Synthesis of a controlled three-faced PAMAM particle†
Mathieu
Arseneault
,
Philippe
Dufour
,
Isabelle
Levesque
and
Jean-François
Morin
*
Département de Chimie and Centre de Recherche sur les Matériaux Avancés (CERMA), Pavillon A.-Vachon, Université Laval, 1045 Ave de la Médecine, Québec, Canada G1V 0A6. E-mail: jean-francois.morin@chm.ulaval.ca; Fax: +1 418-656-7916; Tel: +1 418-656-2812
First published on 14th July 2011
Abstract
We report the synthetic proof-of-concept for a three-faced particle based on a PAMAM dendrimer in which each face bears a different labile protecting group. This new strategy yields a controlled, well-defined and monodisperse dendrimer. To achieve this, a tri-orthogonal strategy was employed through Sonogashira coupling and Cu(I)-catalyzed Huisgen [2 + 3] dipolar cycloaddition (“click” chemistry). The afforded dendrimer is well suited for further development in a variety of fields as each branch can be addressed individually for functionalization.
Introduction
The field of nanomedicine aims to address some of the shortcomings of traditional drugs1 such as bioavailability, the need for synergy between co-drugs and, most importantly, specificity. Often enough, potent drugs are discovered only to be discarded due to severe side effects generated by a lack thereof. The same needs and shortcomings can more or less be applied to the area of in vitro molecular diagnostic tools.2 The ideal molecular architecture for these applications should feature synthetic flexibility to be easily adapted to various needs and a very well defined structure that can bear several different moieties. A considerable wealth of effort has been focused towards creating such scaffolds and among them, polyamidoamide3,4 (PAMAM) dendrimers have been widely studied and used in this regard. This family of dendrimer presents several advantages,1,5–7 mainly biological compatibility due to its molecular subunits being very similar to that of proteins (i.e.amides), its globular shape that helps modulate its behavior in vivo as well as the versatility of the amino terminal groups for further functionalization. Many groups have tackled the problems related to the synthesis of these compounds. Their work can be roughly divided into two approaches, the first one being a statistical path where a high generation PAMAM is linked with a given quantity of each moiety until all available amino groups have been functionalized. While seminal, these previous efforts lacked the high control and synthetic flexibility required to further advance in this field. The second approach is the convergent path where each dendron bearing one type of terminal group is attached to an orthogonal core one after the other. Most attempts yielded a so-called “Janus” particle with two branches.8–11 Recently, work has been published regarding three-faced dendrimers circumventing the orthogonality problems mentioned above.12,13 Simply put, the idea is to build a two-faced “Janus” dendrimer and selectively add another bifunctional dendron onto only one of the initial faces of the dendrimer. While clever, this strategy requires one to build a new dendrimer or dendron for each new terminal moiety. None of these were built using the traditional PAMAM architecture. Thus, the creation of a controlled and monodisperse three branched PAMAM particle serving as a more universal template is the next logical step in this line of work.The main difficulty in building a dendrimer bearing several branches that can be addressed separately lies in the exploration of the available orthogonality, either at the core or at the periphery. Herein we report what we believe to be the first synthesis of a non-statistical three-faced PAMAM particle. The key elements of our synthetic pathway are: (1) the use of three carefully chosen protected alkynes, followed by equally careful sequential deprotection and (2) the use of protected amino-terminated PAMAM dendrons, enabling further decoration with functional groups bearing an electrophilic reactive site. This synthetic proof-of-concept represents a stepping-stone toward such highly defined compound.
Experimental part
Materials
All solvents (ACS grade) were distilled and put through a Vac Atmosphere (CA, USA) solvent purification system. All the reagents were purchased from either Sigma Aldrich Co., TCI America or Oakwood Products and used as received. All equivalents are molar. 3,5-Diiodobromobenzene was synthesized according to previous literature.14GPC analysis was performed using a Jordi DBV 500A in MeOH. 1H and 13C NMR spectra were recorded on a Varian AS400 apparatus in appropriate deuterated solvent solution at 298 K. Chemical shifts were reported as values (ppm) relative to internal tetramethylsilane. High resolution mass spectrometry (HRMS) was performed on a Agilent model 62-10 MS-TOF.General procedure for Sonogashira couplings
Reactions were carried out under inert atmosphere (vacuum then filled with argon or nitrogen at least 3 times) in a flask that was dried with an open flame while under vacuum. Typically, all reagents and solvents that were not sensitive to air or moisture were degassed for 30 to 45 min before being transferred into the aforementioned flask. The alkyne was added with a syringe. The reaction was stirred overnight and then extracted with dichloromethane and washed twice with a saturated solution of NH4Cl. The organic layer was then dried and the purification was done through a silica gel column.General procedure for Huisgen 1,3-dipolar cycloaddition “click” chemistry
Unless otherwise specified, no particular precaution was taken for these reactions. Typically, the azide-containing dendron was dissolved in a MeOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :THF (1:1) mixture. If the solution blurred due to precipitation, MeOH was slowly added until the solution cleared up again. One equivalent of CuSO4·5H2O and one equivalent of sodium L-ascorbate were then added followed by the free alkyne. The reaction was stirred at room temperature for at least 5 days and then filtered and put directly through a LH-20 Sephadex™ column in MeOH with no added pressure. It is noteworthy that all yields reported here could be improved since at every step, mixed fractions needed to be discarded to maximize purity. Optimization would require either a greater quantity of LH-20 Sephadex™ or a different one as the hydrodynamic radius of dendrimers differs than those of other organic compounds. Dialysis has also been tried and found to give disappointing results.
:THF (1:1) mixture. If the solution blurred due to precipitation, MeOH was slowly added until the solution cleared up again. One equivalent of CuSO4·5H2O and one equivalent of sodium L-ascorbate were then added followed by the free alkyne. The reaction was stirred at room temperature for at least 5 days and then filtered and put directly through a LH-20 Sephadex™ column in MeOH with no added pressure. It is noteworthy that all yields reported here could be improved since at every step, mixed fractions needed to be discarded to maximize purity. Optimization would require either a greater quantity of LH-20 Sephadex™ or a different one as the hydrodynamic radius of dendrimers differs than those of other organic compounds. Dialysis has also been tried and found to give disappointing results.
General procedure for dendron functionalization with anhydrides
The amino terminated dendrons were dissolved in MeOH and three equivalents of NaHCO3 per amine were added. The anhydride was then added and the reaction was stirred for 72 h to allow completion. The mixture was then filtered to remove excess of NaHCO3 and concentrated and put directly through a LH-20 Sephadex™ column with no added pressure.Experimental data for compounds 2–12
:1 as the eluent). Yield is 63% (1.3 g, 3.85 mmol). 1H NMR (CDCl3, 400 MHz): 7.82 (s, 1H), 7.71 (s, 1H), 7.54 (s, 1H), 4.48 (d, 2H, J = 6.3 Hz), 1.70 (t, 1H, J = 6.2 Hz). 13C NMR: 140.02, 139.15, 133.95, 126.23, 122.80, 94.00, 90.04, 82.83, 51.70. IR (NaCl): 1398, 1421, 1536, 1573, 3245 (broad band) ν cm−1. HRMS (ESI+): calcd. for C9H6BrIO: 335.8647, found: 335.8645 (M+).
:1 as eluent). Yield is 98% (1.34 g, 4.36 mmol). 1H NMR (CDCl3, 400 MHz): 7.55 (s, 1H), 7.51 (s, 1H), 7.46 (s, 1H), 4.47 (d, 2H, J = 6.3 Hz), 1.65 (t, 1H, J = 6.1 Hz), 0.24 (s, 9H). 13C NMR: 134.82, 134.49, 133.82, 125.38, 124.67, 121.98, 102.57, 97.03, 89.29, 83.64, 51.69. IR (NaCl): 1250, 1424, 1551, 1587, 2155, 2864, 2899, 2959, 3329 (broad band) ν cm−1. HRMS (ESI+): calcd for C14H15BrOSi: 306.0076, found: 306.0081 (M+).
:1 as eluent). Yield is 47% (1.85 g, 4.45 mmol). 1H NMR (CDCl3, 400 MHz): 7.51 (s, 1H), 7.47 (s, 1H), 7.46 (s, 1H), 4.47 (d, 2H, J = 6 Hz), 1.64 (t, 1H, J = 6.3 Hz), 1.18 (s, 21H), 0.24 (s, 9H). 13C NMR: 135.24, 135.09, 134.76, 124.33, 123.96, 123.22, 105.26, 103.33, 96.00, 92.53, 88.47, 84.35, 51.80, 18.77, 11.47. IR (NaCl): 1250, 1412, 1424, 1580, 2151, 2155, 2865, 2893, 2943, 3322 (broad band) ν cm−1. HRMS (ESI+): calcd for C25H36OSi2: 408.2299, found: 408.2186 (M+).
:0.96 as eluent). Yield is 66% (0.916 g, 2.42 mmol). 1H NMR (CDCl3, 400 MHz): 7.51 (m, 3H), 3.08 (s, 1H), 1.12 (s, 21H), 0.25 (s, 9H). 13C NMR: 135.96, 135.36, 135.10, 124.34, 123.96, 122.81, 105.18, 103.26, 96.09, 92.60, 88.47, 84.35, 51.80, 18.77, 11.47. IR (ATR): 1250, 1412, 1464, 1578, 2161, 2866, 2892, 2942, 3303 (broad band) ν cm−1. HRMS (ESI+): calcd for C24H34Si2: 378.2199, found: 378.2080 (M+).
:1 as eluent). Typically, batches of 10 to 15 g of NH2-G2-N3 were made and stored under vacuum to avoid CO2 poisoning of the terminal amines. Similar yields than what was previously reported15 were obtained. 1H NMR (CD3OD, 400 MHz): 3.24 (m), 2.75 (m), 2.58 (br t), 2.36 (br t), 1.72 (qt, J = 6.5 Hz). IR (NaCl): 1250, 1424, 1551, 1587, 2155, 2864, 2899, 2959, 3329 (broad band) ν cm−1. HRMS (ESI+): calcd for C33H68N16O6: 784.55, found: 785.5576 (M + H+).
Results and discussion
The choice of having a single aromatic unit as the core fragment was motivated by several reasons, namely its high level of symmetry and its small size allowing a denser particle surface. Scheme 1 shows the synthesis of the tri-orthogonal core.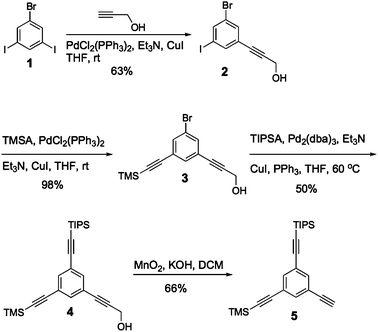 | ||
| Scheme 1 Synthesis of the core fragment. | ||
Compound 2 was obtained through a Sonogashira coupling reaction with less than one equivalent of propargyl alcohol in order to minimize the formation of the byproduct featuring a second propargyl alcohol. Excess of starting material was easily recovered by silica gel chromatography to yield compound 2 in 63% yield. Then, compound 3 was obtained in 98% yield using a chemoselective Sonogashira coupling at room temperature between compound 2 and trimethylsilylacetylene (TMSA). The addition of triisopropylsilylacetylene (TIPSA) onto compound 3 followed an optimization that required heating of the solution at 60 °C before adding both the TIPSA and the catalysts. After 48 hours, the fully protected core was isolated with a 50% yield for the final step. Our initial strategy was to first remove the TMS since it was the most sensitive of our three protecting groups. We threaded along this path successfully until all our efforts to remove propargyl alcohol failed. We hypothesized that it was either the alkalinity of the deprotection reaction or the limited solubility of the substrate once a first dendron has been clicked onto the core. We then proceeded in inverting the deprotection order, starting with the propargyl alcohol. The reaction left the TMS intact as long as the conditions were kept thoroughly anhydrous. Selective deprotection of the alcohol-protected alkyne with activated manganese oxide and KOH afforded compound 5 in a 66% yield.
In parallel, we developed a small library of protected PAMAM dendrons. Because the convergent method of building dendrimers requires highly efficient reactions, we opted for the Cu(I)-catalyzed Huisgen [2 + 3] dipolar cycloaddition (CuAAC) developed by Sharpless et al.16–19 It is noteworthy that click chemistry on a PAMAM dendrimer is not as straightforward as we expected. Several optimizations that have never been performed before on amino-terminated PAMAM dendrimer were necessary to obtain the targeted molecule. Using an optimized strategy developed by Lee et al.,8,9,15,20 compound 6, a G2 PAMAM dendron bearing an azide focal point, was prepared. Our initial idea was to use the common amino protecting group, namely BOC and FMOC, and leave the third dendron on the three-faced dendrimer “naked” with amine end groups. Scheme 2 reports the convenient and efficient synthesis of BOC-NH-G2-PAMAM-N3 dendrons. FMOC protection, on the other hand, was met with little success and the final molecule could never be obtained pure enough because of its very low solubility in common solvents. Needing an alternative for our proof of concept, we turned to another anhydride, namely acetic anhydride (Ac2O). As with BOC, we easily obtained Ac-NH-G2-PAMAM-N3 in similar yields. Every compound reported here was purified through a LH-20 Sephadex™ column in MeOH. Both protected dendrons presented key advantages such as air and heat stability.
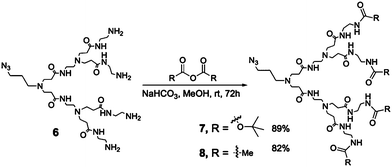 | ||
| Scheme 2 Addition of protecting groups onto PAMAM dendrons. | ||
With all the building blocks in hand, we proceeded to attach BOC-NH-G2-PAMAM-N3 (7) onto compound 5 (Scheme 3). We first tried the classic conditions for CuAAC click reaction with catalytic CuSO4·5H2O and sodium-L-ascorbate, but the reaction could not be brought to completion. The CuAAC mechanism was recently explored in more detail by Shao et al.21 who demonstrated that a low pH was useful to bring the reaction to completion and thus, the catalytic amount of AcOH was very beneficial. On the other hand, it was also reported that the use of pyridine also helped to increase yields of CuAAC reaction.22 Unfortunately, both modifications have been tried on the first click reaction with no observable effect. We tried other sources of copper (CuI and CuBr[PPh3]3, both in catalytic amount) and found that a molar equivalent of both CuSO4·5H2O and sodium-L-ascorbate left to react for 3 days was the only way to consume all the BOC-NH-G2-N3 dendron 7. Purification was done through a LH-20 Sephadex™ column to remove the excess of core molecules. The 1H NMR spectrum of compound 9 presented, as expected, sharp signals at 0.25 and 1.16 ppm for the TMS and TIPS signals respectively while the BOC termini at 1.42 ppm, now the most abundant on the molecule, gave a strong peak. The presence of triazole was confirmed by the appearance of broad singlet at 8.52 ppm. The moderate yield obtained for this step can be attributed in part to the difficulty of removing unreacted compound 5 using the LH-20 Sephadex™ column. The solvents used for the click reaction may also account for lower yield. In fact, it has been demonstrated recently that alcoholic solvents may lead to lower yields in CuAAC reactions.23 Moreover, the compound 9 thus obtained shows greenish coloration attributed to the presence of copper ions trapped inside the dendrimer structure. Indeed, PAMAMs are known to capture ions very efficiently using the various electron pairs within their structure as demonstrated by Tomalia and others.24–28 Although our present objectives pertain to the synthetic proof-of-concept, we are well aware of the toxicity problem that can be caused by the presence of copper ions inside the dendrimer for nanomedicine application. Therefore, we tried to remove the copper ions trapped in the samples after the first CuAAC reaction. Copper removal was attempted by means of AcOEt/NH4OH extractions and was met with a certain measure of success. Unfortunately, the last fractions of the LH-20 Sephadex™ column still presented a slightly green taint, indicating that some copper remained in the samples. Various other combinations of extraction media, notably DCM and saturated NH4Cl, were tried without improvement for the total removal of copper. It is worth mentioning that even the use of a more sterically hindered copper catalyst such as CuBr(PPh3)3 was not sufficient to avoid copper insertion inside the dendrimer structure (result not shown). This is in line with the recent trend of copper-free dendrimer click chemistry.10,29
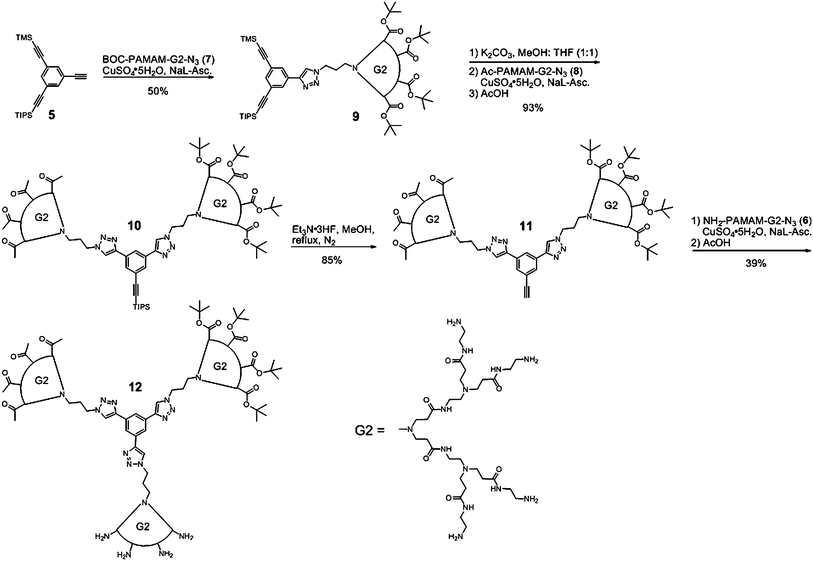 | ||
| Scheme 3 Sequential synthesis of the three faced PAMAM particle. | ||
The next step to the final particle is the synthesis of compound 10, which first involved the removal of the TMS group. This removal required some investigation to determine which reactant was compatible with PAMAM dendrons. We tested AgNO3, KOH and K2CO3 and found that only the last one did not present any reactivity toward compound 6. AgNO3 turned the dendron into a black solid, while aqueous KOH led to a biphasic mixture when added to the dendron. Since compound 9 was poisoned with copper that could not be completely removed at this stage, the removal of TMS was achieved under a nitrogen atmosphere to avoid further oxidative coupling between deprotected alkynes.30 The free alkyne was not isolated but rather generated in situ with the reactants for the click reaction to install Ac-NH-G2-PAMAM, the second third of the particle.
GPC analysis of the second click reaction showed an incomplete transformation (around 50%). Going back to the aforementioned literature, we found that in some cases, addition of AcOH into the reaction mixture can bring the reaction to completion. In our case, more AcOH was needed to first quench all the K2CO3 previously added for the alkyne deprotection. AcOH was added 48 hours after the beginning of the reaction and the mixture was stirred for an additional 16 hours. Unlike the first click reaction used to form compound 9, the addition of AcOH in the second click reaction had a beneficial impact and compound 10 was isolated in 93% yield after purification. The 1H NMR spectrum of compound 10 shows no trace of the TMS protecting group, pointing towards a complete deprotection of the second alkyne. The acetate signal at 1.95 ppm integrated to a third of that of the BOC one, indicating that the right dendrons are in place while the TIPS remained untouched at 1.18 ppm. The next step was to remove the TIPS protecting group on the last protected alkyne. To our surprise, the TIPS group remained unaffected by copious amounts of TBAF (aqueous or in THF). We first hypothesized that, once again, solubility was an issue, thus we tried CsF along with a few drops of water in MeOH, but no reaction occurred. Arguing that compound 10 had tremendous steric hindrance, we moved to Et3N·3HF in MeOH at reflux. HF is the smallest source of fluoride available and once complexed to triethylamine, it becomes almost neutral, thus preserving the BOC groups on compound 10. Very large excess was necessary to drive this reaction to completion and the TIPS removal was confirmed by 1H NMR with the disappearance of the peak at 1.18 ppm. The heat stability of both protected dendrons came into play and no evidence of degradation was observed. We then proceeded to the attachment of the last dendron onto the growing particle. Similar conditions to those used to link Ac-NH-G2-PAMAM were used. The final target (12) was obtained in 39% yield with purity above 95% by GPC (some traces of unreacted dendrons were observed and could be removed with a Jordi DVB 500 preparative column). We hypothesize that steric hindrance is the main factor leading to our moderate yields. This view is supported by the fact that at equal generation, this novel dendrimer bears 50% more terminal groups than an ethylene diamine core at this final step. All moieties presented in this work were characterized by 1H and 13C NMR, ATR-IR as well as high-resolution mass spectrometry (dual ESI) with the exception of compound 11, which was only monitored by 1H NMR.
Conclusions
We have reported the synthesis of a three-faced PAMAM particle aimed towards nanomedicine applications. Each step required thorough optimization and investigation, but the target is now readily obtainable. While Ac-NH-PAMAM was suited for our proof-of-concept, we are well aware that it requires harsh cleavage conditions. This is why work is already well underway to replace it by another protecting group. Future investigations will include selective deprotection and functionalization of each dendron as well as in vitro studies. Higher generation dendrons will also be investigated.Acknowledgements
The authors would like to thank the National Science and Engineering Council of Canada (NSERC) for financial support and Pr. Jean-François Paquin (ULaval) for his advice on TIPS removal. Mathieu Arseneault thanks the NSERC for a PhD scholarship. We are also grateful for professional support from CQMF and CERMA.References
- J. J. Shi, A. R. Votruba, O. C. Farokhzad and R. Langer, Nano Lett., 2010, 10, 3223–3230 CrossRef CAS.
- Y. Hu, D. H. Fine, E. Tasciotti, A. Bouamrani and M. Ferrari, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2011, 3, 11–32 CrossRef CAS.
- D. A. Tomalia, H. Baker, J. Dewald, M. Hall, G. Kallos, S. Martin, J. Roeck, J. Ryder and P. Smith, Polym. J., 1985, 17, 117–132 CrossRef CAS.
- Dendrimers and other Dendritic Polymers, ed. D. A. Tomalia and J. M. J. Fréchet, John Wiley & Sons Ltd, 2001 Search PubMed.
- A. R. Menjoge, R. M. Kannan and D. A. Tomalia, Drug Discovery Today, 2010, 15, 171–185 CrossRef CAS.
- S. D. Caruthers, S. A. Wickline and G. M. Lanza, Curr. Opin. Biotechnol., 2007, 18, 26–30 CrossRef CAS.
- O. Rolland, C. O. Turrin, A. M. Caminade and J. P. Majoral, New J. Chem., 2009, 33, 1809–1824 RSC.
- J. W. Lee, B. K. Kim, H. J. Kim, S. C. Han, W. S. Shin and S. H. Jin, Macromolecules, 2006, 39, 2418–2422 CrossRef CAS.
- J. W. Lee, J. H. Kim, H. J. Kim, S. C. Han, J. H. Kim, W. S. Shin and S. H. Jin, Bioconjugate Chem., 2007, 18, 579–584 CrossRef CAS.
- C. Ornelas, J. Broichhagen and M. Weck, J. Am. Chem. Soc., 2010, 132, 3923–3931 CrossRef CAS.
- E. W. Meijer, E. H. M. Lempens and L. Röglin, Angew. Chem., Int. Ed., 2010, 49, 2–13 Search PubMed.
- M. Weck, C. Ornelas, R. Pennell and L. F. Liebes, Org. Lett., 2011, 13, 976–979 CrossRef.
- A. Sharma, K. Neibert, R. Sharma, R. Hourani, D. Maysinger and A. Kakkar, Macromolecules, 2011, 44, 521–529 CrossRef CAS.
- Y. Zhang, H. H. Shi and Y. Cao, Chin. J. Chem., 2006, 24, 1631–1638 CrossRef CAS.
- J. W. Lee, J. H. Kim, B. K. Kim, J. H. Kim, W. S. Shin and S. H. Jin, Tetrahedron, 2006, 62, 9193–9200 CrossRef CAS.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- G. Franc and A. Kakkar, Chem. Commun., 2008, 5267–5276 RSC.
- G. Franc and A. K. Kakkar, Chem.–Eur. J., 2009, 15, 5630–5639 CrossRef CAS.
- G. Franc and A. K. Kakkar, Chem. Soc. Rev., 2010, 39, 1536–1544 RSC.
- J. W. Lee, H. J. Kim, S. C. Han, J. H. Kim and S. H. Jin, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 1083–1097 CrossRef CAS.
- C. W. Shao, X. Y. Wang, J. M. Xu, J. C. Zhao, Q. Zhang and Y. F. Hu, J. Org. Chem., 2010, 75, 7002–7005 CrossRef CAS.
- V. Aucagne, K. D. Hanni, D. A. Leigh, P. J. Lusby and D. B. Walker, J. Am. Chem. Soc., 2006, 128, 2186–2187 CrossRef CAS.
- S. Lal and S. Diaz-Gonzalez, J. Org. Chem., 2011, 76, 2367–2373 CrossRef CAS.
- M. S. Diallo, L. Balogh, A. Shafagati, J. H. Johnson, W. A. Goddard and D. A. Tomalia, Environ. Sci. Technol., 1999, 33, 820–824 CrossRef CAS.
- M. S. Diallo, L. Balogh, A. Shafagati, D. Swanson, J. H. Johnson, W. A. Goddard and D. A. Tomalia, Abstr. Papers Am. Chem. Soc., 2000, 219, U654 Search PubMed.
- M. Diallo, P. Maiti, T. Cagin and W. A. Goddard, Abstr. Papers Am. Chem. Soc., 2004, 227, U814 Search PubMed.
- M. S. Diallo, W. Arasho, J. H. Johnson and W. A. Goddard, Environ. Sci. Technol., 2008, 42, 1572–1579 CrossRef CAS.
- M. S. Diallo, S. Christie, P. Swaminathan, L. Balogh, X. Y. Shi, W. Um, C. Papelis, W. A. Goddard and J. H. Johnson, Langmuir, 2004, 20, 2640–2651 CrossRef CAS.
- B. Huang, A. Desai, H. Zong, S. Tang, P. Leroueil and J. R. Baker, Jr, Tetrahedron Lett., 2011, 52, 1411–1414 CrossRef CAS.
- P. Siemsen, R. C. Livingston and F. Diederich, Angew. Chem., Int. Ed., 2000, 39, 2633–2657 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed synthesis procedures and characterization. See DOI: 10.1039/c1py00146a |
| This journal is © The Royal Society of Chemistry 2011 |