Cooper, Macromolecules, 2009, 42, 7945 CrossRef CAS.
- M. Destarac, C. Kalai, A. Wilczewska, L. Petit, E. Van Gramberen and S. Z. Zard, ACS Symp. Ser., 2006, 944, 564 CrossRef CAS.
- Y. Y. Tong, R. Wang, N. Xu, F. S. Du and Z. C. Li, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 4494 CrossRef CAS.
- G. Kostov, F. Boschet, J. Buller, L. Badache, S. Brandsadter and B. Ameduri, Macromolecules, 2011, 44, 1841 CrossRef CAS.
- M. J. Monteiro, M. M. Adamy, B. J. Leeuwen, A. M. van Herk and M. Destarac, Macromolecules, 2005, 38, 1538 CrossRef CAS.
- M. P. F. Pepels, C. I. Holdsworth, S. Pascual and M. J. Monteiro, Macromolecules, 2010, 43, 7565 CrossRef CAS.
- M. Destarac, W. Bzducha, D. Taton, I. Gauthier-Gillaizeau and S. Z. Zard, Macromol. Rapid Commun., 2002, 23, 1049 CrossRef CAS.
- T. L. U. Nguyen, K. Eagles, T. P. Davis, C. Barner-Kowollik and M. H. Stenzel, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 4372 CrossRef.
- N. S. Ieong, M. Redhead, C. Bosquillon, C. Alexander, M. Kelland and R. K. O'Reilly, Macromolecules, 2011, 44, 886 CrossRef CAS.
- M. Beija, J. D. Marty and M. Destarac, Chem. Commun., 2011, 47, 2826 RSC.
- D. Taton, A. Z. Wilczewska and M. Destarac, Macromol. Rapid Commun., 2001, 22, 1497 CrossRef CAS.
- A. B. Lowe and C. L. McCormick, Prog. Polym. Sci., 2007, 32, 283 CrossRef CAS.
- A. J. Convertine, B. S. Lokitz, A. B. Lowe, C. W. Scales, L. J. Myrick and C. L. McCormick, Macromol. Rapid Commun., 2005, 26, 791 CrossRef CAS.
- M. Benaglia, J. Chiefari, Y. K. Chong, G. Moad, E. Rizzardo and S. H. Thang, J. Am. Chem. Soc., 2009, 131, 6914 CrossRef CAS.
- M. Benaglia, M. Chen, Y. K. Chong, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 2009, 42, 9384 CrossRef CAS.
- M. L. Coote and D. J. Henry, Macromolecules, 2005, 38, 5774 CrossRef CAS.
- A. Theis, M. H. Stenzel, T. P. Davis, M. L. Coote and C. Barner-Kowollik, Aust. J. Chem., 2005, 58, 437 CrossRef CAS.
- A. Guinaudeau, PhD thesis, University of Toulouse, 2010.
- A. H. E. Muller, R. G. Zhuang, D. Y. Yan and G. Litvinenko, Macromolecules, 1995, 28, 4326 CrossRef.
- E. J. Beckman, Chem. Commun., 2004, 1885 RSC.
- K. Fuchise, R. Sakai, T. Satoh, S. Sato, A. Narumi, S. Kawaguchi and T. Kakuchi, Macromolecules, 2010, 43, 5589 CrossRef CAS.
- L. G. Parada, L. C. Cesteros, E. Meaurio and I. Katime, Polymer, 1998, 39, 1019 CrossRef CAS.
- M. Gordon and J. S. Taylor, J. Appl. Chem., 1952, 2, 493 CAS.
- S. K. Kumar and K. P. Johnston, J. Supercrit. Fluids, 1988, 1, 15 CAS.
- Y. Danten, T. Tassaing and M. Besnard, J. Phys. Chem. A, 2002, 106, 11831 CrossRef CAS.
- S. Kilic, S. Michalik, Y. Wang, J. K. Johnson, R. M. Enick and E. J. Beckman, Macromolecules, 2007, 40, 1332 CrossRef CAS.
- B. Tan, C. L. Bray and A. I. Cooper, Macromolecules, 2009, 42, 7945 CrossRef CAS.
- C. L. Bray, B. Tan, S. Higgins and A. I. Cooper, Macromolecules, 2010, 43, 9426 CrossRef CAS.
- S. Kilic, Y. Wang, J. K. Johnson, E. J. Beckman and R. M. Enick, Polymer, 2009, 50, 2436 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: MALDI-TOF spectra and data on the removal of the xanthate moiety. See DOI: 10.1039/c1py00209k |
| This journal is © The Royal Society of Chemistry 2011 |