Efficient synthesis of benzobisazole terpolymers containing thiophene and fluorene†
Jared F.
Mike
a,
Jeremy J.
Intemann
a,
Min
Cai
b,
Teng
Xiao
b,
Ruth
Shinar
c,
Joseph
Shinar
b and
Malika
Jeffries-EL
*a
aDepartment of Chemistry, Iowa State University, Ames, IA 50011, USA. E-mail: malikaj@iastate.edu; Fax: +1-515-294-0105; Tel: +1-515-294-5759
bAmes Laboratory-USDOE and Department of Physics, Iowa State University, Ames, IA, 50011, USA
cMicroelectronics Research Center and Department of Electrical Engineering, Iowa State University, Ames, IA 50011, USA
First published on 18th July 2011
Abstract
We report the synthesis and lumuminescence properties of three novel polymers composed of 9,9-dioctylfluorene and a donor–acceptor-donor (D–A-D) triad of a benzobisazole moiety sandwiched between two octylthiophenes. The requiste monomers, 2,6-bis(5-bromo-3-octylthiophen-2-yl)-benzobisazoles were obtained efficiently via the Lewis acid catalyzed cyclization of 2-bromo-3-octyl-5-(triethoxymethyl)thiophene and the corresponding diamino diols or dithiols. The polymers were synthesized in excellent yield by the Suzuki coupling reaction between the D–A-D benzobisazole monomers and 9,9-dioctylfluorene bisboronic acid. Alkyl side chains provided the polymers with solubility in common organic solvents, enabling characterization using gel permeation chromatography, 1H NMR, UV-Vis and fluorescence spectroscopy. The polymers have optical bandgaps of 2.43–2.63 eV and HOMO levels at −5.54 to −5.65 eV relative to vacuum as determined by UV visible and photoelectron spectroscopy respectively. Light-emitting diodes using blends of the copolymers in a poly(N-vinyl carbazole) matrix yielded blue-green emission with luminous efficiencies of 0.86 Cd/A at ∼505 nm. This efficient and high-yielding route is a promising approach for the synthesis of polymers containing benzobisazole moieties.
Introduction
As a result of their optical and electronic properties, conjugated polymers are finding widespread use in semiconducting applications such as field-effect transistors, light-emitting diodes and photovoltaic cells.1 The benefits of conjugated polymers include the ability to tune their optical and electronic properties through chemical synthesis and the ability to fabricate devices from them using solution based techniques. Among conjugated polymers, polyfluorenes are widely investigated for use in polymer light-emitting diodes due to their excellent solubility, high solid state photoluminescence quantum yield and good charge carrier mobility. However, poly(9,9-dioctylfluorene) is a blue light emitting polymer with a large band gap (3.1 eV), a low electron affinity (EA = 2.5 eV), and a high ionization potential (IP = 5.6 eV), which makes it difficult to inject both holes and electrons into the polymer.2One way to change the positions of the frontier orbitals is to copolymerize fluorene with electron-deficient comonomers.3 In this regard benzobisazoles, such as benzobisoxazole and benzobisthiazole are promising building blocks for the synthesis of novel materials. These electron-deficient heterocycles are known for improving the charge transport, photoluminescence, and third-order nonlinear optical properties of materials containing them.4–6 Recently, new materials based on a donor–acceptor-donor (D–A-D) triad composed of a benzobisthiazole ring sandwiched between two alkylthiophenes have been reported.7–10 Organic field effect transistors using small molecules based on this D–A-D triad exhibited excellent hole mobility as a result of the materials' high crystalinity.8,10Copolymers composed of this triad and alkylthiophenes also have excellent hole mobility when used in field-effect transistors9 and good performance in photovoltaic cells as a result of the polymers high ionization potential.7
Historically the synthesis of polybenzobisazoles requires strong acids or oxidants and high temperatures, limiting the types of substituents that can be incorporated into the polymer.11,12 Thus, from a synthetic standpoint, a functional triad already containing the benzobisazole moiety is a useful building block as it enables the synthesis of conjugated polymers using transition metal catalyzed cross coupling reactions, instead of condensation polymerization. Unfortunately, the reported methods for the formation of this triad: the Stille coupling of 2,6-dibromo-benzo[1,2-d;4,5-d′]bisthiazole with 2-trimethylstannyl-4-hexylthiophene8 or the acid-catalyzed condensation of 3-octylthiophene-2-carboxylaldehyde with 2,5-diamino-1,4-benzenedithiol dihydrochloride both occur in low yields, minimizing their utility.7Previously, we reported an efficient method for the synthesis of 2,6-disubstituted benzobisazoles using Lewis acid catalyzed cyclization of substituted orthoesters and corresponding diaminodiols or dithiols.13 These new building blocks have been used for the synthesis of novel poly(arylenevinylene)-alt-benzobisazoles.6,14 Based on our earlier success, we set out to develop an efficient approach for the synthesis of new monomers that would enable the synthesis of the polymers containing benzobisazole-arene single bonds.
Results and discussion
Synthesis and physical characterization
The synthetic approach for the D-A-D benzobisazole monomers is shown in Scheme 1. 2,5-Dibromo-3-octylthiophene (1) was synthesized in two steps from 3-bromothiophene according to a literature procedure.15 The Grignard metathesis reaction of 1 affords the intermediate 5-bromomagnesio-2-bromo-3-octylthiophene as the major product.16 Using the method described by Tschitschibabin,17 this intermediate is directly converted to 2-bromo-3-octyl-5-(triethoxymethyl)thiophene 2 without isolation. The orthoester 2 is a stable compound that is readily purified by vacuum distillation. The reaction conditions used for the synthesis of 2 enable the incorporation of a bromine at the 2-position of the thiophene ring at the start of the reaction sequence, reducing the total number of synthetic steps. The Lewis acid catalyzed cyclization of the orthoester 2 with 4,6-diaminoresorcinol 3,112,5-diaminohydroquinone 418 or 2,5-diaminobenzene-1,4-dithiol 512 in THF yielded the corresponding monomers 2,6-bis(4-octylthiophen-2-yl)-benzo[1,2-d; 5,4-d′]bisoxazole 6, 2,6-bis(4-octylthiophen-2-yl)-benzo[1,2-d; 4,5-d′]bisoxazole 7, and 2,6-bis(4-octylthiophen-2-yl)-benzo[1,2-d; 4,5-d′]bisthiazole 8. All of these monomers were solids that were readily purified by recrystalization, with isolated yields ranging from 82 to 86%.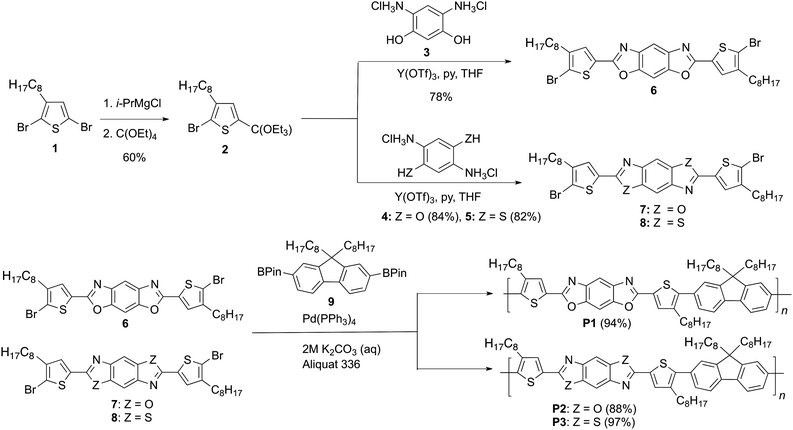 | ||
| Scheme 1 Synthesis of benzobisazole monomers and polymers. | ||
The synthesis of the fluorene-bisthienylbenzobisazole copolymers is also shown in Scheme 1. The polymerization of monomers 6, 7 or 8 with 2,7-bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-9,9-dioctylfluorene 919via the Suzuki cross-coupling reaction afforded the polymers P1, P2 and P3. All polymers were obtained in excellent yields after purification by Soxhlet extraction with methanol followed by hexanes, to remove residual catalyst and low molecular weight material. The higher molecular weight material was soluble in standard organic solvents, such as THF, m-cresol and chloroform at room temperature, facilitating characterization using 1H NMR spectroscopy and gel permeation chromatography (GPC). The 1H NMR spectra for all polymers were in agreement with the proposed polymer structures (Fig. S10–S15, ESI†). The number averaged degree of polymerization (DPn) ranged from 22–86 and all of the polymers showed excellent film forming ability. Thermogravimetric analysis revealed that both polymers were thermally stable with 5% weight loss onsets occurring above 290 °C under air (Fig. S1†). The results are summarized in Table 1.
| Yield (%) | Mwa | Mw/Mn | T d (°C) b | Tg/Tm (°C) | |
|---|---|---|---|---|---|
| a Molecular weights and polydispersity indexes determined by GPCversuspolystyrene standards using THF as the eluent. b Temperature at which 5% weight loss is observed by TGA under N2 with a heating rate of 15 °C/min. | |||||
| P1 | 94 | 80,900 | 3.2 | 295 | 110.3/230.1 |
| P2 | 88 | 74,200 | 2.5 | 292 | 108.1/NA |
| P3 | 97 | 21,500 | 1.8 | 341 | 108.4/257.6 |
Electrochemical properties
The electrochemical properties of the polymers were investigated by cyclic voltammetry (CV) and differential pulse voltammetry. The data were obtained from polymer thin films on a platinum working electrode in acetonitrile, using 0.1 M Bu4NPF6 as the electrolyte and an Ag/Ag+ reference electrode. The onsets were referenced to Fc/Fc+. All of the polymers exhibit measurable and reproducible oxidation and reduction processes. The HOMO levels were also determined using ultraviolet photoelectron spectroscopy (UPS). This technique determines the highest occupied molecular orbital (HOMO) level in organic thin films by bombarding the sample with ultraviolet photons and measuring the kinetic energies of the ejected valence electrons.20 Thus the HOMO values obtained using this method are very accurate. The results are summarized in Table 2. The values obtained for the HOMO levels from the CV waves are in good agreement with the values obtained using UPS. The electrochemical bandgaps are somewhat larger than the optical bandgaps, but are within reasonable limits.| Polymer | Solution | Film | |||||||
|---|---|---|---|---|---|---|---|---|---|
| λabsmax(nm) | λemmax (nm) | λabsmax (nm) | λemmax (nm) | E opt g (eV)a | E ox onset (V)b | E red onset (V) b | HOMO (eV) | LUMO (eV) | |
| a Estimated from the optical absorption edge. b Onset of potentials (vs Fc). c HOMO = −(Eoxonset + 4.8) (eV). d From UPS. e LUMO = −(Eredonset + 4.8) (eV). f LUMO = HOMO (UPS) + Eoptg (eV). | |||||||||
| P1 | 410 | 461 | 410 | 482 | 2.64 | 0.79 | −1.95 | −5.59c/−5.63d | −2.85e/−2.99f |
| P2 | 418 | 471 | 439 | 524 | 2.52 | 0.82 | −2.09 | −5.62c/−5.54d | −2.71e/−3.02f |
| P3 | 434 | 487 | 456 | 538 | 2.43 | 0.67 | −1.87 | −5.47c/−5.59d | −2.93e/−3.16f |
It was anticipated that structural differences of the benzobisazoles would result in different optical properties. The isomeric benzoxazole polymers P1 and P2 have different arrangements of oxygen atoms around the central benzene ring. This modification impacts the symmetry of the monomers, their dipole moments and ultimately impacts the optoelectronic properties of the resulting polymers. Similarly, the benzobisthiazole polymer P3, is structurally analogous to P2 with sulfur atoms replacing the oxygen atoms. Since, sulfur has a lower electronegativity than oxygen and a similar electronegativity to the carbon atom the electron density is more equally shared between sulfur and carbon. Thus the π-orbitals will be more delocalized. Additionally the empty d orbitals of the sulfur atom can contribute to the molecular π-orbitals decreasing the energy of the π–π* transition.5,21 Based on the UPS data it appears that changing the position of the oxygen atoms destabilizes the HOMO level, while slightly stabilizing the LUMO level. Whereas exchanging the oxygen atoms for sulfur stabilizes both the HOMO and LUMO levels.
Overall, the LUMO values of P1, P2, and P3 (−2.71 to −2.85 eV), are also higher than those reported previously for the related PFO3, a copolymer of 9,9-dioctylfluorene and 4,7-dithien-5-yl-2,1,3-benzothiadiazole (−3.53 eV),22 since benzobisazoles are weaker acceptors than benzothiadiazoles. The HOMO values for P1, P2, and P3 of −5.54 to −5.63 eV are more negative than those reported previously for the related poly(9,9-dioctyl-fluorenevinylene-co-benzobisoxazole)s,6 which had HOMO values of −5.29 to −5.36 eV. We attribute this to exchanging the vinylene linkage between the benzobisazole and fluorene moieties for a thiophene ring.
Optical and electronic properties
The photophysical characteristics of the polymers, both as dilute solutions in chloroform and thin films, were evaluated by UV-vis absorption and fluorescence spectroscopy. The normalized absorbance and emission spectra for the polymers as solutions and as thin films are shown in Fig. 1; the data are summarized in Table 2. In solution, the spectra for all the polymers exhibit a single broad absorption band. P2 has a λmax at approximately 418 nm; relative to P2 the λmax for P1 was blue-shifted 8 nm to 410 nm and the λmax of P3 was red-shifted 16 nm to 434 nm. The thin film absorption spectra for all of the polymers were broader than their solution counterparts, and show some weak vibronic coupling. The onsets of absorption for the polymers were obtained from the UV-vis spectra of polymer films and ranged from 471 to 511 nm, resulting in optical band gaps of 2.43–2.64 eV. None of the UV-vis spectra exhibited a second low energy peak typically associated with intramolecular charge-transfer transitions within D-A copolymers.23In solution the fluorescence spectra of the polymers is characterized by one strong emisson peak with a shoulder. The emission spectra of the polymer thin films were considerably red-shifted, relative to the corresponding solution spectra. This phenomenon is most likely caused by geometrical changes between the polymer in solution and in film.
In solution the λabsmax of the benzobisoxazole polymer P1 is blue-shifted relative to the related poly(fluorene-co-benzobisoxazole)s (458 nm), althought the later was measured as a solution in methane sulfonic acid, which protonates the polymer.24 Whereas the λemmax of 468 nm is the same as the reported value of 468 nm. In contrast, the λabsmax of the benzobisthiazole polymer P3 is red-shifted relative to the related poly(fluorene-co-benzobisthiazole) (409 nm).25 The emission spectra for this polymer was not reported. The λabsmax of the benzobisoxazole polymers P1 and P2 are blue-shifted relative to those reported previously for the related poly(fluorenevinylene-co-benzobisoxazole)s (450–470 nm),6 whereas the λemmax of 461–471 nm are similar to the reported values. Indicating that the thiophene π-bridges have little impact on the emission spectra.
Solid-state morphology
The thin films were analyzed by X-ray diffraction. The diffraction patterns for the polymers are shown in (Fig. S3, ESI†) The pattern for P1 does not exhibit any peaks, indicating a lack of regular spacing in the film. X-ray analyses of small molecules containing cis-benzobisoxazole indicates that it has a slight bend along the 2,6 axis.13 The reason for a lack of order in P1 may be due to this bent structure. P3, however, and P2, albeit to a lesser extent, have peaks corresponding to a regular spacing of 15.5 Å. This number is slightly larger than the length of one octyl chain. This suggests a spatial arrangement where octyl chains on neighboring polymers intercalate to some degree. This type of spacing can be seen in other 9,9-dioctylfluorene copolymers.26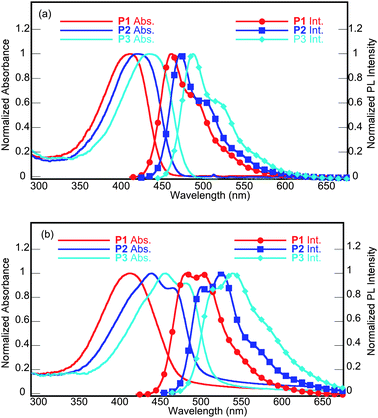 | ||
| Fig. 1 UV-vis absorption and PL spectra of the polymers in: (a) solution (top) and (b) thin film (bottom). | ||
Electroluminescent devices
We first examined the use of the polymers as neat emitting layers in polymer light emitting diodes (PLEDs) with the structure ITO/PEDOT:PSS (60 nm)/P1–P3/BPhen (40 nm)/LiF(1 nm)/Al(100 nm), where ITO is indium tin oxide, PEDOT:PSS is poly(3,4-ethylenedioxy thiophene):poly(4-styrenesulfonate), and BPhen is 4,7-diphenyl-1,10-phenanthroline. All of the polymers are efficient fluorophores in solution, with quantum yields of 0.80, 0.82 and 0.85 for P1, P2, and P3, respectively.Unfortunately, this fluorescence was quenched in the solid state. Thus no electroluminescence (EL) was observed from devices with these polymers as neat emitting films. However, since all these polymers have high fluorescence quantum yield in solution and good solubility in organic solvents, they were suitable candidates for use in guest-host PLEDs.27 We therefore evaluated them as low level dopants in poly(N-vinyl carbazole) (PVK)-based PLEDs using low molecular weight PVK as the host material (average molecular weight: ∼50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000∼100000 g mol−1). The electroluminescent properties of the PLEDs, are summarized in Table 3. The structure of the PLEDs was ITO/PEDOT:PSS(60 nm)/PVK:P1–P3/BPhen(40 nm)/LiF(1 nm)/Al(100 nm) (Fig.2). In these devices, the benzobisazole terpolymers were blended with PVK at different weight ratios. When the concentration of the conjugated polymers in the blend was increased, the overall performance of the devices decreased. This is especially prominent for polymer P1. The highest brightness of the 2 wt. % P1:PVK device was only ∼ 60 Cd/m2 and the 4 wt. % P1:PVK device did not light up. Among all the polymers, P3 showed the best performance with 1860 Cd/m2 at 2 wt.%. The normalized EL spectra of the PLEDs are shown in Fig. 3. As seen, the intensity of the PVK emission, which peaks at ∼420 nm, decreased relative to that of the benzobisazole terpolymers emission when the concentration of the latter increased. This is particularly notable for the 4 wt % P3:PVK PLEDs, where the PVK emission totally vanishes and the P3 emission becomes the only emission. This behavior indicates energy transfer from PVK to the benzobisazole terpolymers or direct carrier injection to the benzobisazole terpolymers. This reduced performance of the polymer is suspected to result from quenching due to aggregation. As a result the fluorescence quantum yield is diminished.28
000∼100000 g mol−1). The electroluminescent properties of the PLEDs, are summarized in Table 3. The structure of the PLEDs was ITO/PEDOT:PSS(60 nm)/PVK:P1–P3/BPhen(40 nm)/LiF(1 nm)/Al(100 nm) (Fig.2). In these devices, the benzobisazole terpolymers were blended with PVK at different weight ratios. When the concentration of the conjugated polymers in the blend was increased, the overall performance of the devices decreased. This is especially prominent for polymer P1. The highest brightness of the 2 wt. % P1:PVK device was only ∼ 60 Cd/m2 and the 4 wt. % P1:PVK device did not light up. Among all the polymers, P3 showed the best performance with 1860 Cd/m2 at 2 wt.%. The normalized EL spectra of the PLEDs are shown in Fig. 3. As seen, the intensity of the PVK emission, which peaks at ∼420 nm, decreased relative to that of the benzobisazole terpolymers emission when the concentration of the latter increased. This is particularly notable for the 4 wt % P3:PVK PLEDs, where the PVK emission totally vanishes and the P3 emission becomes the only emission. This behavior indicates energy transfer from PVK to the benzobisazole terpolymers or direct carrier injection to the benzobisazole terpolymers. This reduced performance of the polymer is suspected to result from quenching due to aggregation. As a result the fluorescence quantum yield is diminished.28
| Dopant | Devicea wt. % | Vonb [V] | Drive Voltage [V] | Current Density [J, mA cm−2] | Brightness [Cd/m2] | Efficiency [Cd/A, (%EQE)c] | λmaxEL[nm] polymer, PVK | rmax | CIE 1931 [x,y] |
|---|---|---|---|---|---|---|---|---|---|
| a Device type structure: ITO/PEDOT:PSS/PVK: poly(arylenebenzobisazole)s/BPhen/LiF/Al. b Turn-on voltage (at which EL is 1 Cd/m2). c EQE = external quantum efficiency. | |||||||||
| PVK | 0.0 | 4.4 | 9.2 | 303 | 296 | 0.44, 0.76 | 422 | 0 | (0.17, 0.07) |
| P1 | 1.0 | 6.0 | 10.0 | 205 | 363 | 0.52, 0.37 | 466, 422 | 4.56 | (0.16, 0.19) |
| 2.0 | 5.4 | 8.2 | 53 | 68 | 0.14, 0.09 | 469 | (0.17, 0.20) | ||
| P2 | 1.0 | 4.0 | 8.6 | 313 | 377 | 0.35, 0.37 | 461, 421 | 0.74 | (0.17, 0.12) |
| 2.0 | 4.6 | 8.8 | 454 | 550 | 0.29, 0.26 | 478, 418 | 0.81 | (0.18, 0.17) | |
| 4.0 | 4.6 | 8.4 | 271 | 362 | 0.14, 0.10 | 483, 421 | 1.46 | (0.20, 0.25) | |
| P3 | 1.0 | 3.8 | 8.2 | 220 | 788 | 0.76, 0.31 | 504, 419 | 2.57 | (0.23, 0.40) |
| 2.0 | 4.3 | 8.8 | 458 | 1862 | 0.86, 0.35 | 504, 418 | 2.24 | (0.23, 0.41) | |
| 4.0 | 3.7 | 8.8 | 864 | 1077 | 0.17, 0.06 | 511 | (0.31, 0.58) |
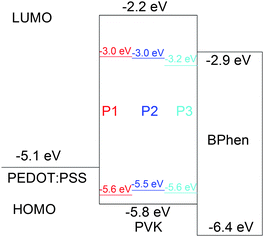 | ||
| Fig. 2 Energy level diagram for the devices. | ||
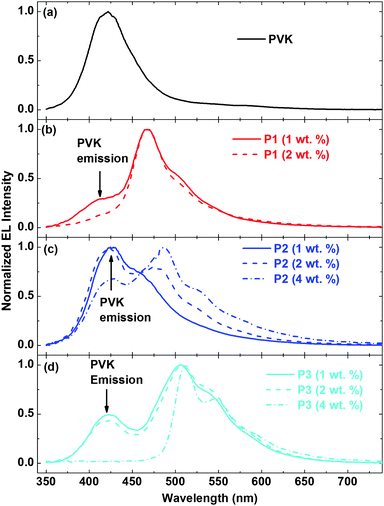 | ||
| Fig. 3 Normalized EL spectra of the OLEDs driven at 57 mA cm−2. | ||
Conclusions
In conclusion, three new benzobisazole terpolymers containing thiophene and fluorene have been synthesized in two steps and in high yields via the Suzuki coupling reaction of fluorene with three new thiophene-benzobisazole-thiophene monomers. The flexible side chains on the thiophene and fluorene ring resulted in polymer which possessed good solubility, while maintaining good thermal stability. Preliminary electroluminescence studies showed that these polymers exhibit moderate brightness in guest-host PLEDs. Although these polymers have comparable electron affinity to previously synthesized benzobisazole/fluorene copolymers, the incorporation of the alkyl thiophene rings into the polymer backbone proved to be detrimental to the polymers' performance in PLEDs. However, the facile synthetic approach described herein will enable the development of new benzobisazole terpolymers with improved properties. The synthesis of such materials is currently ongoing in our laboratories.Experimental section
The starting materials 2,5-dibromo-3-octylthiophene 1,154,6-diamino resorcinol 3,11diaminohydroquinone 4,18diaminobenzene dithiol 5,12 and 2,7-bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-9,9-dioctylfluorene 919 were all synthesized according to previously reported methods. Tetrahydrofuran was dried using an Innovative Technologies solvent purification system. All other compounds were purchased from commercial sources and used without further purification. Nuclear magnetic resonance spectra were obtained on a 400 MHz spectrometer. All samples were referenced internally to the residual protonated solvent. Gel permeation chromatography (GPC) measurements were performed on a GPC separation module equipped with four columns connected in a series (guard, 10000 Å, 1000 Å and 100 Å from American Polymer Service Corporation), a refractive index detector and a UV-Vis detector. Analyses were performed at 35 °C using chloroform as the eluent with the flow rate at 1.0 mL/min. Calibration was based on polystyrene standards. Fluorescence spectroscopy and UV-Visible spectroscopy were performed using polymer solutions in chloroform, and thin films were spun from these solutions. All of the polymers were excited at their respective emission maxima. Quantum yield measurements were taken using Rhodamine B (ϕ = 0.65) in 1-hexanol as a standard (excitation at 410 nm; emission was taken from 420–700 nm).29 All values were corrected for the differences in the refractive index of the solvent. Thermal gravimetric analysis measurements were performed within the temperature interval of 30 °C–650 °C, using a heating rate of 20 °C min−1 under ambient atmosphere. Differential scanning calorimetry was performed with a first scan at a heating rate of 15 °C min−1 to erase thermal history and a second scan to measure transitions from 0 °C to 275 °C under nitrogen. Transitions were also measured with cooling at 15 °C/min. Cyclic voltammograms were performed in 0.1 M tetrabutylammonium hexafluorophosphate using 0.01 M AgNO3 in acetonitrile as the reference electrode. The potential values obtained versus the Ag+ were converted to the ferrocene (Fc) reference. Ultraviolet Photoelectron Spectroscopy measurements were performed using a RKI Instruments Model AC-2 instrument. The measurements were performed on polymer films.
2-Bromo-3-dodecyl-5-(triethoxymethyl)thiophene (S1)
A dry, round bottom flask was equipped with a reflux condenser and addition funnel then placed under argon. The flask was charged with 60 mL of dry diethyl ether and 2,5-dibromo-3-octylthiophene (21.27 g, 60.06 mmol). Isopropyl magnesium chloride (30.1 mL, 60.2 mmol, 2.0 M) was then added dropwise to the flask via the addition funnel and the mixture was brought to reflux. After an hour, metathesis was completed (monitored by GC-MS), and the reaction was cooled to room temperature. Once cool, tetraethyl orthocarbonate (13.86 g, 72.07 mmol) was added dropwise. The mixture was then heated to reflux and stirred overnight. The crude mixture was poured into 200 mL of saturated aqueous ammonium chloride and extracted with 3 × 150 mL diethyl ether. The combined organic layers were washed with brine and dried over sodium sulfate. The solution was filtered and the filtrate concentrated in vacuo. The crude product was purified by Kugel-rohr distillation to yield 5-bromo-2-triethoxymethyl-3-octylthiophene (17.80 g, 42.24 mmol, 70% yield). 1H-NMR (400 MHz, CDCl3): δ 6.85 (s, 1H), 3.43 (q, 6H), 2.50 (t, 2H) 1.57 (m, 2H), 1.57 (m, 10H), 1.18 (t, 9H), 0.86 (t, 3H). 13C-NMR (100 MHz, CDCl3): δ 141.7, 141.3, 127.6, 112.8, 109.8, 58.2, 32.0, 29.7, 29.5, 29.4, 29.3, 22.9, 15.0, 14.3. GC-MS (QP, M+ for C19H33BrO3S): calcd, 420.13; found, 420.35.2,6-Bis(2-bromo-3-octyl-thiophene-5-yl) benzobis[1,2-d:5,4-d′]bisoxazole 6
4,6-Diamino-1,3-dihydroxybenzene bishydrochloride 3 (0.91 g, 4.3 mmol) was placed under argon in a dry round bottom flask. To it was added 4 mL anhydrous DMSO and pyridine (0.67 g, 8.3 mmol); the mixture was stirred until the solids had all dissolved. Meanwhile, a solution of Yttrium(III) triflate (0.11g, 0.21 mmol) in 4 mL dry THF was prepared and heated to 60 °C. The DMSO solution was then transferred dropwise into the THF solution. The reaction was stirred at 60 °C overnight. After stirring, the mixture was cooled and an excess amount of methanol was added to precipitate any dissolved product. The solids were filtered and recrystallized from chloroform and methanol to yield 6 (89%). Mp. 105–106 °C. 1H-NMR (400 MHz, CDCl3): δ 7.80 (s, 2H), 7.62 (s, 2H), 2.62 (t, 4H) 1.65 (t, 4H), 1.28 (m, 20H), 0.88 (t, 3H). 13C-NMR (100 MHz, CDCl3): δ 159.5, 148.4, 144.1, 140.4, 130.9, 128.8, 115.6, 100.8, 32.1, 29.8, 29.7, 29.6, 29.5, 29.4, 22.9, 14.3. HRMS (ESI, [M + H]+ for C32H38Br2N2O2S2): calcd, 705.815; found, 705.814.2,6-Bis(2-bromo-3-dodecyl-thiophene-5-yl)benzo[1,2-d;4,5-d′]bisoxazole 7
2,5-Diamino-1,4-hydroquinone bishydrochloride 4 (0.91 g, 4.3 mmol) was placed under Argon in a dry round bottom flask. To it was added 4 mL anhydrous DMSO and pyridine (0.67 g, 8.3 mmol) and the mixture was stirred until the solids had all dissolved. Meanwhile, a solution of Lanthanum(III) triflate (0.12g, 0.21 mmol) in 4 mL dry THF was prepared and heated to 60 °C. The DMSO solution was then transferred dropwise into the THF solution. The reaction was stirred at 60 °C overnight. After stirring, the mixture was cooled and an excess amount of methanol was added to precipitate any dissolved product. The solids were filtered and recrystallized from chloroform and methanol to yield 7 (78%). Mp. 148–150 °C. 1H-NMR (400 MHz, CDCl3): δ 7.94 (s, 1H), 7.61 (s, 1H), 2.60 (t, 4H) 1.62 (t, 4H), 1.28 (m, 20H), 0.88 (t, 3H). 13C-NMR (100 MHz, CDCl3): δ 159.0, 148.5, 144.0, 130.8, 128.7, 115.4, 109.6, 93.3, 32.1, 29.8, 29.7, 29.6, 29.5, 29.4, 22.9, 14.4. HRMS (ESI, [M + H]+ for C32H38Br2N2O2S2): calcd, 705.815; found, 705.814.2,6-Bis(2-bromo-3-dodecyl-thiophene-5-yl)benzo[1,2-d;4,5-d′]bisthiazole 8
2,5-Diamino-1,4-benzenedithiol bishydrochloride 5 (0.91 g, 4.3 mmol) was placed under Argon in a dry round bottom flask. To it was added 4 mL anhydrous dimethyl acetamide and pyridine (0.67 g, 8.3 mmol) and the mixture was stirred until the solids had all dissolved. Meanwhile, a solution of Ytterbiumm(III) triflate (0.13g, 0.21 mmol) in 4 mL dry THF was prepared and heated to 60 °C. The DMSO solution was then transferred dropwise into the THF solution. The reaction was stirred at 60 °C overnight. After stirring, the mixture was cooled and an excess amount of methanol was added to precipitate any dissolved product. The solids were filtered and recrystallized from chloroform and methanol to yield 8 (84%). Mp. 223–224 °C. 1H-NMR (400 MHz, CDCl3): δ 8.36 (s, 2H), 7.34 (s, 2H), 2.60 (t, 4H) 1.63 (t, 4H), 1.30 (m, 20H), 0.90 (t, 3H). 13C-NMR (100 MHz, CDCl3): δ 161.5, 152.0, 143.8, 136.8, 134.4, 129.7, 115.2, 114.9, 32.1, 29.9, 29.8, 29.6, 29.5, 29.4, 22.9, 14.3. HRMS (ESI, [M + H]+ for C32H38Br2N2S4): calcd, 737.0358; found, 737.0357.General methods for polymer synthesis
A solution of 70 mg Aliquat 336, one of the monomers [6 (353.3 mg, 0.500 mmol), 7 (353.3 mg, 0.500 mmol), or 8 (369.4 mg, 0.500 mmol)] and 2,7-bis(pinacolatoboro)-9,9-dioctylfluorene (321.3 mg, 0.500 mmol) in 10 mL of toluene was prepared. To this solution was added 10 mL of 2M aqueous sodium carbonate. The biphasic system was degassed with Argon for 30 mins before adding Pd(PPh3)4 (11.6 mg, 0.010 mmol, dissolved in 5 mL degassed toluene). The mixture was heated to reflux and stirred for 4 days. After 4 days, the organic layer was separated and precipitated in MeOH. The polymers were then purified by soxhlet extraction first rinsing with MeOH and hexanes before extraction by chloroform. The chloroform was evaporated to yield polymer.P1 (439 mg, 94% yield). 1H-NMR (400 MHz, CDCl3): δ 8.04 (s, 1H), 7.8 (m, 5H), 7.50 (m, 5H), 2.78 (b, 4H), 2.11 (b, 4H), 1.73 (b, 4H), 1.28 − 1.11 (bm, 40H), 0.88 − 0.80 (bm, 16H) 13C-NMR (100 MHz, CDCl3): δ160.0, 151.8, 148.7, 144.4, 140.8, 140.4, 140.3, 132.7, 128.5, 126.7, 123.9, 120.5, 109.3, 93.2, 55.6, 40.7, 32.1, 32.0, 31.1, 30.4, 29.8, 29.7, 29.6, 29.5, 29.4, 29.3, 24.3, 22.9, 22.8, 14.4, 14.3
P2 (410 mg, 88% yield). 1H-NMR (400 MHz, CDCl3): δ 7.86 (m, 5H), 7.52 (m, 5H), 2.79 (b, 4H), 2.11 (b, 4H), 1.73 (b, 4H), 1.30 − 1.12 (bm, 40H), 0.89 − 0.83 (bm, 16H) 13C-NMR (100 MHz, CDCl3): δ160.5, 151.9, 148.7, 144.7, 140.9, 140.7, 140.5, 133.0, 132.9, 128.5, 126.9, 123.9, 120.4, 100.6, 55.7, 40.7, 32.1, 32.0, 31.1, 30.4, 29.8, 29.7, 29.6, 29.5, 29.4, 29.3, 24.3, 22.9, 22.8, 14.3, 14.2
P3 (469 mg, 97% yield). 1H-NMR (400 MHz, CDCl3): δ 8.46 (s, 2H), 7.80 (d, 2H), 7.61 − 7.52 (m, 6H), 2.78 (b, 4H), 2.05 (b, 4H), 1.73 (b, 4H), 1.30 − 1.12 (bm, 40H), 0.90 − 0.82 (bm, 16H) 13C-NMR (100 MHz, CDCl3): δ162.3, 152.2, 151.9, 143.8, 140.9, 140.4, 134.9, 134.7, 133.2, 131.8, 128.4, 124.0, 120.4, 115.0, 55.7, 40.7, 32.1, 32.0, 31.1, 30.4, 29.8, 29.7, 29.6, 29.5, 29.4, 29.3, 24.3, 22.9, 22.8, 14.3, 14.2
Fabrication and characterization of PLEDs
PLEDs were fabricated on nominally 20 Ω/square, 140 nm-thick ITO-coated glass substrates (Colorado Concept Coatings). The substrates were first cleaned with a detergent and organic solvents; they were then treated in a UV/ozone oven to increase the work function of the ITO and hence facilitate hole injection, as described elsewhere.30 A 60 nm PEDOT:PSS layer was spin-coated on the ITO and then baked in air at 160 °C for 1 h. Blends of PVK and the poly(arylenebenzobisoxazole) copolymers in chlorobenzene solutions were spin-coated on top of the PEDOT:PSS layer in an Ar-filled glovebox; the thickness of the poly(arylenebenzobisoxazole) copolymers layers was 30 nm. The combined concentration of the PVK and polymer dopants was kept constant at 9 mg mL−1; the poly(arylenebenzobisazole)s concentration was varied in the range 0.09 to 0.36 mg/mL. The solution was spin coated at 4000 rpm for 60 s. The fabricated structure was then annealed at 60 °C for 30 min. Following this annealing step, the samples were transferred into a thermal evaporator within the glovebox and the BPhen, LiF, and Al layers were deposited sequentially by thermal evaporation at a base pressure of ∼2 × 10−6 Torr. The PLEDs were characterized by monitoring their electroluminescence (EL) spectra, brightness as a function of the applied voltage, and luminous efficiency.Acknowledgements
We thank Mr. Robert Roggers (ISU) for X-ray analysis. We thank Ms. Dana Drochner and Mr. Timothy Mauldin for thermal analysis. We thank Ms. Atta Gueye, Dr Elena Sheina and Dr Christopher Brown of Plextronics for providing UPS measurements. This work was funded by the National Science Foundation (DMR-0846607), and the 3M foundation. MC and TX were supported by the US Department of Energy, Basic Energy Sciences, Materials Sciences and Engineering Division, under Contract No. DE-AC 02-07CH11358.Notes and references
- X. Zhan and D. Zhu, Polym. Chem., 2010, 1, 409–419 RSC; T. A. Skotheim and J. R. Reynolds, Handbook of conducting polymers, 3rd ed/edited by Terje A. Skotheimand John Reynolds. edn, London, Boca Raton, Fla., 2007 Search PubMed; A. C. Grimsdale, K. Leok Chan, R. E. Martin, P. G. Jokisz and A. B. Holmes, Chem. Rev., 2009, 109, 897–1091 Search PubMed; B. C. Thompson and J. M. J. Frechet, Angew. Chem., Int. Ed., 2008, 47, 58–77 CrossRef CAS; A. Facchetti, Chem. Mater., 2010, 23, 733–758 CrossRef.
- A. P. Kulkarni, X. Kong and S. A. Jenekhe, Macromolecules, 2006, 39, 8699–8711 CrossRef CAS; A. P. Kulkarni, Y. Zhu and S. A. Jenekhe, Macromolecules, 2005, 38, 1553–1563 CrossRef CAS; M. Leclerc, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 2867–2873 CrossRef CAS.
- M. Al-Hashimi, J. G. Labram, S. Watkins, M. Motevalli, T. D. Anthopoulos and M. Heeney, Org. Lett., 2010, 12, 5478–5481 CrossRef CAS; X. Zhang, T. T. Steckler, R. R. Dasari, S. Ohira, W. J. Potscavage Jr, S. P. Tiwari, S. Coppee, S. Ellinger, S. Barlow, J.-L. Bredas, B. Kippelen, J. R. Reynolds and S. R. Marder, J. Mater. Chem., 2010, 20, 123–134 RSC; L. Zhang, C. He, J. Chen, P. Yuan, L. Huang, C. Zhang, W. Cai, Z. Liu and Y. Cao, Macromolecules, 2010, 43, 9771–9778 CrossRef CAS; Z. Li, J. Ding, N. Song, J. Lu and Y. Tao, J. Am. Chem. Soc., 2010, 132, 13160–13161 CrossRef CAS; M. Zhang, H. Fan, X. Guo, Y. He, Z.-G. Zhang, J. Min, J. Zhang, G. Zhao, X. Zhan and Y. Li, Macromolecules, 2010, 43, 8714–8717 CrossRef CAS; X. Guo, H. Xin, F. S. Kim, A. D. T. Liyanage, S. A. Jenekhe and M. D. Watson, Macromolecules, 2010, 44, 269–277; P. Herguth, X. Jiang, M. S. Liu and A. K. Y. Jen, Macromolecules, 2002, 35, 6094–6100 CrossRef CAS.
- M. M. Alam and S. A. Jenekhe, Chem. Mater., 2002, 14, 4775–4780 CrossRef CAS; S. A. Jenekhe, J. A. Osaheni, J. S. Meth and H. Vanherzeele, Chem. Mater., 1992, 4, 683–687 CrossRef CAS.
- B. A. Reinhardt, M. R. Unroe and R. C. Evers, Chem. Mater., 1991, 3, 864–871 CrossRef CAS.
- J. J. Intemann, J. F. Mike, M. Cai, S. Bose, T. Xiao, T. C. Mauldin, R. A. Roggers, J. Shinar, R. Shinar and M. Jeffries-El, Macromolecules, 2011, 44, 248–255 CrossRef CAS.
- E. Ahmed, F. S. Kim, H. Xin and S. A. Jenekhe, Macromolecules, 2009, 42, 8615–8618 CrossRef CAS.
- H. Pang, F. Vilela, P. J. Skabara, J. J. W. McDouall, D. J. Crouch, T. D. Anthopoulos, D. D. C. Bradley, D. M. de Leeuw, P. N. Horton and M. B. Hursthouse, Adv. Mater., 2007, 19, 4438–4442 CrossRef CAS.
- I. Osaka, K. Takimiya and R. D. McCullough, Adv. Mater., 2010, 22, 4993–4997 CrossRef CAS.
- M. Mamada, J.-i. Nishida, S. Tokito and Y. Yamashita, Chem. Lett., 2008, 37, 766–767 CrossRef CAS; G. J. McEntee, F. Vilela, P. J. Skabara, T. D. Anthopoulos, J. G. Labram, S. Tierney, R. W. Harrington and W. Clegg, J. Mater. Chem., 2011, 21, 2091–2097 RSC.
- J. F. Wolfe and F. E. Arnold, Macromolecules, 1981, 14, 909–915 CrossRef CAS.
- J. F. Wolfe, B. H. Loo and F. E. Arnold, Macromolecules, 1981, 14, 915–920 CrossRef CAS.
- J. F. Mike, J. J. Inteman, A. Ellern and M. Jeffries-El, J. Org. Chem., 2010, 75, 495–497 CrossRef CAS; J. F. Mike, A. J. Makowski and M. Jeffries-EL, Org. Lett., 2008, 10, 4915–4918 CrossRef CAS.
- J. F. Mike, A. J. Makowski, T. C. Mauldin and M. Jeffries-El, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1456–1460 CrossRef CAS; J. F. Mike, K. Nalwa, A. J. Makowski, D. Putnam, A. L. Tomlinson, S. Chaudhary and M. Jeffries-El, Phys. Chem. Chem. Phys., 2011, 13, 1338–1344 RSC; A. V. Patil, H. Park, E. W. Lee and S.-H. Lee, Synth. Met., 2010, 160, 2128–2134 CrossRef CAS.
- T. A. Chen, X. Wu and R. D. Rieke, J. Am. Chem. Soc., 1995, 117, 233 CrossRef CAS.
- R. S. Loewe, P. C. Ewbank, J. Liu, L. Zhai and R. D. McCullough, Macromolecules, 2001, 34, 4324–4333 CrossRef CAS; R. S. Loewe, S. M. Khersonsky and R. D. McCullough, Adv. Mater., 1999, 11, 250–258 CrossRef CAS.
- D. Bhaumik, H. H. Jaffe and J. E. Mark, Macromolecules, 1981, 14, 1125–1126 CrossRef CAS.
- M. Inbasekaran and R. Strom, OPPI Briefs, 1994, 23, 447–450 Search PubMed.
- A. P. Zoombelt, S. G. J. Mathijssen, M. G. R. Turbiez, M. M. Wienk and R. A. J. Janssen, J. Mater. Chem., 2010, 20, 2240–2246 RSC.
- T. Miyamae, D. Yoshimura, H. Ishii, Y. Ouchi, T. Miyazaki, T. Koike, T. Yamamoto, Y. Muramatsu, H. Etori, T. Maruyama and K. Seki, Synth. Met., 1997, 84, 939–940 CrossRef CAS.
- M. Zhao, M. Samoc, P. N. Prasad, B. A. Reinhardt, M. R. Unroe, M. Prazak, R. C. Evers, J. J. Kane, C. Jariwala and M. Sinsky, Chem. Mater., 1990, 2, 670–678 CrossRef CAS.
- S. Admassie, O. Ingaas, W. Mammo, E. Perzon and M. R. Andersson, Synth. Met., 2006, 156, 614–623 CrossRef CAS.
- E. Zhou, M. Nakamura, T. Nishizawa, Y. Zhang, Q. Wei, K. Tajima, C. Yang and K. Hashimoto, Macromolecules, 2008, 41, 8302–8305 CrossRef CAS; E. Zhou, K. Tajima, C. Yang and K. Hashimoto, J. Mater. Chem., 2010, 20, 2362–2368 RSC; P. M. Beaujuge, C. M. Amb and J. R. Reynolds, Acc. Chem. Res., 2010, 43, 1396–1407 CrossRef CAS.
- X. Liu, X. Xu, Q. Zhuang and Z. Han, Polym. Bull., 2008, 60, 765–774 CrossRef CAS.
- K. D. Belfield, S. Yao, A. R. Morales, J. M. Hales, D. J. Hagan, E. W. Van Stryland, V. M. Chapela and J. Percino, Polym. Adv. Technol., 2005, 16, 150–155 CrossRef CAS.
- Y. M. Kim, E. Lim, I.-N. Kang, B.-J. Jung, J. Lee, B. W. Koo, L.-M. Do and H.-K. Shim, Macromolecules, 2006, 39, 4081–4085 CrossRef CAS; E. Lim, B.-J. Jung, J. Lee, H.-K. Shim, J.-I. Lee, Y. S. Yang and L.-M. Do, Macromolecules, 2005, 38, 4531–4535 CrossRef CAS; M. Grell, D. D. C. Bradley, G. Ungar, J. Hill and K. S. Whitehead, Macromolecules, 1999, 32, 5810–5817 CrossRef CAS; S. Kawana, M. Durrell, J. Lu, J. E. Macdonald, M. Grell, D. D. C. Bradley, P. C. Jukes, R. A. L. Jones and S. L. Bennett, Polymer, 2002, 43, 1907–1913 CrossRef CAS.
- I. Gontia, S. V. Frolov, M. Liess, E. Ehrenfreund, Z. V. Vardeny, K. Tada, H. Kajii, R. Hidayat, A. Fujii, K. Yoshino, M. Teraguchi and T. Masuda, Phys. Rev. Lett., 1999, 82, 4058 CrossRef CAS; R. Hidayat, S. Tatsuhara, D. W. Kim, M. Ozaki, K. Yoshino, M. Teraguchi and T. Masuda, Phys. Rev. B: Condens. Matter, 2000, 61, 10167 CrossRef CAS.
- B. Liu, W.-L. Yu, Y.-H. Lai and W. Huang, Macromolecules, 2000, 33, 8945–8952 CrossRef CAS; M. R. Andersson, O. Thomas, W. Mammo, M. Svensson, M. Theander and O. Inganas, J. Mater. Chem., 1999, 9, 1933–1940 RSC.
- R. F. Kubin and A. N. Fletcher, J. Lumin., 1982, 27, 455–462 CrossRef.
- Z. Zhou, R. Shinar, A. J. Allison and J. Shinar, Adv. Funct. Mater., 2007, 17, 3530–3537 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available: NMR spectra for all new compounds, TGA curves, DSC traces, cyclic voltammographs and X-ray difffractographs. See DOI: 10.1039/c1py00218j/ |
| This journal is © The Royal Society of Chemistry 2011 |