Synthesis of glycosylated peptides by NCA polymerization for recognition of human T-cells†
Thomas
Stöhr
a,
André-René
Blaudszun
b,
Ute
Steinfeld
b and
Gerhard
Wenz
*a
aOrganic Macromolecular Chemistry, Saarland University, C42 Campus Saarbrücken, D-66123, Saarbrücken, Germany. E-mail: g.wenz@mx.uni-saarland.de; Fax: +49 6813023909; Tel: +49 6813023449
bKIST Europe, Saarland University, E 71 Campus Saarbrücken, D-66123, Saarbrücken, Germany. E-mail: a.blaudszun@kist-europe.de
First published on 28th July 2011
Abstract
Peracetylated sugars (glucose, mannose, galactose) were attached via a thiourea linker to the ε-amino group of α-BOC-lysine and α-Z-lysine. After transformation to the N-carboxyanhydride (NCA), the product was copolymerized with both PEGylated lysine-NCA and ε-TFA-lysine-NCA by both tertiary amine and nickel complex catalysts. The resulting statistical copolypeptides are water-soluble after global deprotection and show an α-helical secondary structure. Fluorescein isothiocyanate (FITC) was then coupled to the free ε-NH2 groups of the lysine repeating units. The galactosylated fluorescent peptides are specifically incorporated in human T lymphocytes at 37 °C, as shown by flow cytometry and fluorescence microscopy. Therefore they are potentially useful for selective staining of cells or targeted drug delivery.
Introduction
The recognitions between different cells, between viruses and cells, and between tissues and cells are controlled by the cooperativity of rather weak ligand–receptor interactions. Among others, carbohydrates like glucose, mannose, galactose, and lactose as well as complex oligosaccharides are well-known ligands for biological receptors based within cell membranes.1,2 Binding interactions between carbohydrate ligands and receptor proteins are indeed highly specific, but generally rather weak compared to other biological interactions.3 Dissociation constants range mostly between 50 μM and 1 mM. Therefore, molecular recognition between biological entities requires the interplay of more than one receptor–ligand interaction.Cooperativity of binding can also be used in artificial systems to improve binding affinities. In the so-called ‘multivalency approach’, many ligands are connected to an inert polymer backbone to reach manifold binding.4 Therefore, many carbohydrate ligands have been attached to polymers,5,6carbon nanotubes,7 and cyclic scaffolds, e.g., cyclodextrins8–10 or spherical molecules such as dendrimers.11 The resulting so-called ‘glycoclusters’ or ‘neoglycoconjugates’ indeed showed improved binding to multivalent receptors, e.g., lectins,12–15in vitro and to cellsin vivo,16 and are potentially useful for anti-adhesion therapy17,18 or targeted delivery.19 Despite the successes of the multivalency approach, some drawbacks still obstruct broad application. Firstly, glycoclusters are often not biodegradable because mostly synthetic scaffolds are used in their synthesis. Secondly, polymers and high molecular weight glycoclusters tend to bind nonspecifically to biological surfaces, i.e., by Coulomb interactions between oppositely charged species.
In the following, we describe a new approach using a helical synthetic polypeptide as the framework for the attachment of carbohydrate ligands. Polypeptides are biodegradable by proteolytic enzymes. Also, the α-helix has already proven its use as a rigid scaffold for the investigation of molecular recognition between attached complementary binding sites.20 Homopolyamino as well as copolyamino acids are readily available through the ring-opening polymerization of N-carboxyanhydrides (NCAs).21,22 Our approach is based on previous work about living coordinative polymerization of NCAs with Ni0 catalysts,23,24 and synthesis of water-soluble monoethylene and diethylene glycol-functionalized polylysines that form α-helices.25Glycopeptides had been already synthesized recently by polymer-analogous modification of polylysine with glycosyl-isothiocyanates26 or by polymerization of C-glycosylated-L-lysine NCAs.27 PEGylation of polymers is known to prevent phagocytosis by the immune system.28 We found a straightforward method for the synthesis of neutral O-glycosylated polylysine derivatives with attached diethylene glycol chains to avoid unspecific binding onto cell surfaces. The specific interactions of these glycosylated peptides were investigated with T lymphocytes because they are very promising therapeutic drug carriers for the treatment of cancer.29
Experimental section
Instrumentation
Materials and synthetic procedures
N ε-Trifluoroacetyl-L-lysine-N-carboxyanhydride 30 and Nε-(3,6,9-trioxa-decanoyl)-L-lysine-N-carboxyanhydride25 (both isolated according to Poché et al.31) as well as 2,3,4,6-tetra-O-acetyl-1-O-trichloracetimidato-α-D-mannopyranose,322,3,4,6-tetra-O-acetyl-1-O-trichloracetimidato-β-D-glucopyranose,322,3,4,6-tetra-O-acetyl-1-O-trichloracetimidato-β-D-galactopyranose,332-(2-isothiocyanatoethoxy)ethanol,34 and (2,2′-bipyridine)(1,5-cyclooctadien)nickel(0) (bpyNi(COD))35 were prepared according to literature procedures. N,N′-Dimethylformamide (DMF) was distilled from P2O5 under reduced pressure and stored over molecular sieves 4 Å under Ar. Tetrahydrofuran (THF) was distilled from sodium and subsequently stored over molecular sieves 4 Å under Ar. Triethylamine was freshly distilled from CaH2. All other reactants and solvents were purchased from commercial suppliers and used without further purification. Water was doubly distilled in a glass apparatus.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :15) to yield 1a as a white solid (5.44 g, 70%). 1H NMR (CDCl3): δ/ppm = 1.96, 2.01, 2.07, 2.12 (4 s, 12H, –(C
:15) to yield 1a as a white solid (5.44 g, 70%). 1H NMR (CDCl3): δ/ppm = 1.96, 2.01, 2.07, 2.12 (4 s, 12H, –(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)OCH3), 3.63–3.85 (m, 8H, –OCH2CH2OCH2CH2NCS), 4.01–4.06 (m, 1H, H-5), 4.07–4.11 (m, 1H, H-6), 4.26 (dd, 1H, H-6′), 4.86 (d, 3JHH = 1.5 Hz, 1H, H-1), 5.24 (dd, 1H, H-2), 5.25 (t, 1H, H-4), 5.33 (dd, 1H, H-3); 13C NMR (CDCl3): δ/ppm = 20.62, 20.66, 20.70, 20.83, 45.26, 62.47, 66.07, 67.30, 68.47, 69.02, 69.37, 69.46, 70.15, 97.68, 133.01, 169.68, 169.85, 169.97, 170.57; FTIR (cm−1) = 1043, 1217, 1738, 2106, 2938, 3241, 3360; ESI-MS (m/z) [M + Na]+ calcd for C19H27NNaO11S, 500.12; found, 500.36.
O)OCH3), 3.61–3.67 (m, 6H, –OCH2CH2OCH2CH2NCS), 3.69–3.97 (m, 3H, –OCH2CH2O, H-5), 4.13 (dd, 1H, H-6), 4.25 (dd, 1H, H-6′), 4.59 (d, 3JHH = 8.0 Hz, 1H, H-1), 4.97 (dd, 1H, H-2), 5.06 (t, 1H, H-4), 5.21 (t, 1H, H-3); 13C NMR (CDCl3): δ/ppm = 20.57, 20.58, 20.67, 20.73, 45.21, 61.86, 68.30, 69.10, 69.31, 70.35, 71.23, 71.76, 72.72, 100.73, 132.71, 169.42, 169.44, 170.23, 170.68; FTIR (cm−1) = 1032, 1208, 1745, 2140, 2201, 2884, 2955, 3238, 3356; ESI-MS (m/z) [M + Na]+ calcd for C19H27NNaO11S, 500.12; found, 500.36.
O)OCH3), 3.62–3.67 (m, 6H, –OCH2CH2OCH2CH2NCS), 3.71–3.97 (m, 3H, –OCH2CH2O, H-5), 4.06–4.18 (m, 2H, H-6), 4.55 (d, 3JHH = 8.0 Hz, 1H, H-1), 5.02 (dd, 1H, H-3), 5.18 (dd, 1H, H-2), 5.37 (dd, 1H, H-4); 13C NMR (CDCl3): δ/ppm = 20.53, 20.62, 20.64, 20.76, 45.26, 61.21, 66.97, 68.75, 69.06, 69.31, 70.37, 70.64, 70.81, 101.24, 132.69, 169.96, 170.07, 170.20, 170.36; FTIR (cm−1) = 1042, 1214, 1741, 2113, 2200, 2875, 2941, 3183, 3357; ESI-MS (m/z) [M + Na]+ calcd for C19H27NNaO11S, 500.12; found, 500.36.
:2:0.6, v/v/v). The combined organic fractions were concentrated in vacuum to a residual volume of about 10 mL, diluted with 150 mL ethyl acetate and washed several times with sodium hydrogen carbonate and sodium chloride to remove the acetic acid. Finally, the organic fraction was dried over anhydrous MgSO4 and the solvent was evaporated under vacuum. The resulting white solid was received in 43% yield. 1H NMR (CDCl3): δ/ppm = 1.39 (m, 2H, –NHCHCH2CH2CH2CH2–), 1.55 (m, 2H, –NHCHCH2CH2CH2CH2–), 1.72 (m, 1H, –NHCHCH2CH2CH2CH2–), 1.82 (m, 1H, –NHCHCH2CH2CH2CH2–), 1.96, 2.02, 2.07, 2.12 (4 s, 12H, –(CO)OCH3), 3.40 (m, 2H, –NHCHCH2CH2CH2CH2–), 3.57–3.76 (m, 8H, –OCH2CH2OCH2CH2NH–), 4.00 (m, 1H, H-5), 4.10 (m, 1H, H-6), 4.24 (m, 2H, H-6′, –NHCH(R)C(O)OH), 4.88 (br, 1H, H-1), 5.06 (m, 2H, –CH2C6H5), 5.20–5.32 (m, 3H, H-2, H-3, H-4), 5.77 (br, 1H, NHCH(R)C(O)OH), 6.40/6.74 (2 br, 2H, –NHC(S)NH–), 7.30 (m, 5H, –CH2C6H5); 13C NMR (CDCl3): δ/ppm = 20.67, 20.72, 20.90, 22.42, 28.31, 31.69, 43.92, 44.33, 53.93, 62.52, 66.11, 66.85, 67.00, 68.47, 69.10, 69.70, 69.86, 70.15, 97.48, 128.01, 128.14, 128.48, 136.15, 156.34, 169.73, 170.41, 170.66, 170.79, 171.17, 211.62; FTIR (cm−1) = 977, 1042, 1135, 1216, 1367, 1436, 1454, 1536, 1739, 2867, 2935, 3344; ESI-MS (m/z) [M + H]+ calcd for C33H47N3O15S, 757.80; found, 757.31.
O)OCH3), 3.46 (m, 2H, –NHCHCH2CH2CH2CH2–), 3.55–3.96 (m, 9H, –OCH2CH2OCH2CH2NH–, H-5), 4.14 (dd, 1H, H-6), 4.26 (m, 2H, H-6′ –NHCH(R)C(O)OH), 4.54 (br, 1H, H-1), 4.97 (m, 2H, H-2), 5.09 (m, 3H, –CH2C6H5, H-4), 5.22 (d, 3H, H-3), 5.60 (d, 1H, –NHCH(R)C(O)OH), 6.33/6.72 (2 br, 2H, –NHC(S)NH–), 7.27–34 (m, 5H, –CH2C6H5); 13C NMR (CDCl3): δ/ppm = 20.57, 20.70, 20.78, 22.26, 28.30, 31.78, 43.82, 44.36, 53.49, 61.86, 67.04, 68.37, 68.91, 69.61, 69.88, 71.58, 71.88, 72.45, 100.83, 128.07, 128.18, 128.52, 136.19, 156.12, 169.61, 170.34, 170.79, 174.81, 211.63; FTIR (cm−1) = 909, 1031, 1118, 1213, 1366, 1455, 1531, 1738, 2871, 2938, 3356; ESI-MS (m/z) [M + H]+, calcd for C33H47N3O15S, 757.80; found, 757.31.
O)OCH3), 3.46 (m, 2H, –NHCHCH2CH2CH2CH2–), 3.54–3.97 (m, 9H, –OCH2CH2OCH2CH2NH–, H-5), 4.10 (dd, 1H, H-6), 4.16 (dd, 1H, H-6′), 4.35 (m, 1H, –NHCH(R)C(O)OH), 4.48 (d, 1H, H-1), 5.04 (dd, 2H, H-3), 5.08 (br, 2H, –CH2C6H5), 5.15 (dd, 1H, H-2), 5.38 (dd, 3H, H-4), 5.65 (d, 1H, – NHCH(R)C(O)OH), 6.37/6.83 (2 br, 2H, –NHC(S)NH–), 7.27–7.33 (m, 5H, –CH2C6H5); 13C NMR (CDCl3): δ/ppm = 20.57, 20.62, 20.67, 20.88, 22.29, 28.32, 31.74, 43.77, 44.23, 53.54, 61.19, 66.91, 67.02, 68.93, 69.17, 69.54, 71.53, 70.69, 101.30, 128.06, 128.18, 128.51, 136.10, 156.13, 170.18, 170.22, 170.56, 175.12, 211.60; FTIR (cm−1) = 955, 1040, 1132, 1175, 1215, 1367, 1436, 1455, 1532, 1738, 2870, 2935, 3353; ESI-MS (m/z) [M + H]+ calcd for C33H47N3O15S, 757.80; found, 757.31.
O)OCH3), 3.18 (m, 2H, –NHCHCH2CH2CH2CH2–), 3.35 (m, 2H, OCH2CH2NH–), 3.45–3.86 (m, 6H, –OCH2CH2OCH2CH2NH–), 4.02–4.07 (m, 3H, H-5, H-6, –NHCH–), 4.25 (dd, 1H, H-6′), 4.84 (m, 1H, H-1), 5.22–5.26 (m, 3H, H-2, H-3, H-4); 13C NMR (CDCl3): δ/ppm = 20.68, 20.85, 23.59, 30.73, 31.12, 37.85, 46.21, 62.37, 66.06, 67.29, 68.40, 69.05, 69.44, 69.80, 71.06, 97.68, 150.90, 169.69, 169.92, 170.65, 211.45; FTIR (cm−1) = 973, 1043, 1134, 1217, 1367, 1432, 1532, 1657, 1740, 1783, 1852, 2934.
O)OCH3), 3.12–3.89 (m, 12H, –NHCHCH2CH2CH2CH2–, –OCH2CH2OCH2CH2NH–, –NHCH–, H-5), 4.07–4.13 (m, 1H, H-6), 4.12–4.25 (m, 1H, H-6′), 4.56 (m, 1H, H-1), 4.94 (m, 1H, H-2), 5.06 (t, 1H, H-4), 5.19 (m, 1H, H-3); 13C NMR (CDCl3): δ/ppm = 20.57, 20.72, 21.00, 23.69, 31.19, 31.44, 36.39, 46.31, 61.93, 68.41, 69.02, 69.93, 71.22, 71.77, 72.78, 100.86, 151.28, 169.27, 169.37, 170.16, 170.23, 170.62, 211.63; FTIR (cm−1) = 907, 1032, 1120, 1213, 1365, 1433, 1454, 1532, 1651, 1742, 1783, 1852, 2867, 2939, 3306.
O)OCH3), 3.27–4.38 (m, 12H, –NHCHCH2CH2CH2CH2–, –OCH2CH2OCH2CH2NH–, –NHCH–, H-5, H-6), 4.53 (m, 1H, H-1), 5.03 (m, 1H, H-3), 5.13 (m, 1H, H-2), 5.37 (m, 1H, H-4); 13C NMR (CDCl3): δ/ppm = 20.54, 20.63, 20.76, 24.04, 31.16, 31.25, 38.74, 46.33, 61.21, 67.01, 68.77, 68.91, 69.96, 70.05, 70.67, 70.84, 101.32, 151.68, 170.09, 170.18, 170.32, 211.62; FTIR (cm−1) = 955, 1043, 1126, 1173, 1215, 1366, 1432, 1531, 1658, 1742, 1783, 1852, 2870, 2936, 3295.
O)OCH3), 3.63–3.85 (m, 8H, –OCH2CH2OCH2CH2NCS), 4.01–4.06 (m, 1H, H-5), 4.07–4.11 (m, 1H, H-6), 4.26 (dd, 1H, H-6′), 4.86 (d, 3JHH = 1.5 Hz, 1H, H-1), 5.24 (dd, 1H, H-2), 5.25 (t, 1H, H-4), 5.33 (dd, 1H, H-3); 13C NMR (CDCl3): δ/ppm = 20.62, 20.66, 20.70, 20.83, 45.26, 62.47, 66.07, 67.30, 68.47, 69.02, 69.37, 69.46, 70.15, 97.68, 133.01, 169.68, 169.85, 169.97, 170.57; FTIR (cm−1) = 1043, 1217, 1738, 2106, 2938, 3241, 3360; ESI-MS (m/z) [M + Na]+ calcd for C19H27NNaO11S, 500.12; found, 500.36.
O)OCH3), 3.61–3.67 (m, 6H, –OCH2CH2OCH2CH2NCS), 3.69–3.97 (m, 3H, –OCH2CH2O, H-5), 4.13 (dd, 1H, H-6), 4.25 (dd, 1H, H-6′), 4.59 (d, 3JHH = 8.0 Hz, 1H, H-1), 4.97 (dd, 1H, H-2), 5.06 (t, 1H, H-4), 5.21 (t, 1H, H-3); 13C NMR (CDCl3): δ/ppm = 20.57, 20.58, 20.67, 20.73, 45.21, 61.86, 68.30, 69.10, 69.31, 70.35, 71.23, 71.76, 72.72, 100.73, 132.71, 169.42, 169.44, 170.23, 170.68; FTIR (cm−1) = 1032, 1208, 1745, 2140, 2201, 2884, 2955, 3238, 3356; ESI-MS (m/z) [M + Na]+ calcd for C19H27NNaO11S, 500.12; found, 500.36.
O)OCH3), 3.62–3.67 (m, 6H, –OCH2CH2OCH2CH2NCS), 3.71–3.97 (m, 3H, –OCH2CH2O, H-5), 4.06–4.18 (m, 2H, H-6), 4.55 (d, 3JHH = 8.0 Hz, 1H, H-1), 5.02 (dd, 1H, H-3), 5.18 (dd, 1H, H-2), 5.37 (dd, 1H, H-4); 13C NMR (CDCl3): δ/ppm = 20.53, 20.62, 20.64, 20.76, 45.26, 61.21, 66.97, 68.75, 69.06, 69.31, 70.37, 70.64, 70.81, 101.24, 132.69, 169.96, 170.07, 170.20, 170.36; FTIR (cm−1) = 1042, 1214, 1741, 2113, 2200, 2875, 2941, 3183, 3357; ESI-MS (m/z) [M + Na]+ calcd for C19H27NNaO11S, 500.12; found, 500.36.
:2:0.6, v/v/v). The combined organic fractions were concentrated in vacuum to a residual volume of about 10 mL, diluted with 150 mL ethyl acetate and washed several times with sodium hydrogen carbonate and sodium chloride to remove the acetic acid. Finally, the organic fraction was dried over anhydrous MgSO4 and the solvent was evaporated under vacuum. The resulting white solid was received in 43% yield. 1H NMR (CDCl3): δ/ppm = 1.39 (m, 2H, –NHCHCH2CH2CH2CH2–), 1.55 (m, 2H, –NHCHCH2CH2CH2CH2–), 1.72 (m, 1H, –NHCHCH2CH2CH2CH2–), 1.82 (m, 1H, –NHCHCH2CH2CH2CH2–), 1.96, 2.02, 2.07, 2.12 (4 s, 12H, –(CO)OCH3), 3.40 (m, 2H, –NHCHCH2CH2CH2CH2–), 3.57–3.76 (m, 8H, –OCH2CH2OCH2CH2NH–), 4.00 (m, 1H, H-5), 4.10 (m, 1H, H-6), 4.24 (m, 2H, H-6′, –NHCH(R)C(O)OH), 4.88 (br, 1H, H-1), 5.06 (m, 2H, –CH2C6H5), 5.20–5.32 (m, 3H, H-2, H-3, H-4), 5.77 (br, 1H, NHCH(R)C(O)OH), 6.40/6.74 (2 br, 2H, –NHC(S)NH–), 7.30 (m, 5H, –CH2C6H5); 13C NMR (CDCl3): δ/ppm = 20.67, 20.72, 20.90, 22.42, 28.31, 31.69, 43.92, 44.33, 53.93, 62.52, 66.11, 66.85, 67.00, 68.47, 69.10, 69.70, 69.86, 70.15, 97.48, 128.01, 128.14, 128.48, 136.15, 156.34, 169.73, 170.41, 170.66, 170.79, 171.17, 211.62; FTIR (cm−1) = 977, 1042, 1135, 1216, 1367, 1436, 1454, 1536, 1739, 2867, 2935, 3344; ESI-MS (m/z) [M + H]+ calcd for C33H47N3O15S, 757.80; found, 757.31.
O)OCH3), 3.46 (m, 2H, –NHCHCH2CH2CH2CH2–), 3.55–3.96 (m, 9H, –OCH2CH2OCH2CH2NH–, H-5), 4.14 (dd, 1H, H-6), 4.26 (m, 2H, H-6′ –NHCH(R)C(O)OH), 4.54 (br, 1H, H-1), 4.97 (m, 2H, H-2), 5.09 (m, 3H, –CH2C6H5, H-4), 5.22 (d, 3H, H-3), 5.60 (d, 1H, –NHCH(R)C(O)OH), 6.33/6.72 (2 br, 2H, –NHC(S)NH–), 7.27–34 (m, 5H, –CH2C6H5); 13C NMR (CDCl3): δ/ppm = 20.57, 20.70, 20.78, 22.26, 28.30, 31.78, 43.82, 44.36, 53.49, 61.86, 67.04, 68.37, 68.91, 69.61, 69.88, 71.58, 71.88, 72.45, 100.83, 128.07, 128.18, 128.52, 136.19, 156.12, 169.61, 170.34, 170.79, 174.81, 211.63; FTIR (cm−1) = 909, 1031, 1118, 1213, 1366, 1455, 1531, 1738, 2871, 2938, 3356; ESI-MS (m/z) [M + H]+, calcd for C33H47N3O15S, 757.80; found, 757.31.
O)OCH3), 3.46 (m, 2H, –NHCHCH2CH2CH2CH2–), 3.54–3.97 (m, 9H, –OCH2CH2OCH2CH2NH–, H-5), 4.10 (dd, 1H, H-6), 4.16 (dd, 1H, H-6′), 4.35 (m, 1H, –NHCH(R)C(O)OH), 4.48 (d, 1H, H-1), 5.04 (dd, 2H, H-3), 5.08 (br, 2H, –CH2C6H5), 5.15 (dd, 1H, H-2), 5.38 (dd, 3H, H-4), 5.65 (d, 1H, – NHCH(R)C(O)OH), 6.37/6.83 (2 br, 2H, –NHC(S)NH–), 7.27–7.33 (m, 5H, –CH2C6H5); 13C NMR (CDCl3): δ/ppm = 20.57, 20.62, 20.67, 20.88, 22.29, 28.32, 31.74, 43.77, 44.23, 53.54, 61.19, 66.91, 67.02, 68.93, 69.17, 69.54, 71.53, 70.69, 101.30, 128.06, 128.18, 128.51, 136.10, 156.13, 170.18, 170.22, 170.56, 175.12, 211.60; FTIR (cm−1) = 955, 1040, 1132, 1175, 1215, 1367, 1436, 1455, 1532, 1738, 2870, 2935, 3353; ESI-MS (m/z) [M + H]+ calcd for C33H47N3O15S, 757.80; found, 757.31.
O)OCH3), 3.18 (m, 2H, –NHCHCH2CH2CH2CH2–), 3.35 (m, 2H, OCH2CH2NH–), 3.45–3.86 (m, 6H, –OCH2CH2OCH2CH2NH–), 4.02–4.07 (m, 3H, H-5, H-6, –NHCH–), 4.25 (dd, 1H, H-6′), 4.84 (m, 1H, H-1), 5.22–5.26 (m, 3H, H-2, H-3, H-4); 13C NMR (CDCl3): δ/ppm = 20.68, 20.85, 23.59, 30.73, 31.12, 37.85, 46.21, 62.37, 66.06, 67.29, 68.40, 69.05, 69.44, 69.80, 71.06, 97.68, 150.90, 169.69, 169.92, 170.65, 211.45; FTIR (cm−1) = 973, 1043, 1134, 1217, 1367, 1432, 1532, 1657, 1740, 1783, 1852, 2934.
O)OCH3), 3.12–3.89 (m, 12H, –NHCHCH2CH2CH2CH2–, –OCH2CH2OCH2CH2NH–, –NHCH–, H-5), 4.07–4.13 (m, 1H, H-6), 4.12–4.25 (m, 1H, H-6′), 4.56 (m, 1H, H-1), 4.94 (m, 1H, H-2), 5.06 (t, 1H, H-4), 5.19 (m, 1H, H-3); 13C NMR (CDCl3): δ/ppm = 20.57, 20.72, 21.00, 23.69, 31.19, 31.44, 36.39, 46.31, 61.93, 68.41, 69.02, 69.93, 71.22, 71.77, 72.78, 100.86, 151.28, 169.27, 169.37, 170.16, 170.23, 170.62, 211.63; FTIR (cm−1) = 907, 1032, 1120, 1213, 1365, 1433, 1454, 1532, 1651, 1742, 1783, 1852, 2867, 2939, 3306.
O)OCH3), 3.27–4.38 (m, 12H, –NHCHCH2CH2CH2CH2–, –OCH2CH2OCH2CH2NH–, –NHCH–, H-5, H-6), 4.53 (m, 1H, H-1), 5.03 (m, 1H, H-3), 5.13 (m, 1H, H-2), 5.37 (m, 1H, H-4); 13C NMR (CDCl3): δ/ppm = 20.54, 20.63, 20.76, 24.04, 31.16, 31.25, 38.74, 46.33, 61.21, 67.01, 68.77, 68.91, 69.96, 70.05, 70.67, 70.84, 101.32, 151.68, 170.09, 170.18, 170.32, 211.62; FTIR (cm−1) = 955, 1043, 1126, 1173, 1215, 1366, 1432, 1531, 1658, 1742, 1783, 1852, 2870, 2936, 3295.
For the synthesis of the terpolymer 5c, Nε-trifluoroacetyl-L-lysine-N-carboxyanhydride (0.062 g, 0.23 mmol), Nε-(3,6,9-trioxa-decanoyl)-L-lysine-N-carboxyanhydride (1.23 g, 3.70 mmol), and the glycosylated L-lysine-NCA 3c (0.30 g, 0.46 mmol) were polymerized with triethyl amine (15 mg, 20 μL, 0.15 mmol) according to the same procedure. The copolymer 5u was synthesized accordingly using triethyl amine (6 mg, 8 μL, 0.06 mmol), Nε-(3,6,9-trioxa-decanoyl)-L-lysine-N-carboxyanhydride (0.92 g, 2.75 mmol) and Nε-trifluoroacetyl-L-lysine-N-carboxyanhydride (0.041 g, 0.15 mmol).
For the synthesis of the terpolymers 5a and 5b, Nε-trifluoroacetyl-L-lysine-N-carboxyanhydride (0.1 g, 0.38 mmol), Nε-(3,6,9-trioxa-decanoyl)-L-lysine-N-carboxyanhydride (2.0 g, 6.16 mmol), and the glycosylated L-lysine-NCA 3a respectively 3b (0.50 g, 0.77 mmol) were polymerized with bpyNi(COD) (30 mg, 0.09 mmol) according to the same procedure.
O)OCH3), 3.21–3.34 (m, –NHCHCH2CH2CH2CH2–, CH3OCH2–), 3.52–4.25 (m, –NHCH(R)C(O)–, –OCH2CH2OCH2CH2OCH2C(O)NH–, –OCH2CH2OCH2CH2NH–, H-5, H-6), 4.84 (m, H-1), 5.25 (m, H-2, H-3, H-4), 7.17 (br, NH), 8.19 (s, NH), 8.80 (s, NH); FTIR (cm−1) = 3292, 3066, 2921, 2867, 1747, 1719, 1650, 1536, 1440, 1369, 1338, 1283, 1224, 1200, 1100, 1047, 981, 933, 849.
O)OCH3), 3.21–3.34 (m, –NHCHCH2CH2CH2CH2–, CH3OCH2–), 3.51–4.26 (m, –NHCH(R)C(O)–, –OCH2CH2OCH2CH2OCH2C(O)NH–, –OCH2CH2OCH2CH2NH–, H-5, H-6), 4.57 (m, H-1), 4.95 (m, H-2), 5.05 (t, H-4), 5.19 (m, H-3), 7.17 (br, NH), 8.20 (s, NH), 8.80 (s, NH); FTIR (cm−1) = 3292, 3067, 2923, 2866, 1754, 1719, 1650, 1537, 1440, 1365, 1339, 1281 1223, 1099, 1034, 931, 849.
O)OCH3), 3.22–3.34 (m, –NHCHCH2CH2CH2CH2–, CH3OCH2–), 3.52–4.12 (m, –NHCH(R)C(O)–, –OCH2CH2OCH2CH2OCH2C(O)NH–, –OCH2CH2OCH2CH2NH–, H-5, H-6), 4.54 (m, H-1), 5.02 (m, H-3), 5.15 (t, H-2), 5.37 (m, H-4), 7.14 (br, NH), 8.19 (s, NH), 8.79 (s, NH); FTIR (cm−1) = 3287, 3067, 2921, 2866, 1749, 1719, 1650, 1534, 1439, 1369, 1339, 1221, 1100, 928, 849.
:1 mixture of 0.25 M aqueous sodium chloride and methanol, 250 mL of a 1:1 mixture of distilled water and methanol, and finally, 1 L of methanol. After evaporation of the solvent and dissolution of the residue in water the product was obtained after lyophilization as an orange, cotton-like solid.
Biochemical procedures
000 events per sample was measured by using a FACSCalibur cytometer (BD Biosciences) and CellQuest Pro software (version 5.1.1, BD Biosciences). Fluorescence intensities of gated T cell populations are represented by histograms and geometric means calculated by CellQuest Pro.
Results and discussion
Our intention was to synthesize glycosylated polypeptides soluble in water. They should be neutral to avoid unspecific interactions with cells. Because polyglutamine of a low degree of polymerization (DP < 20) and copolymers with polyglutamine blocks not longer than twenty are known to be water-soluble,36,37 we synthesized a statistical copolymer poly(glutamine-co-lysine) by copolymerization of a mixture of Nε-trityl-glutamine-NCA and TFA-lysine-NCA (molar ratio 5:1) using NEt3 as the catalyst.21 The lysine moieties were set aside for the attachment of carbohydrate moieties at the ε-amino groups by thiourea linkages. After deprotection of the copolymer, the compound was found to be totally insoluble in water as well as in other solvents. Therefore, all attempts to attach carbohydrate moieties were not successful.
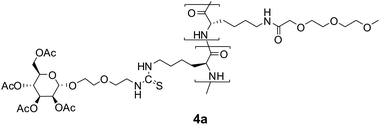
Because we were not aware of any other neutral water-soluble homopolypeptides consisting of natural amino acids, we chose non-natural poly(Nε-(3,6,9-trioxo-decanoyl)-L-lysine) as the backbone, which is known to be soluble in water.25Statistical copolymers 4 of Nε-(3,6,9-trioxo-decanoyl)-L-lysine and a sugar-appended lysine were synthesized by NCA copolymerization of the corresponding NCAs. The sugar-appended lysine-NCA 3 was synthesized in three steps according to Fig. 1 with glycosylation of 2-(2-isothiocyanatoethoxy)ethanol by 2,3,4,6-tetra-O-acetylglycopyranosyl-1-trichloroacetimidate 1 and subsequent coupling of the isothiocyanate group of the products to the ε-amino group of Z-lysine. In all three cases, we observed stereoselective glycosylationtrans to the 2-O-acetyl groups according to the well-known 1,2-trans directing effect of acetyl neighbour groups leading to the α-mannoside, the β-glucoside and β-galactoside, respectively.32 The 1H NMR spectra were consistent with those of related glycosides38,39 and show large coupling constants between H-1 and H-2, 3JHH = 8.0 Hz, characteristic of both, the β-glucoside and the β-galactoside. Finally, direct conversion of the Z-protected glycosylated amino acid 2 to the NCA with dichloromethyl methyl ether was performed.40 The results of the NCA copolymerizations are summarized in Table 1. 1H NMR spectra of both the monomers 3a–c and the corresponding copolymers 4a–c are shown in the ESI†.
:1) in DMF solution leading to polymers 4a, 4b and 4c, respectively
|
|
||||||
|---|---|---|---|---|---|---|
| Initiator | Sugar | Sugar (mol%) | Yield (%) | M w/g mol−1 | PDI | No. |
| a Polymerization in THF solution. | ||||||
| NEt3 | Man | 10.0 | 74 | 40000 |
1.7 | 4a |
| NEt3 | Glc | 10.0 | 70 | 44000 |
2.0 | 4b |
| NEt3 | Gal | 10.0 | 91 | 37400 |
1.6 | 4c |
| Ni0a | Man | 9.1 | 95 | 130000a |
1.2 | 4a′ |
| Ni0 | Glc | 9.6 | 89 | 29400 |
1.4 | 4b′ |
| Ni0 | Gal | 9.1 | 95 | 38000 |
1.2 | 4c′ |
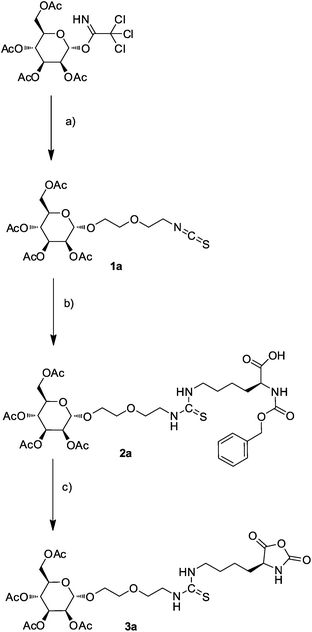 | ||
| Fig. 1 Synthesis of mannose derivative of lysine–NCA 3a; (a) 2-(2-isothiocyanatoethoxy)ethanol, TMS triflate in CH2Cl2, (b) Nα-benzyloxycarbonyl-L-lysine, DIPEA in DMF, (c) dichloromethyl methyl ether in CH2Cl2. | ||
Both employed catalysts, NEt3 and the Ni0 2,2′-bipyridine 1,5-cyclooctadiene complex, furnished the polypeptides in high yields and molecular weights of 30000 to 50000 g mol−1. Higher molecular weights were obtained if THF was used as the reaction medium instead of DMF. The low polydispersity indices (PDIs) of around 1.2 obtained with the Ni0 catalysts are typical for living polymerizations. Therefore, they are well-suited for the synthesis of glycopeptides. These PDIs were significantly lower than the ones obtained with NEt3.
Furthermore, it was desirable to attach fluorescent labels, such as fluorescein, to the glycopolypeptides to allow the detection of any interaction with biological systems. Because synthesis and polymerization of lysine–NCA conjugates with fluorescein were rather difficult, it was decided to synthesize terpolymers with a small amount of TFA protected lysine, which would allow the subsequent attachment of fluorescein in a polymer-analogous reaction. Terpolymerizations of Nε-(3,6,9-trioxo-decanoyl)-L-lysine-NCA with glycosylated L-lysine-NCAs and TFA-lysine-NCA performed well giving rise to soluble polymers in good yields and high molecular weights and narrow PDIs, listed in Table 2. The Ni0 catalysts were superior to NEt3 in terms of the yields, but not in terms of the PDIs. Again, the compositions of the synthesized polymers were identical with the employed feed ratios of the three monomers, allowing for the predictable synthesis of well-defined polymers.
:2:1) in DMF solution leading to polymers 5a, 5b, and 5c, and to final polymers 6a, 6b, 6c, respectively
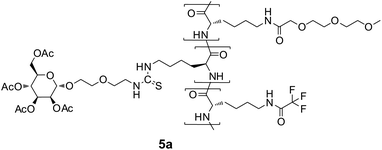
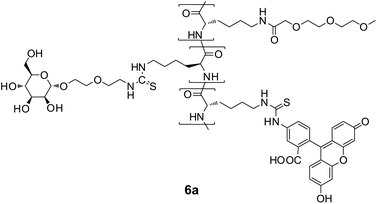
After deprotection of both the sugar residues and the lysine units with NaOCH3, fluorescein isothiocyanate (FITC) was coupled almost quantitatively to the ε-amino groups of the lysine units. The resulting labeled glycopolypeptides 6a–c as well as 6u were soluble in water and showed a bright fluorescence at λ = 520 nm in FBS buffer solution at pH = 7.5. The fluorescence intensities are given in Table 2. According to their IR spectra with typical bands at 1535 and 1650 cm−1 they have α-helical structures.25
The in vitro toxicities of polymers 6a–c as well as 6u were tested with human T lymphocytes using the XTT cell viability assay.41 It was found that natural cell viability remains preserved for polymer concentrations of up to 0.33 mg mL−1, shown in Fig. 2. Also, microscopic investigations revealed that T cells did not change their appearances within 3 d after exposure to solutions of these polymers. From these observations, we conclude that polymers 6a–c are sufficiently biocompatible.
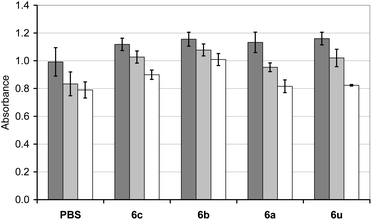 | ||
| Fig. 2 Viabilities of human T lymphocytes (concentration 0.33 mg mL−1) after a 24 h (dark grey bar), 48 h (pale grey bar) and 72 h (white bar) incubation with polymer 6c. | ||
The interactions of polymers 6a–c with human T lymphocytes were investigated by flow cytometry (FCM). The fluorescence intensity distributions only showed a pronounced shift to higher fluorescence intensities for the galactosylated polymer 6c, especially for the incubations at 37 °C, shown in Fig. 3. The uptakes of the fluorescent polymers (Fig. 4) were quantified by calculating the geometric mean of fluorescence intensity over the cell populations and normalized on their different fluorescence intensities taken from Table 2. While there was only little uptake of the fluorescent unsubstituted polymer 6u and the glucose polymer 6b, the mannose polymer 6a and the galactose polymer 6c were readily taken up, especially at 37 °C. Interestingly, this uptake was only observed with activated T lymphocytes, which means that activation of the cells should lead to expression of galactose receptors incorporated in the cell walls. These receptors might be related to L-selectin, which is known to bind the galactose-containing tetrasaccharide sialyl Lewis X and galactose clusters.42,43 The glycoprotein L-selectin (CD62L) is expressed on the surface of T lymphocytes and plays a crucial role during T cell recirculation.44 By binding to its ligands, CD62L initiates the extravasation of T lymphocytes into peripheral lymphoid tissues.
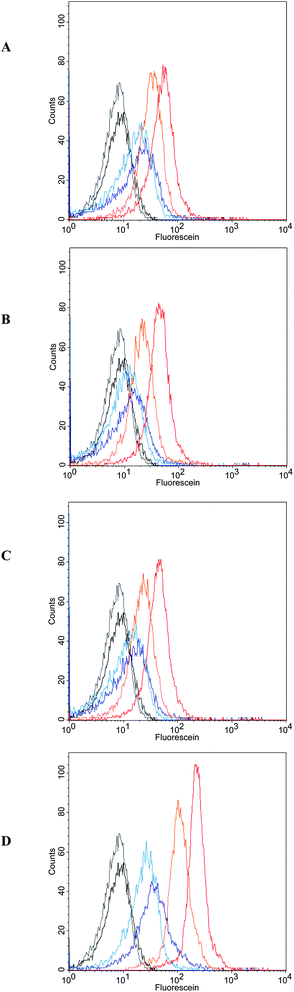 | ||
| Fig. 3 FCM fluorescence intensity distributions of T lymphocytes at 4 °C (black) and at 37 °C (grey), after incubation at 4 °C (pale blue, 2h; dark blue, 8h) and 37 °C (pale orange, 2h; dark orange, 8h) with polypeptides (A) unsubstituted 6u, (B) mannosylated 6a, (C) glucosylated 6b, and (D) galactosylated 6c. | ||
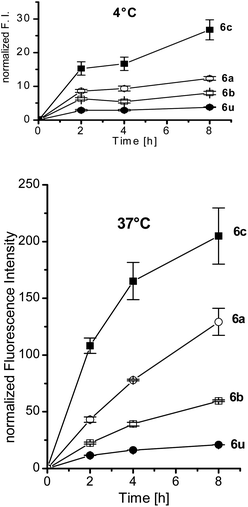 | ||
| Fig. 4 Uptake of the glycopolymers 6a–c and the unsubstituted polymer 6u by T cells at 4 °C and 37 °C, as determined from the geometric mean of FCM fluorescence intensities, normalized by the fluorescence intensity of the polymer. | ||
However, during activation of T lymphocytes, CD62L is down-regulated,45 which is in contrast to our finding of solely activated T lymphocytes taking up polymer 6c significantly. Consequently, L-selectin might not be the desired receptor. Another receptor candidate might be among the members of the galectin family. These mammal lectins exhibit affinity for β-galactosides.46 Galectins can exist as dimers or even pentamers.47 Therefore, extracellularly secreted galectins are able to form lattices with glycoprotein receptors on the cell surface. This process enables the cell to modulate various receptor functions.48 Our galactosylated polymer 6c might bind multivalently to these extracellular galectin lattices. This hypothesis is supported by the experimental results of both galectin-1 and galectin-3 being up-regulated upon T cell activation.49,50
The pronounced temperature dependence of the interaction of the galactosylated polymer with the T lymphocytes might be rationalized by the higher cell membrane mobility at the elevated temperature (37 °C) which should allow receptor-mediated endocytosis, which should lead to an enrichment of the fluorescent polymer 6c within the cells. Fluorescence microscopy studies of the T cells incubated with solutions (conc. 0.10 mg mL−1) of polymers 6a, 6b as well as 6c for 6 h, shown in Fig. 5, indeed demonstrated that only the galactosylated polymer 6c showed significant interactions with the T cells. Most cells took over the green fluorescence of fluorescein. The picture also provides evidence for the fact that the polymer 6c is not only adsorbed at the cell wall but internalized into the cytoplasm of the cell, while the nuclei of the cells stained in blue remained unaffected. This finding is clearly indicative for the uptake of galactosylated polymer 6c within the T lymphocytes by endocytosis. Addition of both galactose and lactose (concentrations up to 0.1 mol L−1) could not inhibit the uptake of polymer 6c in these cells.
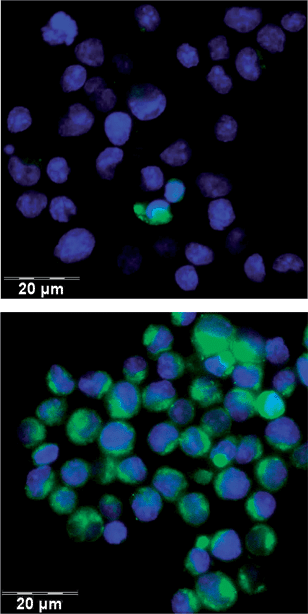 | ||
| Fig. 5 Imaging of fluorescein (green) in the T lymphocytes by fluorescence microscopy after 6 h incubation at 4 °C (upper image) and 37 °C (lower image) with the galactosylated polymer 6c. The cell nuclei were stained blue by DAPI. | ||
Conclusions
PEGylated polylysine is a readily available and water-soluble scaffold for the design of polymeric neoglycoconjugates. PEGylated polylysine with conjugated galactose residues allows the recognition of activated T lymphocytes. It is potentially useful for targeting covalently attached active substances, e.g., drugs, into lymphocytes.Acknowledgements
This work was funded by the project, “Polymere für die zeitkontrollierte Medikamentenfreisetzung in der Immuntherapie” Saarbridge by Saarland Ministerium für Wirtschaft und Wissenschaft.Notes and references
- H. J. Gabius, H. C. Siebert, S. Andre, J. Jimenez-Barbero and H. Rudiger, ChemBioChem, 2004, 5, 741–764 CrossRef.
- C. R. Bertozzi and L. L. Kiessling, Science, 2001, 291, 2357–2364 CrossRef CAS.
- K. N. Houk, A. G. Leach, S. P. Kim and X. Y. Zhang, Angew. Chem., Int. Ed., 2003, 42, 4872–4897 CrossRef CAS.
- M. Mammen, S.-K. Chio and G. M. Whitesides, Angew. Chem., Int. Ed., 1998, 37, 2755–2794 CrossRef CAS.
- D. Rabuka, R. Parthasarathy, G. S. Lee, X. Chen, J. T. Groves and C. R. Bertozzi, J. Am. Chem. Soc., 2007, 129, 5462–5471 CrossRef CAS.
- D. Rabuka, M. B. Forstner, J. T. Groves and C. R. Bertozzi, J. Am. Chem. Soc., 2008, 130, 5947–5953 CrossRef CAS.
- X. Chen, U. C. Tam, J. L. Czlapinski, G. S. Lee, D. Rabuka, A. Zettl and C. R. Bertozzi, J. Am. Chem. Soc., 2006, 128, 6292–6293 CrossRef CAS.
- S. Andre, H. Kaltner, T. Furuike, S. I. Nishimura and H. J. Gabius, Bioconjugate Chem., 2004, 15, 87–98 CrossRef CAS.
- M. Gómez-García, J. M. Benito, D. Rodríguez-Lucena, J.-X. Yu, K. Chmurski, C. Ortiz-Mellet, R. G. Gallego, A. Maestre, J. Defaye and J. M. G. Fernández, J. Am. Chem. Soc., 2005, 7970–7971 CrossRef.
- F. Ortega-Caballero, J. J. Gimenez-Martinez, L. Garcia-Fuentes, E. Ortiz-Salmeron, F. Santoyo-Gonzalez and A. Vargas-Berenguel, J. Org. Chem., 2001, 66, 7786–7795 CrossRef CAS.
- P. R. Ashton, S. E. Boyd, C. L. Brown, S. A. Nepogodiev, E. W. Meijer, H. W. I. Peerlings and J. F. Stoddart, Chem.–Eur. J., 1997, 3, 974–984 CrossRef CAS.
- C. M. Lehr, J. Controlled Release, 2000, 65, 19–29 CrossRef CAS.
- C. Bies, C. M. Lehr and J. F. Woodley, Adv. Drug Delivery Rev., 2004, 56, 425–435 CrossRef CAS.
- D. Neumann, C. M. Lehr, H. P. Lenhof and O. Kohlbacher, Adv. Drug Delivery Rev., 2004, 56, 437–457 CrossRef CAS.
- M. Gomez-Garcia, J. M. Benito, R. Gutierrez-Gallego, A. Maestre, C. O. Mellet, J. M. G. Fernandez and J. L. J. Blanco, Org. Biomol. Chem., 2010, 8, 1849–1860 CAS.
- A. Vargas-Berenguel, F. Ortega-Caballero and J. M. Casas-Solvas, Mini-Rev. Org. Chem., 2007, 4, 1–14 CrossRef CAS.
- J. Kim, Y. Ahn, K. M. Park, D.-W. Lee and K. Kim, Chem.–Eur. J., 2010, 16, 12168–12173 CrossRef CAS.
- M. Ortega-Munoz, J. Morales-Sanfrutos, F. Perez-Balderas, F. Hernandez-Mateo, M. D. Giron-Gonzalez, N. Sevillano-Tripero, R. Salto-Gonzalez and F. Santoyo-Gonzalez, Org. Biomol. Chem., 2007, 5, 2291–2301 CAS.
- Y. Oda, N. Kobayashi, T. Yamanoi, K. Katsuraya, K. Takahashi and K. Hattori, Med. Chem., 2008, 4, 244–255 CrossRef CAS.
- S. Matsumura, S. Sakamoto, A. Ueno and H. Mihara, Chem.–Eur. J., 2000, 6, 1781–1788 CrossRef CAS.
- H. R. Kricheldorf, Angew. Chem., Int. Ed., 2006, 45, 5752–5784 CrossRef CAS.
- G. J. M. Habraken, K. H. R. M. Wilsens, C. E. Koning and A. Heise, Polym. Chem., 2011, 2, 1322–1330 RSC.
- J. N. Cha, G. D. Stucky, D. E. Morse and T. J. Deming, Nature, 2000, 403, 289–292 CrossRef CAS.
- T. J. Deming, Prog. Polym. Sci., 2007, 32, 858–875 CrossRef CAS.
- M. Yu, A. P. Nowak, T. J. Deming and D. J. Pochan, J. Am. Chem. Soc., 1999, 121, 12210–12211 CrossRef CAS.
- R. Wang, N. Xu, F.-S. Du and Z.-C. Li, Chem. Commun., 2010, 46, 3902–3904 RSC.
- J. R. Kramer and T. J. Deming, J. Am. Chem. Soc., 2010, 132, 15068–15071 CrossRef CAS.
- D. E. Owens and N. A. Peppas, Int. J. Pharm., 2006, 307, 93–102 CrossRef CAS.
- U. Steinfeld, C. Pauli, N. Kaltz, C. Bergemann and H.-H. Lee, Int. J. Pharm., 2006, 311, 229–236 CrossRef CAS.
- C. Guillermain and B. Gallot, Macromol. Chem. Phys., 2002, 203, 1346–1356 CrossRef CAS.
- D. S. Poché, M. J. Moore and J. L. Bowles, Synth. Commun., 1999, 29, 843–854 CrossRef.
- R. R. Schmidt and W. Kinzy, Adv. Carbohydr. Chem. Biochem., 1994, 50, 21–123 CrossRef CAS.
- J. M. de la Fuente and S. Penades, Tetrahedron: Asymmetry, 2002, 13, 1879–1888 CrossRef CAS.
- H. Tajima and G. Li, Synlett, 1997, 773–774 CrossRef CAS.
- J. J. Eisch, A. M. Piotrowski, K. I. Han, C. Kruger and Y. H. Tsay, Organometallics, 1985, 4, 224–231 CrossRef CAS.
- S. Chen and R. Wetzel, Protein Sci., 2001, 10, 887–891 CrossRef CAS.
- E. L. Altschuler, N. V. Hud, J. A. Mazrimas and B. Rupp, J. Pept. Res., 1997, 50, 73–75 CrossRef CAS.
- E. K. Woller and M. J. Cloninger, Org. Lett., 2002, 4, 7–10 CrossRef CAS.
- Z.-J. Yin, Q. Li, X.-B. Meng and Z.-J. Li, Carbohydr. Res., 2007, 342, 2729–2734 CrossRef CAS.
- K. Poduska and H. Gross, Chem. Ber., 1961, 49, 527–537 CrossRef CAS.
- D. Scudiero, R. Shoemaker, K. Paull, A. Monks, S. Tierney, T. Nofziger, M. Currens, D. Seniff and M. Boyd, Cancer Res., 1988, 48, 4827–4833 CAS.
- W. S. Somers, J. Tang, G. D. Shaw and R. T. Camphausen, Cell, 2000, 103, 467–479 CrossRef CAS.
- I. Papp, J. Dernedde, S. Enders and R. Haag, Chem. Commun., 2008, 5851–5853 RSC.
- T. A. Springer, Cell, 1994, 76, 301–314 CrossRef CAS.
- C. C. Chao, R. Jensen and M. O. Dailey, J. Immunol., 1997, 159, 1686–1694 CAS.
- S. H. Barondes, V. Castronovo, D. N. W. Cooper, R. D. Cummings, K. Drickamer, T. Feizi, M. A. Gitt, J. Hirabayashi, C. Hughes, K. Kasai, H. Leffler, F. T. Liu, R. Lotan, A. M. Mercurio, M. Monsigny, S. Pillai, F. Poirer, A. Raz, P. W. J. Rigby, J. M. Rini and J. L. Wang, Cell, 1994, 76, 597–598 CrossRef CAS.
- F. T. Liu and G. A. Rabinovich, in Year in Immunology 2, 2010, pp. 158–182 Search PubMed.
- O. B. Garner and L. G. Baum, Biochem. Soc. Trans., 2008, 36, 1472–1477 CrossRef CAS.
- M. B. Fuertes, L. L. Molinero, M. A. Toscano, J. M. Ilarregui, N. Rubinstein, L. Fainboim, N. W. Zwirner and G. A. Rabinovich, Mol. Cell. Biochem., 2004, 267, 177–185 CrossRef CAS.
- H. G. Joo, P. S. Goedegebuure, N. Sadanaga, M. Nagoshi, W. von Bernstorff and T. J. Eberlein, J. Leukocyte Biol., 2001, 69, 555–564 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1py00187f |
| This journal is © The Royal Society of Chemistry 2011 |