Polystyrene grafted polyvinylidenefluoride copolymers with high capacitive performance†
Vijay Kumar
Thakur
a,
Eu Jin
Tan
a,
Meng-Fang
Lin
b and
Pooi See
Lee
*ab
aTemasek Laboratories@NTU, Research Techno Plaza, BorderX Block, 50 Nanyang Drive, Singapore, 637553
bSchool of Materials Science and Engineering, Nanyang Technological University, 50 Nanyang Avenue, Singapore, 639798
First published on 22nd June 2011
Abstract
Electroactive materials are of great interest for high performance capacitive behavior due to their relaxed ferroelectric properties. In this work, we studied the dielectric properties of poly(vinylidene fluoride) (PVDF) grafted with polystyrene (PS) by electron beam radiation induced free radical graft copolymerization reaction in solution. The dielectric constant of polystyrene grafted PVDF copolymers reaches about 90 at 100 Hz at room temperature representing more than seven times increment compared with the pristine PVDF matrix. The dielectric loss of 0.005 at 1 KHz can be achieved. Correlation of the dielectric properties with the graft copolymerization reaction mechanism was discussed. This route represents one of the most effective techniques to synthesize potential graft copolymers with desirable dielectric properties in a wide frequency range for energy storage applications.
Introduction
The continuous increase of energy requirement and exhaustion of fossil fuels has motivated researchers all over the world to improve the efficiency of energy usage as well as seeking new energy sources.1,2 Energy storage capacitors especially high energy density capacitors are devices that could take part of the responsibility to promote wide deployment of portable power in remote or suburb areas where power transmission lines are unavailable or to address power intermittencies. Electrostatic pulse power energy storage has been one of the focuses of interest due to its enormous potential and advantages such as high efficiency, versatile functions, reliability, environmentally friendliness etc.3–5 The necessity of developing high energy density capacitors has led to the development of polymer composite systems that combine the processability and breakdown field strength of the polymer with the high dielectric constant of ceramic fillers.6,7 Poly(vinylidene fluoride) (PVDF) polymer has been proposed as one of the most suitable dielectric material due to the combination of remarkable properties such as high stability, high volume resistivity, low water absorption rate, excellent thermal stability, low dielectric loss and a low shrinking rate.8–12 However, the dielectric permittivity of PVDF is low (7–13 at 10 Hz), so it could not satisfy the need for large capacitance requirements.13–17Copolymers such as PVDF-TrFE and PVDF-HFP have been shown to exhibit enhanced energy storage capabilities due to the relaxed ferroelectric behavior.1,8,10 However, still many improvements are required for these polymers such as higher dielectric constant, reduced dielectric loss and fast discharging. In particular, in the development of polymer composites, the inorganic filler usually has a greater permittivity than that of the polymer matrix, most of the increase in the effective dielectric constant comes through an increase in the average field in the polymer matrix, with very little of the energy being stored in the high permittivity phase.18–20 The huge difference in permittivity between the two phases also gives rise to highly inhomogeneous electric fields. Therefore, novel approach to improve the dielectric permittivity and the energy densities of polymers will lead to increased charging and storage ability of capacitors, and miniaturization of the capacitors could be realized.We have earlier shown the feasibility of grafting the hydrophilic HEMA chains onto PVDF, leading to enhanced dielectric constant.9 To improve the dielectric permittivity and to maintain the good stability of PVDF; we propose graft copolymerization of hydrophobic styrene monomer onto PVDF matrix. Such grafted copolymers would display the advantage of combining the excellent dielectric properties of PVDF and the processability of conventional polymers, and increase the robustness and stability of the resultant capacitors. Graft copolymerization of polymers is capable to form a great number of new materials with unique properties impacted by the ability to tailor surface properties of materials while retaining the bulk properties. Fluoropolymers such as PVDF and PTFE are known as chemically inert materials and the grafting technique is of most interest for the modification of these polymers, because these polymers are difficult to modify by purely chemical methods.21–23 Among numerous methods used for initiating graft copolymerization onto fluoropolymers; X-rays, γ-rays, excimers or Ar+ lasers; ultraviolet light, plasma treatment, electron and ion beam are feasible as fluoropolymers are relatively susceptible to high energy radiation.24–26 Through introduction of new functional groups to the fluoropolymers surface, properties such as hydrophilicity/hydrophobicity, adhesion, compatibility, reduction in crystallinity and conductivity may be attained. The traditional copolymerization reactions for such purposes are quiet tedious involving a number of chemicals along with chemical initiators and are not free from contamination. Herein, we present the effects of electron beam radiation induced graft copolymerization of styrene on the dielectric properties of the PVDF over wide ranges of frequencies. Our aim is to improve the dielectric properties and energy densities of PVDF by grafting with suitable functional groups without addition of inorganic additives, while keeping the cost low. The styrene was selected as the functional monomer as its polymer (polystyrene) has a moderate dielectric constant (2.6) among known liquid monomers and good compatibility with PVDF polymer because of its thermoplastic nature.
Experimental section
PVDF polymer samples used in this work were in powder form having a diameter of 0.1 mm (Solvay Solexis Inc). Styrene, chloroform, methanol and toluene were supplied by (Sigma-Aldrich Chemie GmbH, Germany).Irradiation of PVDF powder samples at different radiation doses (0.2–2.0 M Rad) was carried out at room temperature in vacuum at 175 kV using the Energy Sciences Inc. (ESI) Electron Beam Accelerator as reported earlier.9 After the irradiation of the polymer powder; the samples were immediately used for grafting. The graft polymerization synthesis was performed at a particular temperature for a definite time period under stirring in a heating mantle fitted with a reflux condenser which was sealed. In brief, irradiated PVDF (500 mg) was suspended in a known amount of toluene taken as solvent in a flask. A definite amount of the styrene monomer was added in the flask. Subsequently, nitrogen was purged through the solution for at least 30 min to remove oxygen. After the completion of reaction, the grafted powder (PVDF-g-PS) was filtered and washed many times with toluene, followed by extraction for 72 h with chloroform in Soxhlet apparatus to remove residual styrene monomer and any homopolymer possibly formed. Finally, the PVDF-g-PS powder was dried to constant weight in vacuum oven at 45 °C. The percentage of grafting (Pg) was determined gravimetrically from the initial increase in the weight of the original polymer powder as shown in eqn (1):
 | (1) |
A customized laboratory-scale tape caster was used for the preparation of the grafted as well as the ungrafted polymer films. The slurry was prepared by using 60 mg ml−1 of the PVDF/PVDF-g-PS polymer powder with DMF as the solvent. The slurries were cast onto glass carrier and dried in air at 60 °C for 5 h and in vacuum at 50 °C for 12 h to remove any traces of DMF solvent. The thickness of the tape was measured using a micrometre accurate to 0.001 mm; the weight of the samples was measured using a laboratory scale accurate to 0.01 mg.
Characterizations of the pristine PVDF and PVDF-g-PS samples were done using differential scanning calorimetry (DSC), thermogravimetric analysis (TGA), Fourier transform infrared spectroscopy (FTIR), contact angle measurements, X-ray diffractometry (XRD) and atomic force microscopy (AFM) (see corresponding descriptions in the ESI†). The grafted and ungrafted polymer films were characterized for electrical measurements using a Keithley 4200 semiconductor parameter analyzer and a HP4284 LCR meter in the frequency range of 100 Hz to 1 MHz at room temperatures.9 The electrical breakdown test was carried out using a high-voltage instrument (CS2674A). During the test, the voltage applied on the sample rose rapidly until the samples broke down.
Results and discussion
By using the graft copolymerization technique to modify the polymer surface, the properties can be tailored by judicious choice of monomer. In determining the percentage of grafting and the structural evolution of the grafted polymer, grafting conditions play a major role. The types of radicals formed and the structural changes which take place upon the irradiation of fluoropolymers have been reviewed by a number of researchers.27,28 Therefore, radiation chemistry of the fluoropolymers has not been discussed here and we have focused on the effects of irradiation on the properties of the grafted polymers. However, it is worth drawing attention to some of the common observations. For carrying out grafting onto PVDF polymer (step 1), C–C, C–H and C–F bonds are most susceptible to cleavage under radiation9 (Scheme 1).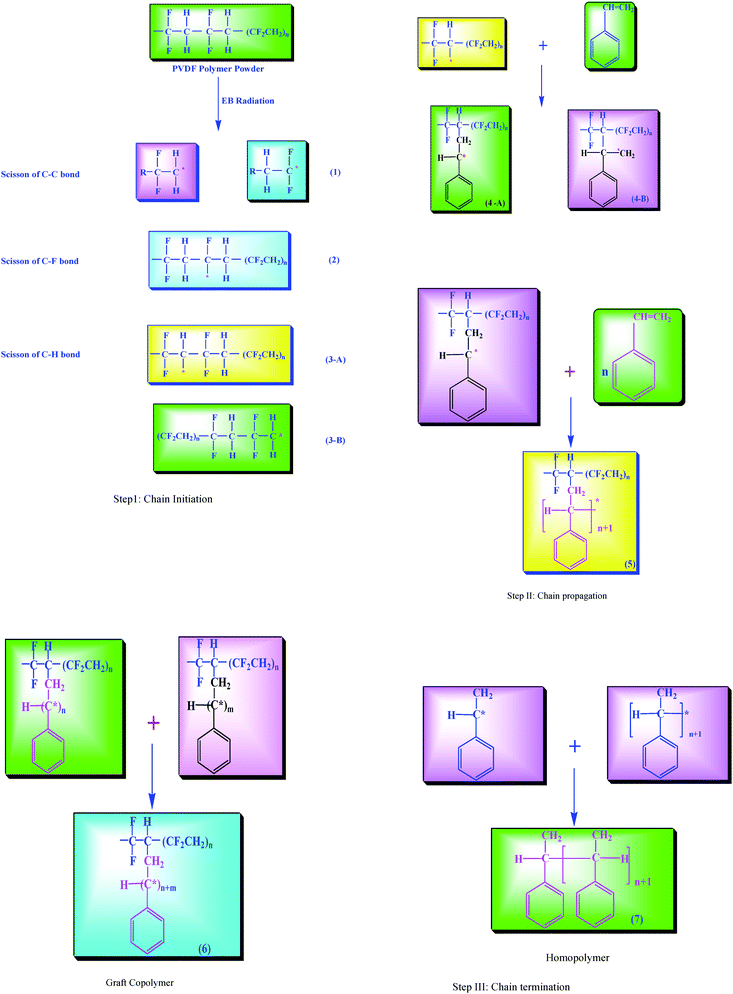 | ||
| Scheme 1 Mechanism for graft copolymerization of styrene onto pristine PVDF polymer. | ||
In the present work, we have used a low radiation dose, so there is utmost possibility of abstraction of H from a C–H bond as compared to a C–F bond. As the bond energy of C–H (413 kJ mol−1) was lower than that of the chemical bond C–F (485 kJ mol−1) and so the covalent interaction of C–H was weaker than that of C–F; C–H was easily broken by the electron beam irradiation treatment in vacuum and then dehydrogenization of PVDF produced a great number of free radicals.
The most suitable active sites to initiate the graft copolymerization in PVDF polymer are the alkyl radical: (i) mid-chain –CF2–C*H–CF2– (ii) and end chain –CF2–C*H2–. As discussed above there are two principal possibilities for the generation of free radical sites on the PVDF polymer backbone through two different routes of hydrogen abstraction, in the present discussion we are taking one possibility, that is, hydrogen abstraction from the mid-chain –CF2–C*H–CF2– as the reaction initiation site (Scheme 1).
Also the grafting of styrene monomer to the pre-irradiated PVDF powder is expected to proceed through two different reactive locations of styrene (Step 4A–B). In this case, we are taking only one possibility of hydrogen abstraction from the mid chain –C6H5–C*H– CH2 (4-A). Upon heating, the hydrogen free radical groups serve to initiate polymerization of the monomer. On the basis of the proposed mechanism, the graft copolymerization and cross-linking reactions have been considered to involve three steps: (I) initiation, (II) propagation, and (III) termination. It is observed from the proposed mechanism that the reactive sites on the backbone polymer can be generated by step (I) the free radicals formed by abstraction of hydrogen from the polymer (step II) initiates the polymerization reaction to produce growing polymeric chain (step III), which can attach to the active sites of the backbone polymer to give the graft copolymer (6). Alternatively, the growing polymeric chains can lead to the formation of homopolymer (7).
Optimization of different reaction parameters for graft copolymerization synthesis
The percentage of grafting of styrene has been optimized by studying the effect of different reaction parameters such as reaction time, temperature, amount of solvent, radiation dose and concentration of the monomer. The optimum values of the above parameters were determined by varying one parameter within certain limits while keeping the values of the other parameters constant. The optimum conditions for maximum percentage of grafting (38.97%) were: radiation dose −1.6 Mrad; monomer concentration—375.77 × 10−2 mol L−1, temperature—60 °C, solvent—10 ml; time—6 h (ESI Table S1†).Evidence of graft copolymerization synthesis
The structural investigations of pristine PVDF before and after irradiation as well as confirmation of graft copolymerization onto styrene grafted poly(vinylidene fluoride) copolymer was unambiguously accomplished by using FTIR spectroscopy, TGA, DSC, contact angle measurement, X-ray diffractometry (XRD), and atomic force microscopy (AFM) (see corresponding description below). The results clearly demonstrate that styrene was successfully grafted into PVDF polymer.In order to investigate the graft copolymer formation in the PVDF-g-PS network, FTIR studies have been carried out. FTIR spectra of the pristine PVDF, PVDF-PS (different percentage of grafting) and polystyrene are shown in (Fig. 1).
 | ||
| Fig. 1 FTIR spectra of pristine PVDF, irradiated PVDF, polystyrene homopolymer, and PVDF-g-PS polymer with different percentage of grafting. | ||
The characteristic peaks appearing at 3000 cm−1, 3021 cm−1 and 2981 cm−1 are assigned to C–H stretching vibration, the asymmetric and symmetric stretching vibrations of the CH2 group of PVDF.9 The infrared spectrum of pristine PVDF, is also characterized by the presence of vibrational bands at 602, 762 and 775 cm −1 that are characteristics of C–F bending, skeletal bending and CH2 rocking, respectively. As can be seen in the spectrum of the irradiated PVDF with 1.6 Mrad, the films after irradiation process do not present significant alterations in the absorption bands, suggesting that the radiation has no effects on the crystalline phases. The grafting of PS onto PVDF was ascertained by the presence of the characteristic absorption peak of the phenyl rings of the PS chains which was absent in pure PVDF. Grafting of polystyrene was confirmed by the presence of benzene ring features: C–H aromatic symmetric stretching vibrations at 3080 and 3024 cm−1 and skeletal C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C in plane-stretching vibrations at, 1452, 1492 and 1600cm−1, respectively.29 The presence of an aromatic out of plane C–H deformation band at 697 cm−1 is due to mono-substitution of the benzene ring. The appearance of these characteristic peaks for phenyl rings indicates that polystyrene has been successfully grafted onto the PVDF powder. Furthermore, with the increase in grafting reaction duration, the IR spectra of the grafted polymers exhibited increased absorbance signals at characteristic band.
C in plane-stretching vibrations at, 1452, 1492 and 1600cm−1, respectively.29 The presence of an aromatic out of plane C–H deformation band at 697 cm−1 is due to mono-substitution of the benzene ring. The appearance of these characteristic peaks for phenyl rings indicates that polystyrene has been successfully grafted onto the PVDF powder. Furthermore, with the increase in grafting reaction duration, the IR spectra of the grafted polymers exhibited increased absorbance signals at characteristic band.
In order to determine the different phases as a result of graft copolymerization we have studied the FTIR spectra of different grafted copolymers along with the pristine PVDF in the range of 2000–400 cm−1 (ESI, Fig. S1†). The bands at 485 and 879 cm−1 are attributed to the amorphous phase of PVDF. The bands at 408, 532, 614, 764, 796, 855 and 976 cm−1 are associated with α phase of PVDF. The bands at 444 and 510 cm−1 are characteristic of the β phase and those at 431, 512, 776, 812 and 833 cm−1 are related to the γ phase. The 840 cm−1 band is common to β and γ phases; a sharp and well-resolved band indicates the β phase, whereas a broad band indicates the γ phase. Our result shows that there is some formation of β phase in all the samples which may also result in improved dielectric properties.30 The formation of β phase in the pristine PVDF and grafted films may be due to processing conditions and the solvent used as reported earlier by Rinaldo and Daniel in their work on the effect of different solvents on phases in PVDF.31
Fig. 2a shows the TGA curves of the pristine PVDF, irradiated PVDF, polystyrene homopolymer and PVDF-g-PS (38.97%) polymer respectively. The PVDF homo polymer is stable up to about 366.00 °C and suffers a weight loss of less than 4% around 440 °C. The weight loss is confined to a single-step degradation pattern and can be attributed to the degradation of the PVDF matrix.9 The irradiation does not significantly alter the decomposition temperature of the pristine PVDF film. The PS homopolymer is thermally stable up to about 133.19 °C and then starts degrading slowly up to 318.27 °C. Then it suffers a major weight loss corresponding to the bulk decomposition of the polymer residue and at 430.71 °C only 6.8% residue is left. The PVDF-g-PS powder shows intermediate weight loss behavior in comparison to that of the pristine PVDF powder and polystyrene. After grafting, the degradation becomes a two-step degradation pattern. The additional degradation step is the degradation of PS graft-chains from 310 to 440 °C.
 | ||
| Fig. 2 (a) TGA analyses of pristine PVDF; irradiated PVDF; polystyrene and PVDF-g-PS (38.97%) polymer powder (b) DSC analyses of pristine PVDF; irradiated PVDF; polystyrene and PVDF-g-PS (38.97%) polymer powder. | ||
The pristine and irradiated PVDF, PS homopolymer and PVDF-g-PS were also characterized for their thermal properties by DSC analysis. The DSC curve of pristine PVDF shows a melting endothermic with melting temperature at 161.27 °C due to the crystal structure present in this polymer (Fig. 2b). The DSC curve for the irradiated films also presents the same profile with melting temperature of 161.44 °C. The melting enthalpy (ΔHm) of pristine PVDF and irradiated PVDF has been found to be 52.73 (J g−1) and 54.38 (J g−1) respectively. The overall percentage crystallinity (XC) of the pristine PVDF and the irradiated PVDF was evaluated from the heat of fusion and has been found to be 50.36 and 51.93 respectively. The slight increase in crystallinity of irradiated PVDF film (51. 93%) may be ascribed to the uncertainty of the measurement or slight cross linking on irradiation. The polystyrene homopolymer which was obtained during the purification of the grafted copolymer showed almost completely an amorphous behaviour. It has been observed that after graft polymerization with polystyrene, the structural symmetry of PVDF is somewhat destroyed, resulting in the lowering of percentage crystallinity.
The melting point (Tm) for PVDF-g-PS (38.97%) copolymer has been found to be 160.16 °C with the melting enthalpy (Hm) of 35.86 (J g−1). The overall percentage crystallinity (XC) of PVDF-g-PS evaluated from the heat of fusion has been found to be 47.81. Indeed the decrease in XC is caused by the introduction of amorphous polystyrene graft-chains which dilute the crystalline zone of the PVDF matrix. This phenomenon is probably associated with the decreasing proportion of the PVDF crystalline phase in the grafted polymer possibly indicating mixing of polystyrene grafts into the crystallizable part of the PVDF melt.
Fig. 3 shows the effect of different grafting percentages on the water contact angles of pristine and grafted PVDF films. PVDF being a hydrophobic polymer possesses a high contact angle with water (98°). PS grafts are also hydrophobic; therefore, grafted polymer films are relatively more hydrophobic. From Fig. 3, it is obvious that the contact angle of grafted PVDF films increased with increasing percentage of grafting. This means the surface hydrophobicity of those grafted films was improved to a significant extent.
 | ||
| Fig. 3 Water contact angles of PVDF films as a function of percentage of grafting. | ||
The highest contact angle of 117° was obtained for the polymer film cast from PVDF-g-PS powder with optimum percentage of grafting (38.97%). The result suggested that styrene was successfully grafted onto PVDF polymer powder and the hydrophobicity of the polymer surfaces increased.
Fig. 4 (a–c) shows the XRD of the pristine PVDF films and grafted copolymers films with minimum and optimum percentage of grafting (17.51 and 38.97%). All the grafted samples along with pristine PVDF recorded some diffraction peaks in the range of 2θ = 16.8 and 21.5. The intensity of the crystalline diffraction peaks in the pristine PVDF was reduced by the introduction of polystyrene grafts and this reduction is a function of the content of polystyrene (% grafting). For all the graft copolymers, the intensity ratio between the crystal peak and the amorphous background signal is low because of their low crystallinity. A trend of decreasing crystallinity with increasing grafting concentration was observed which is in agreement with our DSC results indicating that PS grafting hinders the chain alignment in PVDF for the formation of crystalline region.
 | ||
| Fig. 4 (a–c) XRD of the pristine PVDF and grafted copolymers films having different percentage of grafting. | ||
All α phase peaks decrease within the PVDF-g-PS copolymer. The (100), (020), and (021) peak of α phase decreases in intensity upon grafting. In the contrast, the peak around 20° which corresponds to either α or γ phase is consistently high in intensity. It implies that this peak was not affected much by the grafting. Therefore the peak around 20° could be originated mainly from γ phase with a small mixture of α phase. This infers that grafting of PS into PVDF mainly hinders the crystallization of α phase.
To have a better understanding of the morphology changes of the grafted polymers, the pristine PVDF and PVDF-g-PS (38.97%) copolymers were analyzed by AFM as shown in Fig. 5(a–b). These images demonstrate a distinction between the topography of the ungrafted and grafted films. The surface topography has been found to be altered remarkably with grafted samples. The AFM analysis shows a root mean square (rms) roughness of 6.9 nm and 36.2 nm for pristine PVDF and PVDF-g-PS respectively for a similar film thickness. In addition, the uniform crystallite structures of the PVDF matrix diminish on the grafted polymer surface. This is related to the grafted groups which disrupt the molecular chain orderness and the copolymer surface becomes rougher after the graft reaction.
 | ||
| Fig. 5 (a) AFM images of pristine PVDF film. (b) AFM images of PVDF-g-PS films with optimum percentage of grafting (38.97%). | ||
Dielectric properties measurement of the pristine and grafted PVDF films
For most of the polymers, the low dielectric constant is a critical limiting factor in achieving high energy density electrostatic capacitor applications. We have studied the dielectric properties of the polystyrene grafted PVDF copolymers. The PVDF-g-PS grafted copolymer has been found to exhibit privileged dielectric properties as compared to pristine PVDF polymer. Fig. 6 (a) displays the dielectric constant of pristine PVDF, optimum grafted PVDF-g-PS (38.9 7%) and lowest grafted PVDF-g-PS (17.51%) polymer films as a function of frequency from 100 Hz to 1 MHz at room temperature. The dielectric constant of the PVDF-g-PS polymer substantially increases as compared to the pristine PVDF. For example at 100 Hz, the dielectric constant of PS-g-PVDF film reaches about 90, representing more than 7 times increment compared with the pristine PVDF film and this is one of the highest dielectric constant reported so far in polymer capacitor materials. | ||
| Fig. 6 (a) Dependence of the dielectric constant on various frequencies at room temperature for pristine PVDF, optimum (38.97%) and lowest grafted PVDF-g-PS (17.51%) polymer films. (b) Effect of the percentage of grafting of PS onto the dielectric constant of PVDF at room temperature measured at 100 Hz. | ||
The disordered or amorphous phase in the grafted polymers possibly induced some short-range ordered regions and the interface regions of different phases can contribute to charge metastable accumulation during bias application. The variation of dielectric constant with frequency is about 50% ranging from 100 Hz to 106 Hz for the grafted polymer films. The reduction of capacitance with increased frequency also indicates dynamic resistance of dipolar reorientation against very fast applied signals. This is related to the incorporation of grafted polystyrene (PS) chains in the parent polymer chain. It is known that high-energy electron or ions irradiation have dramatic effects on the polymer properties depending on the radiation dose.32 Since we have used a relatively low radiation dose (1.6 Mrad) in our experiment to initiate the graft copolymerization reaction, the incorporation of radiated species may not alter the dielectric constant of the resultant copolymers, the increment in dielectric constant is directly related to the extent of grafting evidently shown in Fig. 6a.
The amorphous phase in the pristine PVDF and its copolymers and their interaction with the crystalline regions play an important role in dielectric response. As shown in Fig. 6b, the dielectric constant has been found to increase with percentage grafting due to the incorporation of more polystyrene group into the pristine PVDF. A decrease in crystallinity as a result of graft copolymerization (refer to DSC and XRD as discussed earlier) provides a greater chance for the amorphous chains to achieve random disorder and isolation. The improved dielectric behavior of PVDF-g-PS graft copolymers is a result of increase free charge carriers in the amorphous phase and/or charge accumulation at the amorphous/crystalline interface. It is interesting to find that even at the frequency of 1 MHz the dielectric constant of the grafted film still remains higher than pristine PVDF film which satisfies the dielectric requirement for energy storage applications. Previous works by a few researchers have also shown that incorporation of some bulky groups such as TrFE, CTFE, and HFP into PVDF polymers could convert the copolymers into relaxor ferroelectrics resulting in higher dielectric constants due to introduction of defects which created non polar regions.1,8,10
Besides the energy density and dielectric constant of any polymer system, energy loss is a further concern for successful applications of polymers for capacitor applications, especially under a varying electric field. The energy loss is mainly converted into heat and the released heat increases the temperature of the polymer, which limits the application frequency range of the polymer film under an electric field and reduces the breakdown field and reliability for different applications. The energy loss is the dielectric loss, which is related to the reduction of the dipole moments due to the restrictions between the polymer chains and its surroundings during the polarization.
The frequency dependences of the dielectric loss for the pristine PVDF, optimum grafted (38.97%) and lowest grafted PVDF-g-PS (17.51%) polymers are presented in Fig. 7(a). Despite a slight increment in dielectric loss compared to the pristine PVDF films, the PVDF-g-PS dielectric loss achieves at least an order of magnitude improvement compared to other reported PVDF copolymers1,8,10 and polymer composites with fillers.2,20,34–36 The dielectric loss in general consists of two contributions; one due to relaxation effects and other due to conduction. The conduction loss shows a linear log (tan δ) versus frequency plot, while the loss due to relaxation effects shows a maximum at a certain frequency. The peaks in dielectric loss as a function of frequency or temperature plots are related to the “relaxation” processes with specific molecular mechanisms, such as the onset of the glass transition or the movements of some specific molecular groups. In our experiment we obtain the same peak (maximum) due to relaxation effect in the frequency range 104–105 Hz and can be attributed to the strong relaxation effect caused by side-group of polystyrene grafted chains onto the PVDF. However, the possibility of some conduction processes cannot be totally excluded as the polymer film is not an ideal dielectric material and leakage current is inevitable.
 | ||
| Fig. 7 (a) Dependence of dielectric losses on the frequency at room temperature. (b) Effect of the percentage of grafting of PS onto the PVDF at room temperature measured at 100 Hz. | ||
As a result of graft copolymerization of polystyrene onto PVDF, the interactions between the parent polymer chains and bulky styrene will be formed and the molecular motions due to the electric field will be affected resulting in dielectric relaxation.
The dielectric relaxation in PVDF-g-PS may be attributed to the localized fluctuations of the aromatic styrene groups. The αa and αc dielectric relaxation are of interest in our work because they are related to the two main structural phases (i.e., the amorphous and crystalline phases) comprising semi-crystalline PVDF. In the case of PVDF-g-PS copolymer, as a result of graft copolymerization reaction there is more amorphous content and it results in more αa dielectric relaxation. The αa process is ascribed to long range correlated micro-Brownian motion in the amorphous phase. Whether these motions arise in the bulk amorphous phase or in a more constrained amorphous component is an open question. From our results we believe that both relaxations are altered by grafting of polystyrene.
The dielectric loss for grafted copolymers has been found to increase with percentage of grafting (Fig. 7b). A greater fraction of the amorphous component as well as less molecular orientation in the grafted copolymers is the reason for higher loss in the whole frequency range meaning that there are more chain segmental motions in the grafted samples than the pristine PVDF samples. Two prime reasons can be linked to the difference in chain mobilities in the amorphous phase of the grafted copolymers samples and pristine PVDF samples. The first reason is that the grafted copolymer samples have less crystallinity, which means that the molecular mobility of amorphous chains will be less restricted by neighboring crystalline regions. The second reason is that amorphous areas in the pristine PVDF films could have some degree of orientation that would inhibit long range molecular motions due in part to intermolecular forces that would be not as great in the grafted sample. Furthermore as a result of the graft copolymerization, there is disruption of packing of rigid chain polymers, and incorporation of bulky polystyrene molecule results in higher free volume which also contributes to the high dielectric loss. Similar behavior of the grafted polymers to some reported works has been found.33–37
For polymers used in high performance capacitor applications, breakdown strength is an imperative parameter. Under the same operation electric field for the dielectric materials, higher breakdown field means higher reliability and energy storage. Various electrical breakdown mechanisms have been proposed for solid state dielectrics, including thermal breakdown, electromechanical breakdown and electronic breakdown. The breakdown field of the PVDF-g-PS film was measured to be 400 MV m−1, while for the PVDF film; it is 380 MV m−1 (Fig. 8). The similar or better breakdown field observed in the PVDF-g-PS film can be attributed to the better hydrophobic properties of the graft copolymers and enhanced interaction of PVDF polymer with styrene polymer chains in PVDF-g-PS copolymers.
 | ||
| Fig. 8 Breakdown voltage of the pristine PVDF against PVDF-g-PS samples measured at room temperature. | ||
Conclusions
In an effort to improve the dielectric properties of the dielectric polymers, we introduced a new and simple approach to synthesize PVDF-g-PS copolymers. By utilizing electron beam induced free radical graft copolymerization technique, PVDF-g-PS copolymers were successfully synthesized. As a result of graft copolymerization, the grafted polymers exhibit improved dielectric properties such as high dielectric constant and high breakdown field all of which are desirable for high performance capacitors. With respect to the cost effectiveness, a copolymer with sufficient grafting is valuable for potential practical applications. The overall results of this work suggest that the graft copolymerization of PS onto PVDF through electron beam radiation is a promising route to synthesize new materials with higher dielectric properties.Acknowledgements
The authors thank the funding support of project agreement number POD0814080.Notes and references
- B. Chu, X. Zhou and Q. M. Zhang, Science, 2006, 313, 334–336 CrossRef CAS.
- P. Kim, J. Li, N. M. Doss, J. P. Calame, J. P. Tillotson, P. J. Hotchkiss and J. W. Perry, ACS Nano, 2009, 3, 2581–2592 CrossRef CAS.
- R. Schroeder, L. A. Majewski and M. Grell, Adv. Mater., 2005, 17, 1535–1539 CrossRef CAS.
- Z.-M. Dang, Y.-H. Lin and C.-W. Nan, Adv. Mater., 2003, 15, 1625–1629 CrossRef CAS.
- M. Arbatti, X. Shan and Z.-Y. Cheng, Adv. Mater., 2007, 19, 1369–1372 CrossRef CAS.
- J. Li, S. Seok, B. Chu, F. Dogan, Q. Zhang and Q. Wang, Adv. Mater., 2009, 21, 217–221 CrossRef.
- Y. Shen, Y. Lin, M. Li and C.-W. Nan, Adv. Mater., 2007, 19, 1418–1422 CrossRef CAS.
- Q. M. Zhang, V. Bharti and X. Zhao, Science, 1998, 280, 2101–2104 CrossRef CAS.
- V. K. Thakur, E. J. Tan, M.-F. Lin and P. S. Lee, J. Mater. Chem., 2011, 21, 3751–3759 RSC.
- X. Zhou, B. Chu, B. Neese, M. Lin and Q. M. Zhang, IEEE Trans. Dielectr. Electr. Insul., 2007, 14, 1133–1138 CrossRef.
- A. J. Lovinger, Science, 1983, 220, 1115–1121 CAS.
- Y. Bai, Z.-Y. Cheng, V. Bharti, H. S. Xu and Q. M. Zhang, Appl. Phys. Lett., 2000, 76, 3804–3806 CrossRef CAS.
- L. O. Faria, C. Welter and R. L. Moreira, Appl. Phys. Lett., 2006, 88, 192903 CrossRef.
- C. Pecharroman, F. Esteban-Betegon, J. F. Bartolome, S. Lopez-Esteban and J. S. Moya, Adv. Mater., 2001, 13, 1541–1544 CrossRef CAS.
- P. C. P. Watts, W.-K. Hsu, A. Barnes and B. Chambers, Adv. Mater., 2003, 15, 600–603 CrossRef CAS.
- L. Qi, B. I. Lee, S. Chen, W. D. Samuels and G. J. Exarhos, Adv. Mater., 2005, 17, 1777–1781 CrossRef.
- J. Q. Huang, P. Y. Du, L. X. Hong, Y. L. Dong and M. C. Hong, Adv. Mater., 2007, 19, 437–440 CrossRef CAS.
- D. Ma, T. A. Hugener, R. W. Siegel, A. Christerson, E. Martensson, C. Onneby and L. Schadler, Nanotechnology, 2005, 16, 724–731 CrossRef CAS.
- J. Li, J. Claude, L. E. Norena-Franco, S. I. Seok and Q. Wang, Chem. Mater., 2008, 20, 6304 CrossRef CAS.
- P. Kim, S. C. Jones, P. J. Hotchkiss, J. N. Haddock, B. Kippelen, S. R. Marder and J. W. Perry, Adv. Mater., 2007, 19, 1001–1005 CrossRef CAS.
- G. Geuskens, A. Etoc and P. D. Michele, Eur. Polym. J., 2000, 36, 265–271 CrossRef CAS.
- U. Lappan, L. Haussler, G. Pompe and K. Lunkwitz, J. Appl. Polym. Sci., 1997, 66, 2287–2291 CrossRef CAS.
- F. Cardona, G. A. George, D. J. T. Hill, F. Rasoul and J. Maeji, Macromolecules, 2002, 35, 355–364 CrossRef CAS.
- N. Walsby, F. Sundholm, T. Kallio and G. Sundholm, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 3008–3017 CrossRef CAS.
- H. P. Brack, H. G. Buhrer, L. Bonorand and G. G. Scherer, J. Mater. Chem., 2000, 10, 1795–1803 RSC.
- S. Hietala, S. L. Maunu and F. Sundholm, Macromolecules, 1999, 32, 788–791 CrossRef CAS.
- J. S. Forsythe and D. J. T. Hill, Prog. Polym. Sci., 2000, 25, 101–136 CrossRef CAS.
- E. T. Kang and Y. Zhang, Adv. Mater., 2000, 12, 1481–1494 CrossRef CAS.
- L. Li, B. Deng, Y. Ji, Y. Yu, L. Xie, J. Li and X. Lu, J. Membr. Sci., 2010, 346, 113–120 Search PubMed.
- W. Li, Q. Meng, Z. Zhang, Y. Zheng, W. Xia and Z. Xu, Appl. Phys. Lett., 2010, 96, 192905 Search PubMed.
- R. Gregorio Jr. and D. S. Borges, Polymer, 2008, 49, 4009–4016 Search PubMed.
- Y. Nabata, Jpn. J. Appl. Phys., 1990, 29, 2755–2760 Search PubMed.
- D. Rollik, S. Bauer and R. Gerhard-Multhaupt, J. Appl. Phys., 1999, 85, 3282–3288 CrossRef CAS.
- H. von Seggern and S. N. Fedosov, Appl. Phys. Lett., 2002, 81, 2830–2832 CrossRef CAS.
- S. H. Zhang, N. Zhang, C. Huang, K. Ren and Q. M. Zhang, Adv. Mater., 2005, 17, 1897–1901 Search PubMed.
- Z.-M. Dang, L. Wang, Y. Yin, Q. Zhang and Q.-Q. Lei, Adv. Mater., 2007, 19, 852–857 CrossRef CAS.
- J.-K. Yuan, Z.-M. Dang, S.-H. Yao, J.-W. Zha, T. Zhou, S.-T. Li and J. Bai, J. Mater. Chem., 2010, 20, 2441–2447 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Characterizations techniques of the pristine PVDF and PVDF-g-PS films. FTIR spectra of pristine PVDF, irradiated PVDF, and PVDF-g-PS copolymer with different percentages of grafting. Optimization of various reaction parameters for maximum percentage graft copolymerization of styrene onto PVDF polymer powder. See DOI: 10.1039/c1py00225b |
| This journal is © The Royal Society of Chemistry 2011 |