Conjugated polyelectrolyte brushes with extremely high charge density for improved energy transfer and fluorescence quenching applications
Zhiyong
Zhang
,
Xiaomei
Lu
,
Quli
Fan
*,
Wenbo
Hu
and
Wei
Huang
*
Key Laboratory for Organic Electronics & Information Displays (KLOEID) and Institute of Advanced Materials (IAM), Nanjing University of Posts & Telecommunications (NUPT), 9 Wenyuan Road, Nanjing, 210046, Jiangsu, China. E-mail: wei-huang@njupt.edu.cn; iamqlfan@njupt.edu.cn; Fax: + 86 25 8586 6999; Tel: + 86 25 8586 6008
First published on 9th August 2011
Abstract
A conjugated polyelectrolyte brush (PB3) composed of a polyfluorene backbone and poly[2-(dimethylamino)ethyl methacrylate] (PDMAEMA) side chains is synthesized via atom transfer radical polymerization (ATRP), and its properties are investigated and compared with its linear counterpart (P2, poly[9,9′-bis(6-N,N,N-trimethylammoniumhexyl)fluorene] dibromide). Despite the same conjugated backbones for PB3 and P2, the polymer brush architecture of PB3 endows it with an extremely high charge density, consequently give rise to better optical stability, higher water solubility (28 mg mL−1) and higher quantum efficiency (52%) as compared to those of P2; moreover, it induces stronger electrostatic attraction with oppositely-charged analytes, making PB3 contact the energy donor or quencher molecules much more efficiently than P2 does. As such, PB3 can afford not only higher FRET-amplified dye emission but also larger fluorescence quenching constant as compared to P2. With its high water-solubility, the fluorescence of PB3 sustains in the presence of a large amount of ssDNA, showing its optical durability in complicated biological media. As a result, CPE brushes could constitute a new generation of water-soluble fluorescent macromolecules having desirable optical and biochemical properties for various sensing applications.
1. Introduction
Recently, conjugated polyelectrolytes (CPEs) have been widely used in chemical and biological sensing because of their intriguing optoelectronic and biocompatible properties.1–6 These materials have π-electron delocalized conjugated backbones, featuring molecule-wire like light-harvesting structures for rapid intrachain and interchain excitation migration viaelectron transfer (ET) or fluorescence resonance energy transfer (FRET).7,8 Thereby, CPEs can profoundly change their optical properties upon minor perturbation of the external environment, ultimately leading to amplified fluorescence signals as compared to that for small molecule fluorophores.9–11 To promote the level of technologies for efficient, convenient and precise detection of biomolecules such as DNA and proteins, major efforts have been devoted to the development of CPEs with different structures such as poly(p-phenyleneethynylene) (PPE), poly(p-phenylenevinylene) (PPV), poly(thiophene) (PT) and poly(fluorene) (PF) during the past decade.12–21Among CPEs, PF-based derivatives are the most attractive research subjects because of their superior optoelectronic properties and ease of side chain modification at 9-positon.22 In particular, they generally exhibit higher photoluminescence (PL) quantum efficiencies in aqueous media as compared to other CPEs, making them more powerful for sensing applications. However, their quantum efficiencies are still constrained below 40%,9 which are significantly lower than those in organic solvents.23,24 Such weakened PL quantum efficiencies in aqueous media are mainly ascribed to the polymer aggregation caused by the hydrophobic nature of aromatic backbones and the low density of charge side chains.25–28 Apart from determining water-solubility, charged side chains of CPEs also manipulate their interactions with oppositely charged biomolecules during the assay operation. Despite the importance of charged side chains, the ratio of the fluorene unit (hydrophobic segments) to the number of charges on their side chains (hydrophilic segments) in most CPEs is relatively low (≤2). To improve the photophysical and biochemical properties of CPEs for sensing applications, it is of significance to develop CPEs with high charge densities. In addition, investigation of these CPEs should be beneficial to further revealing their structure–properties relationship and in turn exploring their full potential in chemical and biological sensing.
Polymer brushes refer to a kind of unique macromolecules with a linear polymeric backbone and densely grafted long side chains.29 Atom transfer radical polymerization (ATRP) has been utilized to synthesize polymer brushes using monomers such as N-isopropylacrylamide (NIPPA), 2-(dimethylamino)ethyl methacrylate, (DMAEMA) and tert-butyl acrylate (TBA). In this regard, our group has particular interest in developing polymer brushes comprising conjugated polymers as the backbone. We found that by virtue of the shielding effect of coil segments, the interchain interactions between conjugated backbones are efficiently reduced, which substantially inhibit the formation of low-energy defects (e.g. excimers and aggregates) and consequently result in much higher efficient optoelectronic properties as compared to their linear counterparts.30–33 However, water-soluble CPE-based polymer brushes have been rarely developed to date. By attaching highly-charged polymer to the conjugated backbone viaATRP, it should be feasible to generate CPEs with high charge density that overcome the disadvantages of linear polyelectrolytes to have more desirable properties for various sensing applications.
In this article, we design and synthesize a water-soluble CPE brush (PB3, Scheme 1) comprising a PF backbone and poly(2-(dimethylamino)ethyl methacrylate) (PDMAEMA) as the grafted side chain for improved FRET and fluorescence quenching sensing applications. This polymer is synthesized viaATRP from a PF macro initiator, which has an extremely high charge density, an average charge number of 74 per fluorene unit (FU). To reveal the advantage of polymer brush structure over linear structure, we also synthesize its linear counterpart (P2, Scheme 1). We find that due to the high charge density arising from PDMAEMA graft side chains, PB3 exhibits excellent water-solubility (∼28 mg mL−1), much higher quantum yield (52%) and much higher stability in solution as compared to those of P2. More importantly, we also systematically investigate and compare their FRET and fluorescence quenching properties in aqueous media. The results illustrate that the high charge density of PB3 induces stronger electrostatic attraction with oppositely-charged analytes, enabling it to contact the energy donor or quencher molecules much more efficiently than P2 does. Under such circumstances, PB3 can bring in not only higher FRET-amplified dye emission and but also a larger fluorescence quenching constant as compared to P2. As such, this study provides a novel polymer brush approach to develop CPEs with greatly improved chemical and physical properties for sensing applications.
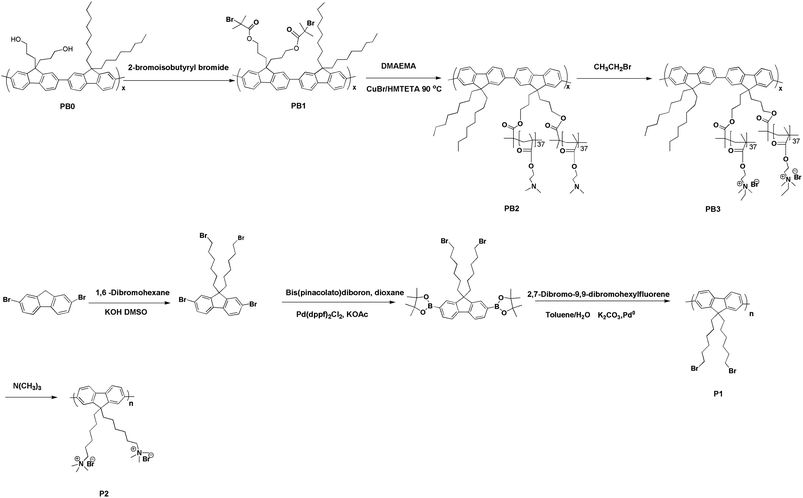 | ||
| Scheme 1 Synthetic routes towards PB3 and P2. | ||
2. Results and discussion
2.1 Synthesis and characterization
The synthetic routes towards PB3 and P2 are shown in Scheme 1. According to our previous report34PB0 was obtained by heating the mixture of 2,7-dibromo-9,9-bis(3-hydroxylpropyl)-fluorene and 9,9-dioctylfluorene-2,7-bis(trimethylene boronate) in toluene at 85 °C for 72 h using Pd(PPh3)4 as the catalyst. Treatment of PB0 with 2-bromoisobutyryl bromide gives PB1. Macroinitiator (PB1), the number-average molecular weight and polydispersity are 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300 and 1.6, respectively, corresponding to a degree of polymerization of ∼10. ATRP of macroinitiator (PB1) with DMAEMA was accomplished in o-dichlorobenzene with 1,1,4,7,10,10-hexamethyl triethylenetetramine (HMTETA) as the ligand and CuBr as the catalyst. The resulting solution was passed through a column of neutral alumina, reduced and then precipitated in n-hexane, producing a light yellow solid. PB2 was treated with bromoethane in THF at 50 °C to obtain PB3. The chemical structures of PB2 and PB3 were characterized by 1H NMR and 13C NMR The sample was characterized by high performance liquid chromatography (HPLC) in water (0.1 M NaCl) using Oat β-glucan standards to obtain a molecular weight (Mn = 136800, Mw/Mn = 1.5), corresponding to a degree of polymerization of ∼744. This reveals that the number of monomers per FU is close to 74. As shown in Fig. 1, all the peaks of side chains can be clearly observed. The ratio of integrated areas of methyl (–NCH2CH3) (1.31 ppm) and methylene (–CH2N) (3.09 ppm) peaks in 1H NMR showed that the degree of quaternization is higher than 95%. However, the signals of protons assigned to conjugated backbones are more difficult to discern. This phenomenon may be attributed to two factors. Firstly, as compared to the much greater number of protons on the side chains, there are relatively few protons on the fluorene rings. Secondly, restricted mobility may also result in peak broadening, making the signals from the protons of fluorene unit substantially weaker.35
300 and 1.6, respectively, corresponding to a degree of polymerization of ∼10. ATRP of macroinitiator (PB1) with DMAEMA was accomplished in o-dichlorobenzene with 1,1,4,7,10,10-hexamethyl triethylenetetramine (HMTETA) as the ligand and CuBr as the catalyst. The resulting solution was passed through a column of neutral alumina, reduced and then precipitated in n-hexane, producing a light yellow solid. PB2 was treated with bromoethane in THF at 50 °C to obtain PB3. The chemical structures of PB2 and PB3 were characterized by 1H NMR and 13C NMR The sample was characterized by high performance liquid chromatography (HPLC) in water (0.1 M NaCl) using Oat β-glucan standards to obtain a molecular weight (Mn = 136800, Mw/Mn = 1.5), corresponding to a degree of polymerization of ∼744. This reveals that the number of monomers per FU is close to 74. As shown in Fig. 1, all the peaks of side chains can be clearly observed. The ratio of integrated areas of methyl (–NCH2CH3) (1.31 ppm) and methylene (–CH2N) (3.09 ppm) peaks in 1H NMR showed that the degree of quaternization is higher than 95%. However, the signals of protons assigned to conjugated backbones are more difficult to discern. This phenomenon may be attributed to two factors. Firstly, as compared to the much greater number of protons on the side chains, there are relatively few protons on the fluorene rings. Secondly, restricted mobility may also result in peak broadening, making the signals from the protons of fluorene unit substantially weaker.35
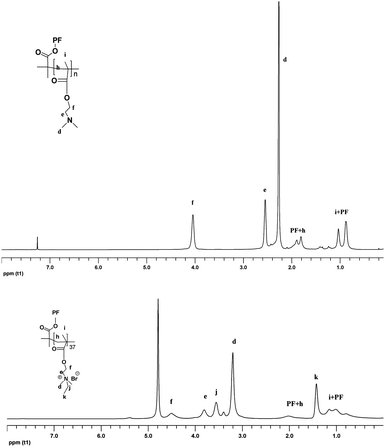 | ||
| Fig. 1 1H NMR spectra of PB2 and PB3 in CDCl3 and D2O respectively. PF refers to fluorene units and alkyl chains. | ||
2,7-Bis[9,9′-bis(6-bromohexyl) fluorene and 2,7-bis[9,9′-bis(6-bromohexyl) fluoreneyl]-4,4,5,5-tetramethyl[1,3,2] dioxaborolane were synthesized according to previous literature.36 The Suzuki cross-coupling polymerization at a feed ratio of 1:1 provided the neutral polymer (P1). The sample was characterized by gel permeation chromatography (GPC) in THF using polystyrene standards. The number-average molecular weight and polydispersity of P1 are 12,000 and 1.8, respectively, which shows P1 has a degree of polymerization (DP) close to PB1. The quaternization degree of P2 could be determined by 1H NMR spectra. After a treatment with trimethylamine, the polymer P2 exhibited a well-resolved peak at 3.05 ppm corresponding to the methyl groups adjacent to the nitrogen atom. The relative integrals of the methyl groups and the peak at 3.30 ppm (–CH2–) are nearly 4:1. In addition, there were no split peaks arising from the quanternized (high field) and unquanternized components. Therefore, the quaternization degree of P1 was estimated to be higher than 90%. P2 is soluble in a number of polar solvents such as methanol and water.
The solubility limits of PB3 and P2 in water at 25 °C are ∼28 and 8 mg mL−1, respectively. The much higher solubility of PB3 relative to P2 stems from the presence of cationic graft side chains that offer much higher charge density. To further study the solubility status, dynamic light scattering (DLS) analysis was performed for P2 and PB3 at [FU] = 1 μM. The mean diameters are measured to be 116 nm and 30 nm for P2 and PB3, respectively. The data indicate that PB3 has a well-extended chain conformation in aqueous solution, while P2 forms large aggregates due to its poor water-solubility. With the much better water-solubility and less aggregation, PB3 is anticipated to outperform P2 in sensing applications.
2.2 Optical properties
Table 1 shows optical data for P2 and PB3. The UV-vis absorption and PL spectra of PB3 are depicted in Fig. 2 (a) and (b), respectively. The concentration for PB3 based on every two FU is 1 μM. In comparison with the absorption maximum for PB3 in solution, its absorption maximum in film is not only slightly red-shifted by 7 to 387 nm, but also its absorption profile becomes broad. Meanwhile, the emission spectrum of PB3 in film is also red-shifted to 423 nm as compared to that of PB3 in solution. In comparison with PB3, the UV-vis absorption spectra of P2 in film and in solution are located at 384 and 385 nm, while its PL spectrum in film has minor change in emission maximum as in solution (see Fig. 3). The much larger red-shifted absorption and emission maxima for PB3 in film and in solution as compared with those for P2 indicate that the grafted side chains offer PB3 a greater flexible conformation in aqueous solution and thus a more pronounced planarity of fluorene ring in film, resulting in the increase of the effective conjugation length and thus red-shifted the absorption and PL spectra.37![(a) UV–vis absorption spectrum of PB3 in PBS. (b) PL emission spectrum of PB3 in PBS (20 mM, pH = 7.4). [PB3] = 1 μM. Excitation at 380 nm.](/image/article/2011/PY/c1py00213a/c1py00213a-f2.gif) | ||
| Fig. 2 (a) UV–vis absorption spectrum of PB3 in PBS. (b) PL emission spectrum of PB3 in PBS (20 mM, pH = 7.4). [PB3] = 1 μM. Excitation at 380 nm. | ||
![(a) UV–vis absorption spectrum of P2 in PBS. (b) PL emission spectrum of P2 in PBS (20 mM, pH = 7.4). [P2] = 1 μM. Excitation at 380 nm.](/image/article/2011/PY/c1py00213a/c1py00213a-f3.gif) | ||
| Fig. 3 (a) UV–vis absorption spectrum of P2 in PBS. (b) PL emission spectrum of P2 in PBS (20 mM, pH = 7.4). [P2] = 1 μM. Excitation at 380 nm. | ||
To better know the difference in photophysical properties between P2 and PB3, we studied their fluorescence response to varying amounts of THF in phosphate-buffer saline (PBS). As shown in Fig. 4 (a), the emission intensity of P2 increases gradually upon the addition of THF. When the THF content is up to 30%, the emission maximum of P2 at 431 nm is shifted to 421 nm, while the strongest emission intensity is observed. Moreover, further increasing the THF content to 90% results in obvious red shift in the emission maximum, while the emission intensity is gradually decreased. These observations are ascribed to the presence of two different aggregation condition.38 One is the poor solubility of P2 in PBS. As confirmed by DLS, hydrophobic and π–π interactions of P2 occur in aqueous solution, which lower the emission of P2 in PBS. Thus, the addition of THF into P2 aqueous solution weakens interchain contacts, leading to reduced self-quenching and increased emission intensity. Another aggregate species is generated from the interactions between positively charged quaternary amine groups and anionic counterions. Fig. 4 (b) shows that the emission intensity of PB3 in PBS increases gradually with changing THF content from 10% to 40%. However, there are no obvious shifts in the emission maxima of PB3 due to its lower interchain interactions and much higher water solubility, which are significantly different from the observation for P2 upon the addition of THF. When the THF content is increased to 80%, the emission intensity begins to decrease and at the same time the emission maximum at 419 nm is shifted to 423 nm. This is caused by the fact that the amount of THF forces the production of one aggregate due to reduced solubility of PB3 in PBS/THF.38 The experiments above further indicate that grafted side chains with higher charge densities can significantly inhibit the formation of optically-detrimental aggregates in aqueous solution.
![(a) Emission maxima and PL intensity of P2 as a function of THF content in PBS. (b) Emission maxima and PL intensity of PB3 as a function of THF content in PBS. [P2] = [PB3] = 1.0 μM, Excitation at 380 nm.](/image/article/2011/PY/c1py00213a/c1py00213a-f4.gif) | ||
| Fig. 4 (a) Emission maxima and PL intensity of P2 as a function of THF content in PBS. (b) Emission maxima and PL intensity of PB3 as a function of THF content in PBS. [P2] = [PB3] = 1.0 μM, Excitation at 380 nm. | ||
The PL quantum efficiencies for P2 and PB3 in PBS (20 mM) were measured to be 28% and 52%, respectively, using quinine sulfate in 0.1 M H2SO4 (quantum efficiency = 55%) as the standard. Moreover, the PL spectra of PB3 ([FU] = 1 μM) is still stable in 20 mM phosphate-buffer saline (pH = 7.4) for 12 months, while great intensity loss is observed for P2 solution after storing for the same time. The durable brightness of PB3 again reflects that the incorporation of charged polymeric side chains into the CPEs substantially is an effective way to construct high-performance fluorescent materials.
2.3 FRET study
To investigate the polymer brush over its linear counterpart in terms of FRET, PL experiments using ssDNA-FAM (5′-FAM-AGT TGG AGG TGA-3′) as the anionic energy-acceptor for PB3 and P2 were performed in 20 mM PBS (pH = 7.4) at [ssDNA-FAM] = 2.5 × 10−8 M (based on strands). The samples were incubated for 5 min and the PL spectra were measured. FAM was chosen as the label because of the efficient overlap between its absorption spectra and the emission spectra of both polymers, which should favor FRET. FAM emission was collected at 524 nm upon excitation of the polymer at 380 nm so as to observe FRET-induced dye emission.Changes in the PL spectra of ssDNA-FAM upon addition of P2 and PB3 are depicted in Fig. 5 (a) and (b), respectively. It is worth noting that under these conditions, FAM emission cannot be observed for both systems. FAM emission intensity as a function of both polymer concentrations is shown in Fig. 6. It is clear that the FAM emission intensity gradually increases with the successive addition of polymers for both systems, but the degree of increase in FAM emission intensity for PB3/ssDNA-FAM is significantly larger than that for P2/ssDNA-FAM within the tested polyelectrolyte concentration. As shown in Fig. 7, when P2 and PB3 concentrations were fixed at 75 nM, respectively, FAM emission from PB3/ssDNA-FAM was 3 times more intense than that from P2/ssDNA-FAM. Thereby, FRET experiments indicate that PB3 serves as better donor relative to P2 to favor FRET in aqueous solution.
![(a) The emission spectrum of P2/ssDNA-FAM solution upon increasing of [P2]. (b) The emission spectrum of P2/ssDNA-FAM solution upon increasing of [PB3]. The arrows indicate increasing [P2/PB3]. Excitation at 380 nm.](/image/article/2011/PY/c1py00213a/c1py00213a-f5.gif) | ||
| Fig. 5 (a) The emission spectrum of P2/ssDNA-FAM solution upon increasing of [P2]. (b) The emission spectrum of P2/ssDNA-FAM solution upon increasing of [PB3]. The arrows indicate increasing [P2/PB3]. Excitation at 380 nm. | ||
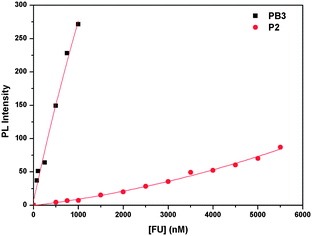 | ||
| Fig. 6 FAM emission intensity as a function of the polyelectrolyte concentration for PB3/ssDNA-FAM and P2/ssDNA-FAM. | ||
![Emission spectra of PB3 and P2 in the presence or absence of ssDNA-FAM ([ssDNA-FAM] = 2.5 × 10−8 M, [P2] = 75 nM, [PB3] = 75 nM).](/image/article/2011/PY/c1py00213a/c1py00213a-f7.gif) | ||
| Fig. 7 Emission spectra of PB3 and P2 in the presence or absence of ssDNA-FAM ([ssDNA-FAM] = 2.5 × 10−8 M, [P2] = 75 nM, [PB3] = 75 nM). | ||
To confirm that polyelectrolyte brushes have much stronger interactions with oppositely charged target molecules, acceptor (FAM) quenching experiments were performed under the same conditions as in the FRET experiments. The intrinsic emission properties of FAM were measured by direct excitation at its absorption maximum (480 nm). A 65% decrease in FAM emission intensity is observed for PB3/ssDNA-FAM and a 68% decrease is observed for P2/ssDNA-FAM (Fig. 8). On the basis of the previous studies, the acceptor quenching originates from the increased local dye concentration induced by DNA compaction. With the formation of complexes between CPEs and DNA due to the electrostatic interactions, acceptors are brought into close proximity, leading to increased local dye concentration.29,39 In this respect, charge densities of the polycation can directly determine the extent of association between CPEs and DNA. Therefore PB3 with higher charge densities per FU forms much tighter complexes with ssDNA as compared to P2, but it does not lead to more efficient dye-self fluorescence quenching for PB3, which should mainly be ascribed to three-dimensional conformation of PB3. It is found that the emission maximum of FAM at 518 nm with a series of [PB3] is red-shifted to 523 nm relative to its emission in the absence of PB3, while no obvious shift in the emission maximum of FAM can be observed for the P2/ssDNA-FAM at [P2] = 1 μM, indicating that PB3 and ssDNA-FAM have more stronger contacts; upon increasing the P2 concentration to 4.5 μM, the red-shifted FAM emission peak begins to emerge. This is because the complex formation between P2/PB3 and ssDNA-FAM can change the polarity near FAM.39 In addition to charge density, the extended conformation of PB3 in aqueous media as proven by DSL should also facilitate the complexation with ssDNA-FAM, as grafted side chains could act as long arms with large interacting region to favor the capture of target molecules.
![(a) Emission of ssDNA-FAM in the presence of P2. (b) Emission of ssDNA-FAM in the presence of PB3 (excitation at 480 nm) [ssDNA] = 2.5 × 10−8 M).](/image/article/2011/PY/c1py00213a/c1py00213a-f8.gif) | ||
| Fig. 8 (a) Emission of ssDNA-FAM in the presence of P2. (b) Emission of ssDNA-FAM in the presence of PB3 (excitation at 480 nm) [ssDNA] = 2.5 × 10−8 M). | ||
The above data clearly show that PB3 outperforms P2 in FRET. To be a good energy donor in a complicated biological system, the solution stability upon complexation is also required. Thus, we study the fluorescent responses of PB3 upon addition of ssDNA. As shown in Fig. 9b, the PL spectrum of PB3 is not significantly influenced in the presence of ssDNA. This phenomenon is essentially different from the previous report that CPEs fluorescence is greatly quenched upon addition of ssDNA due to the polymer aggregation (Fig. 9a).40 This improvement should be mainly attributed to the intrinsically high water-solubility of PB3 that makes the ssDNA/PB3 complexes stable enough in aqueous solution without forming the optically-detrimental defects. This result further reveals that PB3 holds a great promise for FRET applications in physiological systems.
![(a) Emission spectrum of P2 upon a series of [ssDNA] ([P2] = 1 μM). (b) Emission spectrum of PB3 upon a series of [ssDNA] ([PB3] = 1.](/image/article/2011/PY/c1py00213a/c1py00213a-f9.gif) | ||
| Fig. 9 (a) Emission spectrum of P2 upon a series of [ssDNA] ([P2] = 1 μM). (b) Emission spectrum of PB3 upon a series of [ssDNA] ([PB3] = 1. | ||
2.4 PL quenching
Having found that PB3 is superior to P2 in FRET, it is also important to compare their fluorescence quenching properties. Sodium anthraquinone-2,6-disulfonate (AQS) and K3Fe(CN)6 were chosen as the small molecular quenchers to investigate the fluorescence quenching of P2 and PB3via a photoinduced electron transfer mechanism. Due to the difference in molecular structure, AQS and K3Fe(CN)6 have their typical characteristics. AQS features large hydrophobic planar aromatic rings, whereas Fe(CN)63− has much stronger binding with oppositely charged target molecules because of more negative charges.41,42The fluorescence quenching of P2 and PB3 by AQS and Fe(CN)63− were carried out in PBS. The PL spectra of P2 and PB3 upon the dropwise addition of AQS were depicted in Fig. 10 and the Stern–Volmer (SV) quenching plots were shown in the inset of Fig. 10. The PL spectra of P2 and PB3 upon the dropwise addition of Fe(CN)63− were depicted in Fig. 11 and the Stern–Volmer (SV) quenching plots were shown in the inset of Fig. 11. For the AQS system PB3 emission intensity is quenched much more efficiently than P2 at the tested concentrations of AQS. At low quencher concentrations (<1.4 μM) for PB3 and any tested concentrations of quenchers for P2 the SV plots are linear and KSV values are 2.0 × 106 M−1 and 5.6 × 105 M−1, respectively. For the Fe(CN)63− system the SV plots are nearly linear at any tested concentrations of quenchers and KSV values are 1.1 × 106 M−1 for PB3 and 7.4 × 105 M−1 for P2.
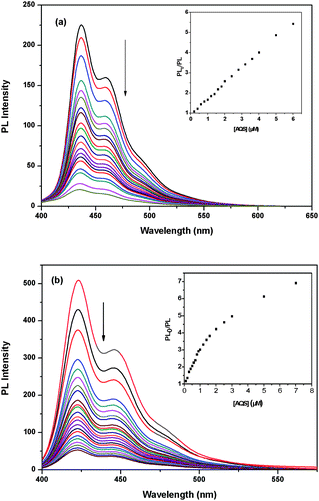 | ||
| Fig. 10 (a) PL quenching of P2 by AQS in PBS. The inset shows the Stern–Volmer plots of P2. (b) PL quenching of PB3 by AQS in PBS. The inset shows the Stern–Volmer plots of PB3. | ||
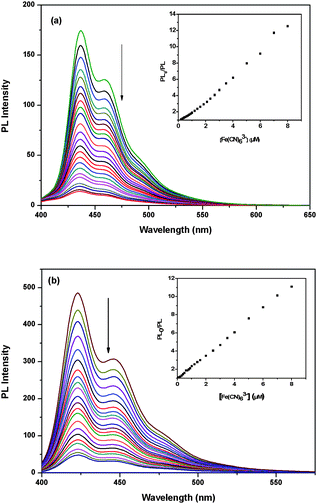 | ||
| Fig. 11 (a) PL quenching of P2 by Fe(CN)63− in PBS. The inset shows the Stern–Volmer plots of P2. (b) PL quenching of PB3 by Fe(CN)63− in PBS. The inset shows the Stern–Volmer plots of PB3. | ||
According to the previous studies, the quenching efficiency is closely dependent on ion-pair association of cationic polymer and anionic quenchers.41,42 The quenchers are associated with polymer chains and hence excitons are able to rapidly diffuse along the chain to the quenchers to result in fluorescence quenching. Similar to the observation in FRET, the higher quenching efficiencies of PB3 by both quenchers relative to those of P2 should originate from the high charge density of PB3, which allow it not only to form tighter complexes with quenchers but also capture more quencher molecules.
3. Conclusions
In conclusion, a CPE brush (PB3) and its linear counterpart (P2) were synthesized viaATRP and Suzuki coupling polymerization, respectively. Despite having the same conjugated backbones, PB3 exhibits better optical stability, more excellent water solubility and higher quantum efficiencies as compared to P2. These property advantages should benefit from its higher charged density on the grafted side chains that inhibits interchain π–π stacking. Furthermore, PB3 can serve as a better energy donor relative to P2 to amplify the dye emission via FRET. More importantly, the fluorescence of PB3 sustains in the presence of a large amount of ssDNA, showing its optical durability in complicated biological media. On the other hand, PB3 also gives high fluorescence quenching constants relative to those of P2 when using both AQS and Fe(CN)63− as the quenchers. As a result, CPE brushes have advantageous properties over linear CPEs in terms of both FRET and fluorescence quenching based sensing applications.From the materials viewpoint, the ease of ATRP allows one to finetune both component and length of conjugated backbone and grafted side chains of CPE brushes, providing the feasibility to further improve their sensing performance. For instance, multicolor CPE brushes could be developed simply by doping a small amount of benzothiadiazole into their backbone. From the practical perspective, biorecognition elements such as folic acid can be copolymerized into graft side chains in the course of ATRP, paving the way to integrate useful biochemical features into CPE brushes. Thus, this work not only develops a new generation of CPEs that overcomes the intrinsic drawbacks of linear CPEs (e.g. low charge density, poor water solubility, low brightens and weak association with target molecules), but also highlights a new direction on the molecular level to diversify the properties and advance the applications for water-soluble conjugated polymers.
4. Experimental
4.1 General methods
NMR spectra were recorded on a Bruker Ultra Shield Plus 400 MHz NMR (1H: 400 MHz, 13C: 100 MHz). The UV-visible absorption spectra were recorded on a Shimadzu UV-3600 UV-VIS-NIR spectrophotometer. PL spectra were measured using a RF-5301PC spectrofluorophotometer. Gel permeation chromatography (GPC) analysis of the neutral polymers was conducted on Shim-pack GPC-80X columns with THF as the eluent and polystyrenes as standard. The software package provided by Shimadzu Instruments was used for data analysis. Agilent 1100 high performance liquid chromatography (HPLC) analysis of polyelectrolyte was carried out in water (0.1 M NaCl) at room temperature with a Waters differential refractometer equipped with TSK-gei GMPWXL (7.8 mm × 300 mm, 13 μm). The flow rate was 0.5 mL min−1. Calibration was performed with Oat β-glucan standards. Quantum yields were measured using quinine sulfate as the standard, with a quantum yield of 55% in H2SO4 (0.1 M). DNA concentrations were determined by measuring the UV-vis absorbance at 260 nm in 3 mL quartz cuvettes) at room temperature. The optical properties, FRET and quenching experiments of PB3/P2 by quenchers were generally studied in dilute solutions (c = 1 μM, based on every two fluorene unit). The quenchers were successively added to the aqueous solutions of the polyelectrolytes, which was in a 3 mL quartz cuvette. All of the PL, UV experiments were carried out at room temperature. DLS measurements were carried out using a Brookhaven Instruments Corporation 90 Plus instrument with λ = 532 nm, and the particle diameters were calculated by nonnegative least squares (NNLS) method.4.2 Materials
All chemical reagents used were purchased from Sigma-Aldrich, Acros, and Alfa and were used as-received. 2,7-Dibromo-9,9′-bis(6-bromohexyl)-fluorene was synthesized according to previous literature.43 2,7-Dibromo-9,9′-bis(3-hydroxylpropyl)–fluorene, poly[(9,9′-bis(3-hydroxylpropyl)-fluorene)-alt-(9,9′-dioctylfluorene)] (PB0), macroinitiators (PB1) were synthesized according to our previous reports.334.3 Synthesis
:1) as the eluent. The final product is a white solid. Yield: 48%. 1H NMR (CDCl3, 400 MHz): 7.83–7.81 (m, 2H), 7.73–7.72 (m, 4H), 3.25 (t, 4H), 2.03–1.99 (m, 4H), 1.65–1.58 (m, 4H), 1.39 (s, 24H), 1.17–1.19 (m, 4H), 1.07–0.99 (m, 4H), 0.58–0.51 (m, 4H); 13C NMR (CDCl3, 100 MHz): 150.09, 143.91, 133.82, 128.78, 119.48, 83.78, 55.06, 39.94, 33.96, 32.67, 28.96, 27.74, 24.97, 23.38.
PB0: 1H NMR (CDCl3, 400 MHz): 7.67–7.86 (br, 12H), 3.47 (br, 4H), 2.13–2.27 (br, 8H), 1.13–0.81 (m, 32H); 13C NMR (THF-d8, 100 MHz): 152.12, 141.07, 140.79, 132.55, 129.06, 127.42, 126.64, 126.57, 121.80, 121.56, 120.41, 62.54, 55.82, 40.81, 37.23, 32.34, 30.62, 29.80, 28.43, 23.05, 13.98.
PB1: 1H NMR (CDCl3, 400 MHz): 7.67–7.86 (br, 12H), 4.02 (br, 4H), 2.35–2.13 (br, 8H), 1.86 (s, 12H), 1.25–0.82 (m, 32H); 13C NMR (CDCl3, 100 MHz): 171.54, 151.86, 150.06, 141.11, 140.18, 132.80, 128.80, 127.23, 126.34, 121.34, 120.32, 120.10, 65.82, 66.03, 64.58, 40.54, 36.62, 31.84, 30.74, 30.12, 29.32, 23.92, 23.86, 22.84, 14.10.
Acknowledgements
This work was financially supported by the national Basic Research Program of China under Grants 2009CB930600, the National Natural Science Foundation of China under Grants 90406021 and 20874048 and the Fok Ying-Tong Education Foundation under Grant 111051 as well as Program for New Century Excellent Talents in University under Grants NCET-10-0179 and Specialized Research Fund for the Doctoral Program of Higher Education under Grant 20093223110003 and NY211003.Notes and references
- F. Xia, X. L. Zuo, R. Q. Yang, Y. Xiao, D. Kang, A. Vallée-Bélisle, X. Gong, A. J. Heeger and K. W. Plaxco, J. Am. Chem. Soc., 2010, 132, 1252 CrossRef CAS.
- A. Satrijo and T. M. Swager, J. Am. Chem. Soc., 2007, 129, 16020 CrossRef CAS.
- D. T. McQuade, A. H. Hegedus and T. M. Swager, J. Am. Chem. Soc., 2000, 122, 12389 CrossRef CAS.
- H. A. Ho and M. Leclerc, J. Am. Chem. Soc., 2004, 126, 1384 CrossRef CAS.
- H. A. Ho and M. Leclerc, J. Am. Chem. Soc., 2003, 125, 4412 CrossRef CAS.
- K. Doré, S. Dubus, H. A. Ho, I. Lévesque, M. Brunette, G. Corbeil, M. Boissinot, G. Boivin, M. G. Bergeron, D. Boudreau and M. Leclerc, J. Am. Chem. Soc., 2004, 126, 4240 CrossRef CAS.
- F. He, Y. L. Tang, S. Wang, Y. L. Li and B. D. Zhu, J. Am. Chem. Soc., 2005, 127, 12343 CrossRef CAS.
- S. W. Thomas, G. D. Joly and T. M. Swager, Chem. Rev., 2007, 107, 1339 CrossRef CAS.
- X. R. Duan, L. B. Liu, F. D. Feng and S. Wang, Acc. Chem. Res., 2009, 43, 260.
- P. Howes, M. Green, J. Levitt, K. Suhling and M. Hughes, J. Am. Chem. Soc., 2010, 132, 3989 CrossRef CAS.
- F. K. Wang and G. C. Bazan, J. Am. Chem. Soc., 2006, 128, 15786 CrossRef CAS.
- B. S. Gaylord, A. J. Heeger and G. C. Bazan, J. Am. Chem. Soc., 2003, 125, 896 CrossRef CAS.
- S. Wang, B. S. Gaylord and G. C. Bazan, J. Am. Chem. Soc., 2004, 126, 5446 CrossRef CAS.
- B. Liu and G. C. Bazan, J. Am. Chem. Soc., 2004, 126, 1942 CrossRef CAS.
- S. J. Dwight, B. S. Gaylord, J. W. Hong and G. C. Bazan, J. Am. Chem. Soc., 2004, 126, 16850 CrossRef CAS.
- B. Liu, B. S. Gaylord, S. Wang and G. C. Bazan, J. Am. Chem. Soc., 2003, 125, 6705 CrossRef CAS.
- M. J. Martínez-Tomé, R. O. Esquembre, R. Mallavia and C. R. Mateo, Biomacromolecules, 2010, 11, 1494 CrossRef CAS.
- C. H. Fan, K. W. Plaxco and A. J. Heeger, J. Am. Chem. Soc., 2002, 124, 5642 CrossRef CAS.
- K.-Y. Pu, R. Y. Zhan and B. Liu, Chem. Commun., 2010, 46, 1470 RSC.
- K.-Y. Pu and B. Liu, J. Phys. Chem. B, 2010, 114, 3077 CrossRef CAS.
- C. H. Fan, J. W. S.Wang Hong, G. C. Bazan, K. W. Plaxco and A. J. Heeger, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 6297 CrossRef CAS.
- U. Scherf and E. J. W. List, Adv. Mater., 2002, 14, 477 CrossRef CAS.
- H. P. Wang, P. Lu, B. L. Wang, S. Qiu, M. R. Liu, M. Hanif, G. Cheng, S. Y. Liu and Y. G. Ma, Macromol. Rapid Commun., 2007, 28, 1645 CrossRef CAS.
- B. Zhu, Y. Han, M. H. Sun and Z. S. Bo, Macromolecules, 2007, 40, 4494 CrossRef CAS.
- M. A. Monteserín, H. D. Burrows, A. J. M. Valente, R. Mallavia, R. E. Di Paolo, A. L. Maçanita and M. A. J. Tapia, J. Phys. Chem. B, 2009, 113, 1294 CrossRef CAS.
- J. Choi, C. R. Ruiz and E. E. Nesterov, Macromolecules, 2010, 43, 1964 CrossRef CAS.
- K.-Y. Pu and B. Liu, Adv. Funct. Mater., 2009, 19, 277 CrossRef CAS.
- K.-Y. Pu, L. P. Cai and B. Liu, Macromolecules, 2009, 42, 5933 CrossRef CAS.
- K.-Y. Pu, K. Li and B. Liu, Adv. Funct. Mater., 2010, 20, 2770 CrossRef CAS.
- X.-F. Yu, Lu, S. C. Ye, T. C. Li, T. X. Liu, S. Y. Liu, Q. L. Fan, E.-Q. Chen and W. Huang, Macromolecules, 2006, 39, 1364 CrossRef CAS.
- S. Lu, Q. L. Fan, S. Y. Liu, S.-J. Chua and W. Huang, Macromolecules, 2002, 35, 9875 CrossRef CAS.
- S. Lu, Q. L. Fan, S.-J. Chua and W. Huang, Macromolecules, 2002, 36, 304.
- K.-Y. Pu, Y. Chen, X. Y. Qi, C. Y Qin., Q. Q. Chen, H. Y. Wang, Y. Deng, Q. L. Fan, Y. Q. Huang, S. J. Liu, W. Wei, B. Peng and W. Huang, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 3776 CrossRef CAS.
- Z. Y. Zhang, Q. L. Fan, P. F. Sun, L. L. Liu, X. M. Lu, B. Li, Y. W. Quan and W. Huang, Macromol. Rapid Commun., 2010, 31, 2160 CrossRef CAS.
- M. F. Wang, S. Zou, G. Guerin, L. Shen, K. Q. Deng, M. Jones, G. C. Walker, G. D. Scholes and M. A. Winnik, Macromolecules, 2008, 41, 6993 CrossRef CAS.
- K.-Y. Pu and B. Liu, Macromolecules, 2008, 41, 6636 CrossRef CAS.
- M. Elbing, A. Garcia, S. Urban, T.-Q. Nguyen and G. C. Bazan, Macromolecules, 2008, 41, 9146 CrossRef CAS.
- S. Wang and G. C. Bazan, Chem. Commun., 2004, 2508 RSC.
- B. Liu, B. S. Gaylord, S. Wang and G. C. Bazan, J. Am. Chem. Soc., 2003, 125, 6705 CrossRef CAS.
- K.-Y. Pu, Z. Fang and B. Liu, Adv. Funct. Mater., 2008, 18, 1321 CrossRef CAS.
- Y. Q. Huang, Q. L. Fan, X. F. Liu, N. N. Fu and W. Huang, Langmuir, 2010, 26, 19120 CrossRef CAS.
- M. B. Ramey, J. Hille, M. F. Rubner, C. Y. Tan, K. S. Schanze and J. R. Reynolds, Macromolecules, 2005, 38, 234 CrossRef CAS.
- H. Y. Woo, D. Vak, D. Korystov, A. Mikhailovsky, G. C. Bazan and D.-Y. Kim, Adv. Funct. Mater., 2007, 17, 290 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |