Fluorescent carbazole dendrimers for the detection of explosives
Guoqiang
Tang
a,
Simon S. Y.
Chen
a,
Paul E.
Shaw
*a,
Katalin
Hegedus
b,
Xin
Wang
a,
Paul L.
Burn
*a and
Paul
Meredith
*a
aCentre for Organic Photonics and Electronics, The University of Queensland, Brisbane, Queensland 4072, Australia. E-mail: p.burn2@uq.edu.au; meredith@physics.uq.edu.au; p.shaw3@uq.edu.au
bMolecular and Health Technologies, CSIRO, Melbourne, Victoria 3168, Australia
First published on 4th August 2011
Abstract
Three generations of fluorescent carbazole dendrimers with spirobifluorene cores are studied as model chemosensor systems for the detection of nitroaromatic explosives via fluorescence quenching. Stern–Volmer measurements in solution with a series of nitrated analytes including the 2,4,6-trinitrotoluene (TNT) byproduct 2,4-dinitrotoluene (DNT) and the plastic explosives taggant 2,3-dimethyl-2,3-dinitrobutane (DMNB) showed an increase in affinity and hence quenching efficiency between the first and second generation dendrimers. In spite of the differences in the solution Stern–Volmer constants the solid state quenching response to the analytes was found to be independent of generation with the exception of 1,4-dinitrobenzene (DNB), where the quenching decreases with increasing generation. It was found that it was necessary to heat the films to release the analytes with the temperature required dependent on the analyte and/or dendrimer generation. These two results show that a simple solution Stern–Volmer analysis is not always sufficient for qualifying film sensing performance and that the drive to develop sensing materials with high solution Stern–Volmer constants for real applications needs to be reconsidered.
Introduction
The detection of chemical explosives is critical for military operations, public security and environmental monitoring. However, the detection of nitroaromatic explosives such as TNT represents a significant challenge due to the low vapor pressures of these compounds.1,2 As TNT is a principal explosive used in landmines there is also an urgent need for a portable detection system.3 Though there are established detection technologies for explosives such as ion mobility spectrometry and mass spectrometry, such detectors are bulky and unsuitable for incorporation into a portable device.4 Trained canines are still widely used but they are expensive to train and maintain. Fluorescence quenching is a promising alternative method for the detection of explosives with devices incorporating sensing units containing conjugated polymers already on the market.5,6Materials for the detection of nitroaromatic explosives by fluorescence quenching must fulfill two basic criteria. First, they must be fluorescent with a good photoluminescence quantum yield (PLQY) in the solid state. Second, if quenching is to occur by electron transfer to the explosive analyte, the excited electron of the sensing compound must be higher in energy than the electron affinity of the explosive with an energy offset greater than the exciton binding energy.7 In addition to these two characteristics the chemical structure needs to be compatible with that of the explosive to allow the two to interact and facilitate charge transfer. Whilst conjugated polymer fluorescence based sensors work, an understanding of what limits their performance is complicated by the non-uniformity of the materials. For example, it is not easy to determine the number of effective chromophores in a polymer chain. In addition, the morphology of the films, which governs the ingress of the analyte into the film, is strongly dependent on the molecular weight and polydispersity of the polymer, which can vary from batch to batch. In contrast, light-emitting dendrimers can be monodisperse with the number of chromophores simply dialed in by varying the dendrimer generation.8 Hence, dendrimers offer a promising approach for designing and understanding materials that can sense explosive analytes with the potential for tuning to enable the detection of specific compounds.9,10
In common with the detection studies using conjugated polymers most reports on dendritic materials for sensing have focused on solution measurements.9–12 However, two key studies have shown that first generation fluorescent dendrimers can detect nitroaromatic compounds by fluorescence quenching in thin films13 and when incorporated into an organic laser.14 However, the effect of dendrimer generation on the solid state sensing capabilities of fluorescent dendrimers has not been investigated. Guo et al. performed an investigation in solution on four generations of flexible partially conjugated fluorescent dendrimers optimized for two-photon absorption and response to TNT.12 They observed an increase in the quenching efficiency with generation but it is not clear whether this effect would be mirrored in thin films, which are required for practical applications.
In this paper we report the impact of dendrimer generation for carbazole-based dendrimers with 9,9′-spirobifluorene cores and 9,9-di-n-propylfluorene surface groups (Fig. 1) as sensing materials for the detection of nitroaromatic analytes and the plastic explosive taggant DMNB by fluorescence quenching. Measurements in solution show that the quenching efficiency increases between the first and second generation with only a minor increase in moving to the third generation dendrimer. At room temperature the analytes caused a rapid loss of film fluorescence, which could be recovered by heating.
 | ||
| Fig. 1 Structures of three dendrimers, defined as G1, G2 and G3. | ||
Results and discussion
Synthesis and physical properties
The synthesis of the three generations of dendrimer was straightforward (Scheme 1). Each generation of carbazole dendron15 was reacted with 2,7-dibromo-9,9′-spirobifluorene under Buchwald–Hartwig amination conditions to give the corresponding dendrimer. The first, second, and third generation dendrimers were isolated in yields of 80%, 68%, and 58% respectively. One of the important reasons to use the convergent route for forming dendrimers is to ensure that a single compound is formed. Gel permeation chromatography showed that the dendrimers were monodisperse, that is, they were discrete macromolecules. Thermal gravimetric analysis showed that the dendrimers were stable to temperatures greater than 350 °C. | ||
| Scheme 1 i Pd2(dba)3, NaOtBu, xylenes, heat, Ar. For carbazolyl and fluorenyl connectivity refer to Fig. 1. | ||
Photophysical properties
The absorbance and fluorescence spectra for each dendrimer in solution and film are shown in Fig. 2. In both phases (solution and thin film) there are some clear and common differences between the three generations. The peak absorbance shifts to shorter wavelengths as the generation increases with the biggest change occurring between the G1 and the G2. The blue shift in the peak absorbance is primarily due to the spectrum being strongly weighted by the dendron absorption. The fluorescence spectrum of G1 in solution shows clear vibronic structure while the spectra of G2 and G3 are broader and featureless with a slightly red shifted peak. We assign the broad features in the emission of G2 and G3 as aggregate or excimer emission caused by intramolecular interactions of the chromophores. In moving to the solid state there is a red shift of all the emission spectra relative to solution, indicating a planarisation of the chromophores, which is often seen for conjugated materials. For all three dendrimers there is an extended long wavelength tail which is indicative of excimer/aggregate emission in the solid state. For G1 the increase in the intensity of the vibronic shoulder also contributes to the broadening. The effect of these intramolecular (in solution) and intermolecular (solid state) interactions can be seen in the photoluminescent quantum yield (PLQY) measurements (Table 1). G1 has a PLQY of 65 ± 3% in solution with G2 and G3 having lower PLQYs around 20%. We attribute the decrease in PLQY for G2 and G3 relative to G1 to intramolecular chromophore interactions that lead to the quenching of the luminescence. In moving to the solid state the PLQY of G1 decreases to 31 ± 3% while the film PLQYs of G2 and G3 are similar to their solution measurements suggesting that for the two higher generations any additional intermolecular interactions in the solid state do not cause extra quenching over and above the intramolecular interactions in solution. | ||
| Fig. 2 Absorbance and emission spectra of the three dendrimer generations in solution (solid line) and in film (dashed line). | ||
| Dendrimer | Solution | Film |
|---|---|---|
| G1 | 65% ± 3% | 31 ± 3% |
| G2 | 21% ± 2% | 20 ± 2% |
| G3 | 18% ± 2% | 15 ± 2% |
Energy levels
Before embarking on quenching studies it is necessary to determine whether the energy levels of the sensing materials and analytes are such that it would be favourable for an electron to be transferred from the excited sensor to the analyte leading to fluorescence quenching. However, it is important to note many of the techniques used to determine the relevant energy levels come with significant uncertainty and hence only a rough estimate can be obtained. In addition, while solution measurements will give one set of energies these may be different to the solid state for numerous reasons including solvent interactions. In this work we have studied quenching of the sensor fluorescence both in solution (Stern–Volmer analysis) and the solid state and hence calculate the energy levels of the dendrimers in solution and the solid state using a combination of cyclic voltammetry, photoelectron spectroscopy in air (PESA), and UV-visible and fluorescence spectroscopy.Cyclic voltammetry measurements in solution showed that each of the dendrimers undergo a chemically reversible oxidation. The E1/2s were 0.67 V, 0.62 V and 0.61 V (against the ferrocenium/ferrocene couple) for G1, G2, and G3 respectively. Chemically reversible reductions were not seen in the cyclic voltammetry experiments and hence to estimate the reduction potential in solution we use a combination of the UV-visible and fluorescence spectra. For G1 the absorption and fluorescence spectra follow the mirror image rule and hence we calculate the lowest energy transition as being halfway between the long wavelength shoulder (364 nm, 3.4 eV) in the absorption and the (0,0) transition (386 nm, 3.2 eV) in the fluorescence spectra, that is, 3.3 eV. Subtracting this energy from the oxidation E1/2 of G1 gives a reduction potential of −2.6 V. For G2 and G3 the mirror image rule does not hold due to the intramolecular chromophore interactions. However, since they contain similar chromophores to G1 they should also have a reduction potential around −2.6 V. This reduction potential is ≥0.4 V more negative than all the analytes (DNT: −1.5 V, DNB: −1.2 V, p-nitrotoluene (pNT): −1.7 V, benzophenone (BP): −2.1 V, DMNB: −2.2 V) (against the ferrocenium/ferrocene couple) investigated and given that the ‘exciton binding energy’ is often reported as being of order 0.3 ± 0.1 eV16 it was expected from an electronic perspective that all three dendrimers should have their fluorescence quenched by the analytes. It is worth noting that some aromatic molecules that might be considered potential quenchers, such as benzene and naphthalene, have significantly more negative reduction potentials than the nitroaromatics and therefore exciton dissociation will not occur.17 To determine the relevant energy levels in the film we used a combination of PESA and the film absorption and fluorescence spectra for G1 for which the mirror image rule still essentially holds. PESA measurements for all three dendrimers gave the ionization potential and adding the optical gap energy as before leads to an estimation of the energy of the excited electron of 2.4 eV. By assuming that the solvent effects of the analytes in solution are similar to that of ferrocene in the electrochemical experiments, and taking into account that the measured ionization potential for ferrocene is 4.8 eV18 the electron affinity for each of the analytes is estimated to be DNT (3.3 eV), DNB (3.6 eV), pNT (3.1 eV), BP (2.7 eV), and DMNB (2.6 eV). Therefore, from an electronic point of view the response in films to BP and DMNB may be weaker than the nitroaromatic analytes.
Fluorescence quenching in solution
Having established that the dendrimers have the potential to be sensors for explosive analytes we first undertook steady-state solution Stern–Volmer measurements. Each dendrimer was tested with the five analytes: DNT, DNB, pNT, DMNB, and BP. DNT is a byproduct of TNT and is much more volatile than TNT itself (180 p.p.b. compared to 7.5 p.p.b. at 25 °C),3,19 making it an ideal model for evaluating potential detection in actual devices. DNB has a similar electron affinity to TNT and a low vapor pressure (0.02 p.p.m. at 25 °C).1pNT possesses a lower electron affinity than TNT and a much greater vapor pressure (200 p.p.m. at 25 °C).1DMNB is a volatile taggant (2.7 p.p.m. at 25 °C)20 added to commercial plastic explosives to aid their detection by canines, but its lower electron affinity compared to the nitroaromatic analytes means it is a less efficient electron acceptor and, therefore, harder to detect by fluorescence quenching.21 BP is a non-nitrated electron acceptor with a high vapor pressure (2.0 p.p.m. at 25 °C) and was adopted as a control representative of many chemicals found in everyday products. The Stern–Volmer equation for a single quenching process is given by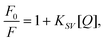 | (1) |
All five analytes resulted in increased quenching of the fluorescence of the dendrimers when the concentration of the analyte was increased. By plotting the change in fluorescence intensity against the analyte concentration the Stern–Volmer plots for each analyte were obtained, which are shown in Fig. 3. Fits to the data using equation (1) were used to calculate KSV values, which are summarized in Fig. 4. For each analyte there is a significant increase in the Stern–Volmer constant between the G1 and G2 with only a small difference between the values for the G2 and G3. Moreover, the nitroaromatic compounds all produce larger KSV values than either the control analyte BP or the taggant DMNB. The overall trend is consistent with the differences in electron affinities between the analytes. The values of the Stern–Volmer constants are overall higher than what has been reported for conjugated polymers23 but lower than the ∼1400 M−1 constant reported for a fourth generation dendrimer with TNT by Guo et al.12 The high Stern–Volmer constant for the G2 and G3 with the analyte DMNB is noteworthy as responses with polymer systems are generally much lower.
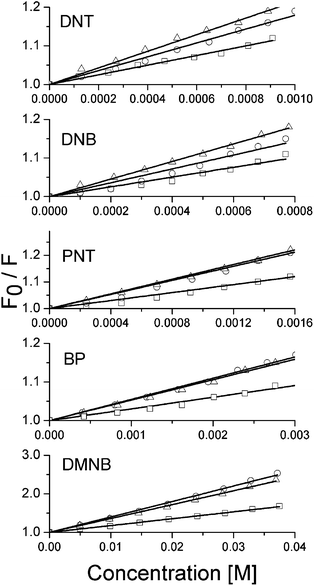 | ||
| Fig. 3 Stern–Volmer plots for the G1 (squares), G2 (circles) and G3 (triangles) with the five analytes. The solid lines show fits to the data with equation (1). | ||
 | ||
| Fig. 4 Stern–Volmer constants for the dendrimers with the five analytes. | ||
Solid state fluorescence quenching
The characterization of the quenching efficiency with Stern–Volmer constants is the general method used for comparing sensing materials and analytes, but is limited by the fact that all measurements are performed in solution. An actual detector is likely to incorporate a solid state sensing element and, therefore, intermolecular interactions, film morphology and steric effects are going to influence the response of a sensor film to a target molecule. The question that therefore arises is whether there is a direct correlation between the performance of a sensing compound in solution and in the solid state. To evaluate the behavior of the dendrimers in the solid state, the change in fluorescence intensity of thin films upon exposure to saturated vapors of all five analytes was measured.Fig. 5 shows the first 300 s of the fluorescence intensity of thin films of G1, G2 and G3 with thicknesses of ∼25 nm exposed to saturated DNT, DNB, pNT, BP and DMNB vapors. Also shown are the fluorescence decays in air, which display a gradual signal decrease due to photodegradation. A low photodegradation rate is important for real applications. In the presence of saturated DNT vapor there is a rapid decrease of the fluorescence intensity with approximately 30% quenching in the first 10 s. This response is comparable to what has previously been reported for a range of conjugated polymer thin films sensors exposed to saturated DNT vapour.1,23–25 Interestingly, there is little difference between the three generations of dendrimer despite the differences in Stern–Volmer constants observed in solution. From a practical point of view this is important, as first generation dendrimers are easier to synthesize. In addition, the results also strongly suggest that a very high solution Stern–Volmer constant is not actually necessary for the detection of analytes in the solid state. A similar trend is seen with the analytes pNT, DMNB and BP, all of which exhibit similar responses to the three dendrimer generations. The exception is DNB, where the quenching response decreases with increasing generation. This is in direct contrast to the measurements in solution where quenching efficiency increased with generation and highlights the importance of solid state factors such as steric effects and film morphology. The dendrimer generation dependence seen in the quenching by DNB may be related to the much lower vapor pressure of the analyte, which could impair diffusion into the film particularly if the analyte is strongly binding to the sensing dendrimers.1 It is important to note that the vapor pressures of the different analytes vary over four orders of magnitude and this is the reason why pNT, which has the highest vapor pressure, results in the largest quenching response. It is also worth noting that although analytes such as DMNB result in a modest loss of fluorescence it is still a measurable change and, therefore, sufficient for detection in a sensor.
 | ||
| Fig. 5 Film quenching kinetics of G1 (solid line), G2 (dashed line) and G3 (dotted line) with the five analytes. The reference kinetics for the loss of fluorescence due to photodegradation in air is included in the topmost panel. | ||
Solid state fluorescence recovery
An important consideration for a real time, practical, and reusable sensor is the release of the analyte after the exposure and the recovery of the fluorescence. In spite of its importance there are only a few reports that show the recovery of the fluorescence after exposure to analyte vapour,3,20,23 which may be due to the fact that recovery is often a slow process for many materials. Measurements reported by Zhao and Swager using Fido show rapid responses and recovery times but it is not stated whether their measurements were performed at room temperature.23Fig. 6 shows the fluorescence spectra of the G2 films prior to exposure to the analytes, immediately after a 10 min exposure to saturated analyte vapor and then after 3 min under a flow of nitrogen at room temperature. For DNT and BP there was no significant fluorescence recovery, indicating that the analyte molecules are tightly bound within the film. In the case of DNB a small amount of fluorescence recovery was observed, which could be explained by there simply being less analyte in the film due to the lower vapor pressure of this analyte. It is possible that for the analytes with a higher vapor pressure an excess of the analyte is absorbed into the film, making recovery of the fluorescence harder to achieve. Although pNT results in strong rapid quenching of the film fluorescence due to its high vapor pressure, the nitrogen flow did result in a partial recovery of the fluorescence, which would indicate that at least a portion of pNT is not strongly bound within the film. The same behavior was observed for the G1 and G3 films. | ||
| Fig. 6 The fluorescence recovery characteristics of thin films of the G2 showing the initial fluorescence (solid line), the fluorescence following a 10 min exposure to the analyte (dashed line), the fluorescence after 3 min in a nitrogen flow at room temperature (dotted line) and the fluorescence after heating under a nitrogen flow (dash-dot line). | ||
The fact that the films do not recover at room temperature has important design ramifications for a real time sensor. The commercially available FIDO detection system uses a heated conjugated polymer sensing element which we believe is required to increase the rate of analyte desorption. We therefore investigated the effect of temperature on the recovery of the fluorescence of the films. The photoluminescence recovery characteristics of the dendrimers at different temperatures are shown in Fig. 7. The fluorescence of films saturated with DNT and BP were found to recover quickly when heated between 60–80 °C for G1 and 80–100 °C for G2 and G3. This result suggests that both analytes are more strongly bound to the higher generation dendrimers, which is consistent with the measured KSVs. For the films exposed to pNT, the fluorescence recovered quickly from 40–60 °C irrespective of generation indicating a weaker binding interaction. DNT and DNB have more nitro substituents than pNT and should have stronger electrostatic interactions with the dendrimers that would slow their diffusion into and out of the film.1pNT having a smaller electrostatic interaction is also consistent with the rapid fluorescence quenching of dendrimer thin films and their partial recovery under flowing nitrogen. DMNB displayed no obvious changes in the fluorescence recovery rate with temperature, which may be due to weak binding of the analyte to the dendrimers. With DNB the fluorescence returned between 60 °C and 80 °C in all three generations.
 | ||
| Fig. 7 Temperature dependent fluorescence recovery of thin films of the G1 (top), G2 (middle) and G3 (bottom) dendrimers after exposure to each of the five analytes. | ||
The fluorescence spectra of the recovered G2 films after heating are also shown in Fig. 6. For each analyte there is a clear recovery of the majority of the fluorescence, indicating that most if not all of the analyte has been removed.
Conclusion
We have reported a detailed study of the performance and underlying photophysics of three generations of carbazole dendrimers as sensing materials for the detection of nitroaromatic explosives by fluorescence quenching. Measurements in solution reveal high Stern–Volmer constants for all three dendrimer generations with nitrated analytes with a significant increase in sensitivity between the first and the higher generation dendrimers. Moreover, thin films of all three generations are strongly quenched by the nitroaromatic analytes. With the exception of DNB there was no significant difference in the quenching between the three generations in the solid state. Comparisons between the results from solution and film measurements confirm the limitations of Stern–Volmer analysis in characterizing sensing compounds for solid state detectors. It is important to recognize that in a practical sense the solid state thin film properties are what define the performance in vapor sensing systems. Heating of the analyte-exposed dendrimer films under a nitrogen flow resulted in the recovery of the majority of the initial fluorescence. These results show that fluorescent dendrimers are excellent models for sensors based on fluorescence quenching. Finally, we have shown that light-emitting dendrimers are a promising technology for the detection of explosives with key features of sensitivity and the potential for re-usable elements.Experimental
All commercial reagents were used as received unless otherwise noted. Xylenes were distilled from sodium and benzophenone under a nitrogen atmosphere before use.Structural and physical characterisation
1H NMR spectra were recorded on a Bruker Avance 500 (500 MHz) spectrometer. Chemical shifts (δ) are reported in parts per million (ppm) relative to the residual solvent peak. Multiplicities are reported as singlet (s), doublet (d), triplet (t), and multiplet (m) and coupling constants (J) quoted to the nearest 0.5 Hertz (Hz). Assignments of peaks are as follows: Carb H = carbazole H with G1, G2, and G3 denoting the generation number; surf Fl H = surface fluorenyl H; core Fl H = spiro-bifluorene H, where a fluorene or carbazole proton cannot be definitively assigned as surface or core they are denoted as Fl H and Carb H respectively. Gel permeation chromatography was carried out on a Polymer Laboratories PL-GPC 50 using PLgel Mixed-A columns (600 mm + 300 mm lengths, 7.5 mm diameter) from Polymer Laboratories calibrated with polystyrene narrow standards (Mp = 162–6.0352 × 106) in tetrahydrofuran with toluene as the flow marker. The tetrahydrofuran was pumped at a rate of 1 cm3 min−1 at 40 °C. Mass spectra were recorded on an Applied Biosystems Voyager-DE STR instrument with a matrix-assisted laser-desorption ionisation-time of flight (MALDI-TOF MS; positive-ion, reflectron) setup, with (E)-2-[3-(4-t-butylphenyl)-2-methyl-2-propenylidene] malononitrile (DCTB) as the matrix. Loading of the sample on the analysis stage was done by first spotting a solution of matrix in dichloromethane onto the plate, and when dried, a solution of dendrimer in dichloromethane-light petroleum ether mixture (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) was spotted, and finally the matrix solution was deposited on top. Mass units (m/z) are presented in Daltons, and intensities are quoted in percentages of the base peak. Thermal gravimetric analysis (TGA) and differential scanning calorimetry (DSC) were performed on a Mettler Toledo TGA/DSC/STARe and Mettler Toledo DSC/STARe System, respectively, with the former analysed under nitrogen flow (20 mL min−1) from 25 to 600 °C at 10 °C min−1 in aluminum crucibles. Melting points were carried out on a Gallenkamp melting point apparatus and are uncorrected. Elemental analyses were carried out using a Carlo Erba NCHS Analyser Model NA 1500 instrument.
:1) was spotted, and finally the matrix solution was deposited on top. Mass units (m/z) are presented in Daltons, and intensities are quoted in percentages of the base peak. Thermal gravimetric analysis (TGA) and differential scanning calorimetry (DSC) were performed on a Mettler Toledo TGA/DSC/STARe and Mettler Toledo DSC/STARe System, respectively, with the former analysed under nitrogen flow (20 mL min−1) from 25 to 600 °C at 10 °C min−1 in aluminum crucibles. Melting points were carried out on a Gallenkamp melting point apparatus and are uncorrected. Elemental analyses were carried out using a Carlo Erba NCHS Analyser Model NA 1500 instrument.
Absorbance and fluorescence measurements
For determining the molar extinction coefficients UV-Visible spectra were recorded on a Varian Cary 5000 UV-Vis-NIR Spectrophotometer in spectroscopic grade tetrahydrofuran. For the fluorescence measurements all dendrimer solutions were prepared with spectroscopic grade tetrahydrofuran with a peak absorbance of less than 0.2. The absorbance spectra were recorded with a Varian Cary 5000 spectrophotometer. Fluorescence spectra for solutions were measured with a Horiba Jobin-Yvon Fluoromax 4 system with the dendrimer solutions excited at the peak of the absorbance.Photoluminescence quantum yield
The solution PLQYs of the dendrimers were measured relative to a reference of quinine sulfate solution in 0.5 M sulfuric acid.26 The excitation wavelength was 310 nm with the measured emission spectra corrected for self-absorption. All solutions were prepared in spectroscopic grade tetrahydrofuran with no degassing. The thin film PLQY were measured in an integrating sphere using the method described by Greenham et al.27 The 325 nm emission line from a HeCd laser was used as the excitation source. The laser power was attenuated to be in the range of 200–300 μW with a beam diameter at the sample of approximately 1 mm. To minimize the impact of photodegradation the interior of the integrating sphere was flushed with nitrogen.Stern–Volmer measurements
For each dendrimer a 50–100 cm3 volume of dilute solution with peak absorbance of less than 0.2 in tetrahydrofuran was prepared. This solution was used to dissolve measured quantities of the analytes to give solutions of known concentration. The use of the dendrimer solution to dissolve the analytes meant that the dendrimer concentration remained constant when the dendrimer and analyte solutions were mixed. Stern–Volmer measurements were performed by measuring the fluorescence and absorbance spectra of 2.5 cm3 of dendrimer solution in a cuvette before and after a series of 25 μL additions of the analyte solution. The results were corrected for absorption of the fluorescence by the dendrimer-analyte solution and direct absorbance of the excitation by the analyte. Both these effects if left uncorrected would result in an overestimation of the Stern–Volmer constants.Film quenching measurements and recovery
Film photoluminescence was recorded with a Jobin-Yvon Fluorolog Tau3 system. The dendrimer films were spin-coated on glass or fused silica substrates at 2000 rpm from solutions with a concentration of 7 mg cm−3. Film thicknesses were measured with a Veeco Dektak 150 Surface Profilometer and found to be in the range of 20–25 nm. For the thin film fluorescence quenching measurements a small quantity of the analyte was placed beneath some cotton wool inside a sealed cuvette and left overnight to allow the analyte vapor to reach equilibrium (fully saturated). All the measurements were performed at room temperature to ensure a constant vapor pressure. The measurement consisted of two parts: a fluorescence spectrum before and after exposure to the analyte vapor and a kinetic scan during the exposure. For each measurement four films were used with the resulting PL intensity kinetics averaged to give a single decay. For the kinetic scan the dendrimer films were mounted onto a modified cuvette cap, which provided some control of the position of the film inside the cuvette. The fluorescence signal was measured at the emission peak (395 nm for the G1, 400 nm for the G2 and 410 nm for the G3) for ten minutes. To determine whether the analyte could be displaced from the film and the fluorescence recovered the films were flushed with nitrogen for 3 min and the fluorescence spectrum measured again.Temperature dependent recovery
Tests were carried out by placing the dendrimer films onto a heating stage in a custom-built metal chamber incorporating a UV LED emitting at a wavelength of 365 nm and an optical window through which the fluorescence was transmitted via a fibre optic cable to an Ocean Optics USB2000 spectrograph. A temperature controller was used in this measurement to control the stage temperature. The temperature was changed in 20 °C steps every 10 min up to 120–140 °C before the sample was allowed to cool. The fluorescence signal was integrated over the emission range of 415 nm to 450 nm with an integration time of 300 milliseconds for the G1, and 415 nm to 500 nm with an integration time of 600 milliseconds for the G2 and G3. Nitrogen was blown into the chamber at a rate of 5 L min−1 for the duration of the measurement.Synthesis
:7) as the eluent to give G1as a cream solid (421.8 mg, 80%). M.p. 312–314 °C; Found: C, 91.3; H, 6.7; N, 1.8. C125H110N2 requires: C, 91.5; H, 6.7; N, 1.7; δH (500 MHz, CDCl3) 8.42 (4 H, bs, G1-Carb H), 8.22 (2 H, d, J = 8, core Fl H), 7.82–7.62 (24 H, m, Fl H and/or Carb H), 7.43–7.26 (20 H, m, surf and core Fl H), 7.07 (4 H, d, J = 8, core Fl H), 2.08–1.97 (16 H, bm, CH2), 0.83–0.66 (40 H, brm, CH2 and CH3); λmax(THF)/nm 361sh (log ε/dm3mol−1cm−1 5.04), 335 (5.19), 318sh (5.15), 309sh (5.11), 299sh (5.05), 270 (5.02), 226sh (5.44); m/z [MALDI-TOF, DCTB] Anal. Calcd for C125H110N2: 1638.9 (71%), 1639.9 (100%), 1640.9 (70%), 1641.9 (32%), 1642.9 (11%), 1643.9 (3%). Found: 1638.7 (60%), 1639.7 (100%), 1640.7 (63%), 1641.7 (26%), 1642.7 (9%), 1643.7 (3%) [M+]. Mn = 1741, Mv = 1806, Mw = 1817, p.d. = 1.04.
:7 to 2:3) as eluent to give G2 as a white powder (73 mg, 68%). M.p. 315–318 °C; Found: C, 90.8; H, 6.7; N, 2.6. C119H116N2 requires: C, 90.8; H, 6.7; N, 2.6; δH (500 MHz, CDCl3) 8.51 (8 H, brs, G2-Carb H), 8.36 (2 H, d, J = 8.0, core Fl H), 8.34 (4 H, brs, G1-Carb H), 7.93 (2 H, dd, J = 1.5, J = 7.5, core Fl H), 7.91 (2 H, d, J = 7.5, core Fl H), 7.82–7.67 (40 H, m, surf Fl H and G2-Carb H), 7.65 (4 H, d, J = 9.0, G1-Carb H), 7.60 (4 H, d, J = 9.0, G1-Carb H), 7.52–7.45 (10 H, m, G2-Carb H and core Fl H), 7.39–7.27 (28 H, m, surf Fl H, and core Fl H), 7.17 (2H, d, J = 7.5, core Fl H), 2.00–1.96 (32 H, bm, CH2), 0.82–0.66 (80 H, m, CH2 and CH3); λmax(THF)/nm 338sh (log ε/dm3mol−1cm−1 5.52), 321 (5.56), 309sh (5.51), 270 (5.27), 233 (5.56); m/z [MALDI-TOF, DCTB] Anal. Calcd for C119H116N2: 3291.7 (25%), 3292.7 (71%), 3293.7 (100%), 3294.7 (93%), 3295.7 (65%) 3296.7 (36%), 3297.7 (17%), 3298.8 (6%), 3299.8 (2%). Found: 3291.1 (23%), 3292.2 (76%), 3293.1 (100%), 3294.1 (95%), 3295.1 (66%), 3296.1 (36%), 3297.1 (19%) [M+]. Mn = 3320, Mv = 3504, Mw = 3537, p.d. = 1.07.
:3) as eluent. The main fraction was collected and the solvent removed in vacuo. The residue was dissolved in dichloromethane and precipitated by pouring into methanol. The precipitate was collected at the filter to give G3 as a cream solid (128 mg, 58%). Mp. decomp 337 °C; Found: C, 90.1; H, 6.65; N, 3.0. C497H434N14 requires: C, 90.4; H, 6.6; N, 3.0; δH (500 MHz, CDCl3) 8.59 (4 H, bs, G1-Carb H), 8.52 (16 H, bs, G3-Carb H), 8.43 (10 H, m, G2-Carb H and Fl H), 8.01 (2 H, d, J = 7.5, Fl H), 7.96 (2 H, d, J = 7.5, Fl H), 7.85 (4 H, d, J = 8.5, Carb H or Fl H), 7.80–7.77 (102 H, m, Carb H and/or Fl H), 7.55 (18 H, m, G3-Carb H and Fl H), 7.43–7.24 (52 H, m, surf and core Fl H), 2.08–1.91 (64 H, m, CH2), 0.82–0.60 (160 H, m, CH2 and CH3); λmax(THF)/nm 339sh (log ε/dm3mol−1cm−1 5.86), 319 (5.96), 308sh (5.93), 299sh (5.86), 270 (5.72), 231 (5.94); m/z [MALDI-TOF, DCTB] Anal. Calcd for C497H434N14: 6602.7 (100%). Found: 6601.7 (100%) [M+]; Mn = 5164, Mv = 5450, Mw = 5502, p.d. = 1.06.
Acknowledgements
Professor Paul Burn is the recipient of an Australian Research Council Federation Fellowship (Project FF0668728). Professor Paul Meredith is the recipient of a Smart State Senior Fellowship from the Queensland State Government. The work was funded by the Australian Research Council under the Discovery Program (DP0986838). Guoqiang Tang is a recipient of a University of Queensland International Postgraduate Research Scholarship and the Centre for Organic Photonics and Electronics is a strategic initiative funded by the University of Queensland.References
- J. S. Yang and T. M. Swager, J. Am. Chem. Soc., 1998, 120, 11864 CrossRef CAS.
- S. J. Toal and W. C. Trogler, J. Mater. Chem., 2006, 16, 2871 RSC.
- T. Caron, M. Guillemot, P. Montmeat, F. Veignal, F. Perraut, P. Prene and F. Serein-Spirau, Talanta, 2010, 81, 543 CrossRef CAS.
- D. S. Moore, Rev. Sci. Instrum., 2004, 75, 2499 CrossRef CAS.
- M. Fisher, M. laGrone and J. Sikes, Detection and Remediation Technologies for Mines and Minelike Targets Viii, Pts 1 and 2, 2003, 5089, 991 CAS.
- M. Fisher, J. Sikes and M. Prather, Sensors, and Command, Control, Communications, and Intelligence(C31) Technologies for Homeland Security and Homeland Defense Iii, Pts 1 and 2, 2004, 5403, 409 Search PubMed.
- C. P. Chang, C. Y. Chao, J. H. Huang, A. K. Li, C. S. Hsu, M. S. Lin, B. R. Hsieh and A. C. Su, Synth. Met., 2004, 144, 297 CrossRef CAS.
- S.-C. Lo and P. L. Burn, Chem. Rev., 2007, 107, 1097 CrossRef CAS.
- H. Cavaye, P. E. Shaw, X. Wang, P. L. Burn, S.-C. Lo and P. Meredith, Macromolecules, 2010, 43, 10253 CrossRef CAS.
- D. A. Olley, E. J. Wren, G. Vamvounis, M. J. Fernee, X. Wang, P. L. Burn, P. Meredith and P. E. Shaw, Chem. Mater., 2011, 23, 789 CrossRef CAS.
- J. Liu, Y. Zhong, P. Lu, Y. Hong, J. W. Y. Lam, M. Faisal, Y. Yu, K. S. Wong and B. Z. Tang, Polym. Chem., 2010, 1, 426 RSC.
- M. Guo, O. Varnavski, A. Narayanan, O. Mongin, J. P. Majoral, M. Blanchard-Desce and T. Goodson, J. Phys. Chem. A, 2009, 113, 4763 CrossRef CAS.
- H. Cavaye, A. R. G. Smith, M. James, A. Nelson, P. L. Burn, I. R. Gentle, S.-C. Lo and P. Meredith, Langmuir, 2009, 25, 12800 CrossRef CAS.
- S. Richardson, H. S. Barcena, G. A. Turnbull, P. L. Burn and I. D. W. Samuel, Appl. Phys. Lett., 2009, 95, 063305 CrossRef.
- K. A. Knights, S. G. Stevenson, C. P. Shipley, S.-C. Lo, S. Olsen, R. E. Harding, S. Gambino, P. L. Burn and I. D. W. Samuel, J. Mater. Chem., 2008, 18, 2121 RSC.
- S. F. Alvarado, P. F. Seidler, D. G. Lidzey and D. D. C. Bradley, Phys. Rev. Lett., 1998, 81, 1082 CrossRef CAS.
- K. Meerholz and J. Heinze, J. Am. Chem. Soc., 1989, 111, 2325 CrossRef CAS.
- P. I. Djurovich, E. I. Mayo, S. R. Forrest and M. E. Thompson, Org. Electron., 2009, 10, 515 CrossRef CAS.
- T. F. Jenkins, D. C. Leggett, P. H. Miyares, M. E. Walsh, T. A. Ranney, J. H. Cragin and V. George, Talanta, 2001, 54, 501 CrossRef CAS.
- S. W. Thomas III, J. P. Amara, R. E. Bjork and T. M. Swager, Chem. Commun., 2005, 4572 RSC.
- S. W. Thomas III, G. D. Joly and T. M. Swager, Chem. Rev., 2007, 107, 1339 CrossRef.
- J. R. Lakowicz, Principles of fluorescence spectroscopy, Springer, New York, 2006 Search PubMed.
- D. Zhao and T. M. Swager, Macromolecules, 2005, 38, 9377 CrossRef CAS.
- Y. Liu, R. C. Mills, J. M. Boncella and K. S. Schanze, Langmuir, 2001, 17, 7452 CrossRef CAS.
- J. Kumar, A. Kumar, M. K. Pandey, R. Anandakathir, R. Mosurkal, V. S. Parmar and A. C. Watterson, Sens. Actuators, B, 2010, 147, 105 CrossRef.
- J. N. Demas and G. A. Crosby, J. Phys. Chem., 1971, 75, 991 CrossRef.
- N. C. Greenham, I. D. W. Samuel, G. R. Hayes, R. T. Phillips, Y. A. R. R. Kessener, S. C. Moratti, A. B. Holmes and R. H. Friend, Chem. Phys. Lett., 1995, 241, 89 CrossRef CAS.
- W. F. Jiang, H. L. Wang, A. G. Wang and Z. Q. Li, Synth. Commun., 2008, 38, 1888 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |