DOI:
10.1039/C1PY00201E
(Paper)
Polym. Chem., 2011,
2, 2306-2315
A facile one-step synthesis to ionic liquid-based cross-linked polymeric nanoparticles and their application for CO2 fixation†
Received
8th May 2011
, Accepted 28th June 2011
First published on 21st July 2011
Abstract
Highly cross-linked polymeric nanoparticles (CLPNs) were prepared via a facile one-step synthesis, and these nanoparticles are effective catalysts for CO2 cycloaddition with epoxides. In this study, CLPNs were synthesized by radical copolymerization of 4-vinylbenzyl-tributylphosphorous chloride (PIL) and ethylene glycol dimethacrylate (EGDMA) in selective solvent, such as C1∼C5 alcohols. The results revealed that spherical nanoparticles with the mean diameter range of 10–100 nm could be prepared in these alcohols, and the size of the CLPNs could be facilely tuned by the feed ratio of EGDMA to PIL. The CLPNs were characterized using dynamic light scattering, scanning electron microscopy, transmission electron microscopy, atomic force microscopy, thermogravimetric analysis, atom absorbance spectrometry, and Fourier transform infrared spectroscopy techniques. In addition, CLPNs can be exploited as highly active and selective catalyst for the cycloaddition of CO2 to epoxides. The effects of parameters, such as reaction temperature, pressure, reaction time, and catalyst amount, on the cycloaddition reaction were investigated. As a result, excellent yield (100%) and selectivity (100%) of cyclic carbonates could be achieved under mild conditions (0.1 g CLPNs, 3.0 MPa CO2, 140 °C and 3 h) without the addition of any solvents or co-catalysts. Unexpectedly, the CLPNs could be dispersed in the products homogeneously, which resulted in the extremely high activity and selectivity. At the same time, the CLPNs could be easily separated by filtration after pouring the reaction mixture into toluene. CLPNs could be recycled six times with less than 1.5% loss of catalytic activity.
Introduction
Ionic liquids (ILs), which have been widely promoted as green and ‘designable’ solvents, are attracting considerable attention due to their chemical stability, low flammability, negligible vapour pressure, high ionic conductivity, and wide electrochemical window.1 Depending on their unique properties, ILs can be used in a wide variety of research fields, such as solvents for organic synthesis and polymerization reactions, catalysts, electrochemistry, and media for extraction processes.2 In recent years, ILs have presented great potential as catalysts for CO2 coupling with epoxides.3 Although the ability of ILs has been demonstrated successfully in many reactions, the chemical industry still prefers to use solvent-free and metal-free as well as reusable catalyst systems because of the ease of separation and the possibility to use a fixed-bed reactor. On the basis of economic criteria and possible toxicological concerns, a useful alternative is to immobilize ILs onto suitable supports.4 As a result, various heterogeneous IL-based catalysts for the cycloaddition reaction of CO2 to epoxides have been explored.5
Polymeric nanoparticles have received increasing attention in both theoretical and applied research fields due to their potential applications as nanocarriers for catalysts, molecules with electronic and photonic functions, biological and medical species, drug carriers, etc.6 Until now, several routes have been explored to prepare polymeric nanoparticles, such as emulsion polymerization, dispersion polymerization, self-assembly, etc.7 In order to improve the stability of polymeric nanoparticles, much effort has been devoted to stabilize the micellar structure by cross-linking the core or the shell formed in selective solvents.8 Recently, some one-pot synthetic strategies were explored to prepare stable polymeric nanoparticles.9 A useful alternative to this widely used process is dispersion polymerization, which can be regarded as a special type of precipitation polymerization conducted in the presence of a suitable polymeric stabilizer.10 This versatile process can be conducted in many solvents, including water, and typically produces micrometre sized latex particles.11 However, there still are some disadvantages in current approaches used to prepare stable polymeric nanoparticles, such as the surfactants or stabilizers are indispensable and it is difficult to remove them from the surface of the nanoparticles, several steps, special reagents or manipulation are usually necessary, in other cases macroinitiator or block copolymer must be presynthesized.12
In our previous study, we demonstrated a facile one-step synthesis to cross-linked polymeric nanoparticles (CLPNs) by conventional radical copolymerization of the cross-linker, ethylene glycol dimethacrylate (EGDMA), and 4-vinylbenzyl-triphenylphosphorous chloride in selective solvent.13 Moreover, CLPNs could also be obtained by cross-linking copolymerization of EGDMA and imidazole-based ionic liquids.14 As discussed in the papers, the cross-linking copolymerization to CLPN was via a dispersion polymerization process. In a typical dispersion polymerization, the monomer, initiator, and polymeric stabilizer are all initially soluble in the polymerization medium, while the targeted polymer is not soluble. As the polymerization progresses, the monomer is converted into insoluble polymer and phase separation occurs. The polymeric stabilizer adsorbs onto the nascent microscopic nuclei and prevents macroscopic precipitation by a steric stabilization mechanism. Polymerization takes place mainly within the monomer-swollen droplets and, at the end of the polymerization, latex particles are obtained as a stable colloidal dispersion.15
In the present study, we explored the synthesis of highly cross-linked polymeric nanoparticles (CLPNs) via conventional radical copolymerization of EGDMA and IL monomer, 4-vinylbenzyl-tributylphosphorous chloride (PIL), in different selective solvents. The effects of feed ratio of EGDMA and PIL on the size of nanoparticles were investigated, and the reactivity ratio of the monomers was calculated according to the Fineman-Ross method. More importantly, tributylphosphorous (PBu3)-based CLPNs presented excellent activity and selectivity in the cycloaddition reaction of CO2 to epoxides. It can be seen that the facile one-step synthesis is a universal approach for the preparation of IL-based CLPNs.
Experimental
Materials
Carbon dioxide with a purity of 99.99% was provided from a commercial source. Azobisisobutyronitrile (AIBN) was purchased from Shanghai Chemical Reagent Company and recrystallized from methanol before use. Propylene oxide, styrene oxide, allyl glycidyl ether, ethylene glycol dimethacrylate (98%), cyclohexene oxide, and 4-vinylbenzyl chloride were brought from Aladdin Reagent Co. Other reagents, such as epichlorohydrin (ECH), methanol, ethanol, iso-propanol (IPA), N,N-dimethylformamide (DMF), tetrahydrofuran (THF), ethyl acetate, diethyl ether, toluene, and tributylphosphine (PBu3) were A.R. grade. All these reagents were used as received. Absolute methanol, ethanol, and iso-propanol were prepared according to the standard process.
Synthesis of 4-vinylbenzyl-tributylphosphorous chloride
The following representative example describes the synthesis of 4-vinylbenzyl-tributylphosphorous chloride.134-Vinylbenzyl chloride (4.03 g, 26.4 mmol), PBu3 (5.36 g, 26.5 mmol) and acetone (50 mL) were introduced into a dried flask with a magnetic stirrer. The mixture was stirred at 60 °C under N2 for 48 h. Then, PIL monomer was obtained by filtration after pouring the mixture into diethyl ether and washed with diethyl ether. The solid was dried overnight under vacuum at room temperature to give a white power (Yield: 78.6%). M.P. 148–150 °C. PIL was characterized by 1H and 13C NMR (as shown in Fig. S1 and S2 in the ESI†). 1H NMR (D2O, 400 MHz, δ (ppm)): 0.68 (t, 9H), 1.12–1.36 (m, 6H), 1.88 (s, 6H), 3.39 (t, 6H), 5.11 (d, 2H), 5.65 (d, 1H), 6.45–6.63 (m, 2H), 7.0–7.4 (m, 4H); 13C NMR (D2O, 100 MHz, δ (ppm)): 12.8, 17.3, 17.8, 22.7, 23.5, 25.5, 26.1, 115.3, 127.2, 128.1, 130.3, 136.0, 137.4.
Synthesis of cross-linked polymeric nanoparticles
CLPNs were prepared by conventional radical copolymerization of EGDMA and PIL using AIBN as initiator in selective solvents. The following example describes the synthesis of CLPNs using 200 mol% EGDMA comonomer based on PIL. This protocol is representative of all CLPN syntheses. EGDMA (1 g, 5 mmol), PIL (0.89 g, 2.5 mmol) and AIBN (0.012 g, 0.075 mmol) were dissolved in methanol (70 ml), and the mixture was stirred at 70 °C for 48 h. The reaction mixture was precipitated from diethyl ether (150 ml) and the products were washed with THF and water. The products were dried under vacuum for 24 h at 50 °C. (Yield: 76.0%).
Typical procedure for the synthesis of cyclic carbonate from ECH and CO2
All the cycloaddition reactions were carried out in a 50 ml stainless steel reactor with a magnetic stirrer and an automatic temperature control system. After appropriate amounts of epoxide and catalyst were charged into the reactor, CO2 was introduced and the pressure was kept constant until the reaction was completed. Then the reactor was heated to the desired temperature. After the reaction time, the reactor was cooled to 0 °C by immersion into iced water. Then CO2 was released through a cold trap with DMF to capture the reactants and products entrained by CO2. Because the catalyst was well-dispersed in the mixture, the reaction mixtures were poured into 20 ml toluene. Then the catalyst was recovered by filtration. The resulting filtrate together with the absorbent was analyzed by GC (GC2010AF). The retention time of the products were compared with available authentic standards. In the recyclability experiments of CLPN catalyst, several parallel experiments in the first run were conducted to provide enough catalysts for the next run. The cyclic carbonates were analyzed at room temperature on a Bruker 400 MHz NMR spectrometer using CDCl3 as solvent.
Characterization
1H NMR spectra were recorded on a Brucker AM 400MHz spectrometer at 25 °C. Analytical GC was recorded on a Shimadzu GC-7A. Fourier transform infrared (FT-IR) spectra were recorded on a DIGIL FTS3000 spectrophotometer using KBr tablets. Thermogravimetric analyses (TGA) were measured on a Perkin Elmer TG/TGA 6300 at a heating rate of 10 °C min−1. The morphology of the product was observed by scanning electron microscopy (SEM, JSM 6700F, Japan), transmission electron microscopy (TEM, Philips CM120), and atomic force microscopy (AFM, Nanoscope IV microscopy). All the samples were prepared according to the standard methods. Dynamic light scattering (DLS) measurements were performed at 25 °C and a scattering angle of 90° on a commercial laser light scattering (ALV/SP-125) equipped with an ALV-5000 multi-τ digital time correlator and ADLAS DPY425 solid-state laser (output power = 22 mW at λ = 632.8 nm). The content of P element was determined by atom absorbance spectrometry using a Perkin Elmer optima 4300 DV as emission spectrometer (USA). Zeta potential of the products were measured with a Malvern Zetasizer Nano ZS90 (Malvern instruments Ltd., UK). ζ-potential values were calculated by the Dispersion Technology Software provided by Malvern according to the Henry equation, and each sample was analyzed in triplicate.
Results and discussion
Preparation of CLPNs and the formation mechanism
As discussed above, the preparation of CLPNs was via a dispersion polymerization process. IL chains in the copolymer played the role of polymeric stabilizer due to their solvophilicity. Moreover, CLPNs could be stabilized because of the electrostatic repulsion between the polymeric nanoparticles. Thus, no macroscopic precipitation could be produced. A plausible formation mechanism for CLPNs is illustrated in Scheme 1.
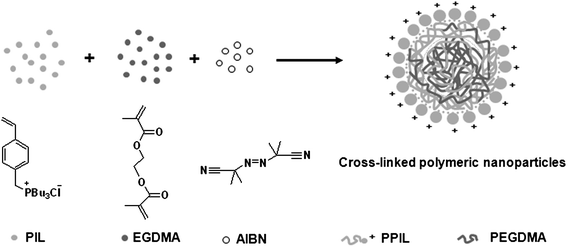 |
| | Scheme 1 Schematic illustration of the CLPN formation mechanism based on PPIL-co-PEGDMA. | |
In this study, PBu3-based ionic liquid (PIL), 4-vinylbenzyl-tributylphosphorous chloride, was prepared through the reaction between 4-vinylbenzyl chloride and PBu3. Its structure was examined by 1H and 13C NMR (see Fig. S1 and S2 in the ESI†). Studies indicated that EGDMA, PIL, and PIL′s homopolymer (PPIL), were soluble in solvents such as C1∼C5 alcohol. At the same time, the homopolymer of EGDMA, PEGDMA, was insoluble. In the present case of cross-linking copolymerization of EGDMA and PIL, nanogel rather than macroscopic precipitation was obtained due to both the stabilization of PPIL chains and electrostatic repulsion interaction between CLPNs. In this strategy, solvent played the key role for the successful synthesis of CLPNs. Therefore, cross-linking copolymerizations of EGDMA and PIL in different solvents were investigated firstly, and the results were summarized in Table 1. Initially, the solutions were homogenous and transparent, but as the polymerization processed, the reaction mixture became opaque. Emulsion with no trace of any precipitation was obtained when the cross-linking copolymerization was conducted in C1∼C5 alcohols (as shown in Fig. S3 in the ESI†). It was demonstrated that CLPNs with the mean diameter range of 10–50 nm could be prepared in these alcohols with the feed ratio of EGDMA and PIL of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. The diameter of CLPN prepared in ethanol was the smallest, which may be because the solubility of PPIL in ethanol was better than methanol and iso-propanol. It was also found that CLPNs prepared in absolute alcohols were smaller than the ones in alcohols with a trace of water. The results show that the solubility of PIL in the solvents present significant influence on the size of the CLPNs.
:1. The diameter of CLPN prepared in ethanol was the smallest, which may be because the solubility of PPIL in ethanol was better than methanol and iso-propanol. It was also found that CLPNs prepared in absolute alcohols were smaller than the ones in alcohols with a trace of water. The results show that the solubility of PIL in the solvents present significant influence on the size of the CLPNs.
To inspect the formation mechanism of CLPNs in more detail, the zeta potential of the CLPNs was measured and the results are summarized in Table 2. It can be seen that all CLPNs are positive charged. It can be concluded that PIL chains are distributed mainly on the surface of the CLPNs which would be a benefit for stabilizing the nanogels due to their solvophilicity and electrostatic repulsion interactions. The positive charges on the surface of CLPNs decrease with the increase of PIL in the feed ratio. This may be because small particles can be stabilized with fewer charges. Compared with triphenylphosphine (PPh3)-based CLPNs, there are fewer positive charges in PBu3-based CLPNs.13 It is probably attributed to the greater solvophilicity of PBu3-based PIL. As a result, fewer charges were required to stabilize the nanoparticles. The nanogels were very stable in these solvents and could be stored for several months without any sedimentation. However, the copolymer precipitated from the solution when the cross-linking copolymerization was conducted in acetone. It is because both PPIL and PEGDMA are insoluble in acetone. Therefore, cross-linked polymer could not be stabilized in acetone.
| Entry |
EGDMA:PIL |
Yield (%) |
ζ (mV) |
R
h (nm) |
PDI |
|
Polymerization conditions: ([EGDMA] + [PIL])/[AIBN] = 100:1, solvent methanol (70 ml), polymerization temperature = 70 °C, polymerization time = 48 h. Rh = the average hydrodynamic radius of the resultant nanoparticles. PDI: polydispersion.
|
| 1 |
15:1 |
80.0 |
44.7 |
58.5 |
0.34 |
| 2 |
12:1 |
80.3 |
38.2 |
35.7 |
0.32 |
| 3 |
10:1 |
78.4 |
28.8 |
35.2 |
0.23 |
| 4 |
5:1 |
78.2 |
23.2 |
19.5 |
0.21 |
| 5 |
2:1 |
72.4 |
17.7 |
12.3 |
0.27 |
| 6 |
1:1 |
59.2 |
15.9 |
8.4 |
0.23 |
| 7 |
1:2 |
55.1 |
3.4 |
4.7 |
0.29 |
| 8 |
1:5 |
53.3 |
4.2 |
5.5 |
0.28 |
The formation mechanism of one-step synthesis of CLPNs could also be shown by the copolymerization of EGDMA and PIL with molar ratio from 15:1 to 1:5. As shown in Fig. S4a (in the ESI†), transparent and colorless polymeric solution was obtained when the molar ratio of EGDMA to PIL was 1:1 or less, which resulted from the increase in PPIL chains in the cross-linked copolymer causing the increase in solubility of the copolymer in methanol. However, the presence of nanoparticles in these transparent solutions could be proven by the light scattering method using a laser pointer (as shown in Fig. S4b†). When the molar ratio of EGDMA to PIL was 2∼15, solutions with uniform opalescence could be obtained, which illustrated the formation of stable NPs in methanol. The cross-linked copolymer would precipitate from the solution when the molar ratio of EGDMA to PIL was more than 15, which resulted from too many solvophobic EGDMA segments in the copolymer. In addition, the average hydrodynamic radius of the resultant nanoparticles decreased with the increase of PIL in the feed ratio (as shown in Table 2). It is reasonable because PIL plays the role of emulsifier due its solubilization.
According to the dispersion polymerization mechanism, more stabilizer in the system will result in a decrease of the particles size. In addition, compared with PPh3-based PIL, PBu3-based PIL was more solvophilic. Therefore, the nanogel produced in selective solvent could be stabilized more effectively by PBu3-based PIL than PPh3-based PIL, and CLPN with smaller size obtained. In addition, there was a broad distribution of diameter in CLPN.
Determination of reactivity ratios of EGDMA and PIL in methanol
We have found that imidazole-based CLPNs could also prepared conveniently via this one-step synthesis in selective solvent. However, the cross-linked copolymer precipitated from the solvent after the copolymerization was completed. SEM images illustrated that CLPNs had the diameter range of 300–500 nm, which was much larger than PIL-based CLPNs.14 To inspect the copolymerization in more detail, the monomer reactivity ratios were determined by the application of conventional linearization methods, such as the Fineman-Ross Function,16 by using AIBN as the initiator in methanol. For the Fineman-Ross (FR) method, copolymerizations were performed with different initial feed ratios while maintaining the monomer conversion below 10%. The copolymer compositions (f) were determined by atom absorbance spectrometry. According to the FR method, the monomer reactivity ratios can be obtained as follows:
where r1 and r2 correspond to the reactivity ratios of EGDMA and PIL, respectively. The FR parameters G and H are defined as follows:
| H = F2/f and G = F(f − 1)/f |
With
where M1 and M2 are the monomer molar compositions in the feed, and m1 and m2 are the copolymer molar compositions.
The plot of G vs. H gave a straight line and the reactivity ratios r1 and r2 were calculated from the slope and intercept, respectively (Fig. 1). The rEGDMA and rPIL for this system was 2.15 and 0.1. These values mean that the EGDMA radical at the growing chain end reacts about 2 times faster with EGDMA than with PIL. From the reactivity values calculated, it is evident that PIL prefers cross propagation while EGDMA prefers self-propagation. It was presumably because the electrostatic repulsion had influence on the self-propagation of PIL. Since the value of rEGDMA is greater than one and rPIL is less than one, this suggests the presence of a lower amount of PIL units in the copolymer than in the feed. Moreover, product of rEGDMA and rPIL is also less than one, which indicates that the system leads to random distribution of monomer units with longer sequence of EGDMA units in the copolymer chains. When EGDMA was copolymerized with smaller molar ratio of PIL, the relative copolymerization reactivity of PIL is usually evaluated by 1/rEGDMA, which represents the preference of addition of PIL over that of EGDMA toward the same growing polymer chain end radical. For this system, 1/rEGDMA was 0.47. The deviation of 1/rEGDMA from 1.0 can be attributed to the effect of both diffusion control and incompatibility, both of which seem to be significant in this system.17
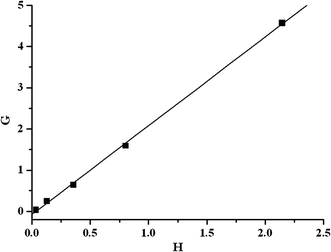 |
| | Fig. 1 Fineman-Ross plot for the radical copolymerization of EGDMA with PIL. | |
Morphology of cross-linked polymeric nanoparticles
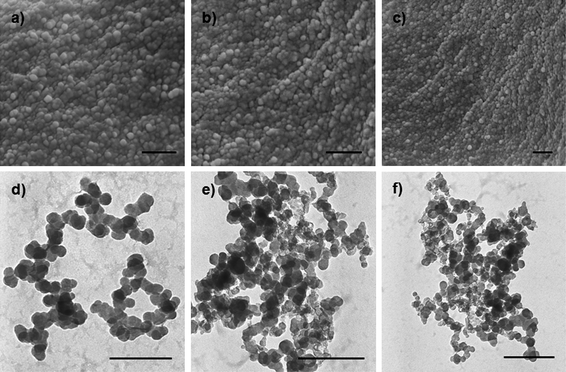
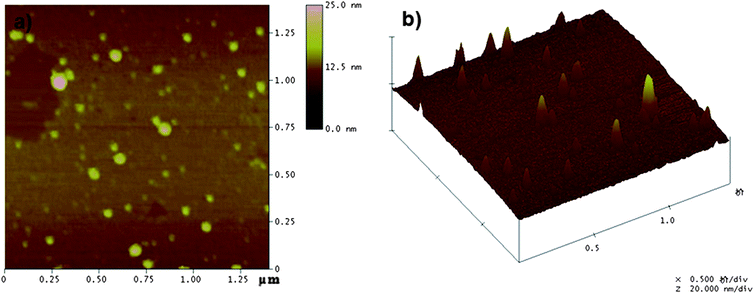
Fig. 2 represents the typical SEM and TEM images of CLPNs prepared with different feed ratio of EGDMA and PIL. According to these images, spherical nanoparticles with the average diameter of about 10–30 nm were synthesized in methanol. The particles in the SEM and TEM images were smaller than the results measured by DLS (see Table 2). This was because the latter were highly swollen in methanol. According to the TEM images, the average size decreased with the increase of PIL in the feed ratio. The results were consistent with the DLS results which were because small particles could be stabilized by fewer charges. The heights of these dried CLPNs were in the range of 5–15 nm, as judged by tapping-mode AFM three dimensions images (Fig. 3b) acquired using Nanoscope IV scanning probe microscopy. In addition, it can be observed that there was a broad distribution of diameter in CLPNs from Fig. 3a.
 |
| | Fig. 2
SEM (a, b, c) and TEM (d, e, f) images of CLPNs with the molar ratio of EGDMA to PIL of a) 5:1; b) 2:1; c) 1:2; d) 5:1; e) 2:1; f) 1:2. The scale bar is 200 nm. | |
 |
| | Fig. 3
AFM images of CLPNs with the molar ratio of EGDMA and PIL of 2:1. (a) Top view; (b) 3D image. | |
Table 3 Performance of various catalysts for the cycloaddition reaction of CO2 to ECHa
| Entry |
Catalyst
|
P contentb [wt%] |
Yield [%] |
Selectivity [%] |
|
Reaction conditions: ECH 3 ml, catalyst 0.1 g, 3 MPa CO2, 140 °C, 3 h.
Absolute amount of phosphorus determined by atomic absorbance spectrometry.
The molar ratio of EGDMA to PIL.
Synthesized by homopolymerization of PIL.
Calculated on the monomeric unit basis.
|
| 1 |
10:1c |
0.31 |
99.5 |
99.8 |
| 2 |
5:1 |
1.56 |
99.7 |
100 |
| 3 |
2:1 |
2.88 |
100 |
100 |
| 4 |
1:1 |
3.64 |
99.5 |
100 |
| 5 |
1:2 |
5.23 |
98.3 |
100 |
| 6 |
1:5 |
5.31 |
99.4 |
100 |
| 7 |
PIL
|
8.7e |
93.3 |
100 |
| 8 |
PPIL
d |
8.7e |
90.8 |
100 |
Table 4 Performance of CLPN catalysts prepared in different solvents for the cycloaddition reaction of CO2 to ECHa
A disadvantage associated with using CO2 as a reagent in organic synthesis is the potential dangers resulting from operating at high temperature and pressure, especially in the presence of heterogeneous catalytic system.18 Therefore the dependence of the yields of cyclic carbonate on the reaction conditions were investigated. The results indicate that our catalyst system can operate very efficiently under mild conditions. The influence of temperature on the cycloaddition reaction is depicted in Fig. 4. The reaction is temperature-sensitive: cyclic carbonate yield varies from 96.7% to 100% with temperature increasing from 120 °C to 140 °C. However, further increasing the reaction temperature causes a decrease in both the catalytic activity and selectivity slightly. The decrease of cyclic carbonate yield and selectivity is plausibly due to the side reactions at high temperature, such as ECH polymerization,19 cyclic carbonate isomerization to acetone and ring opening to propylene glycol by water.20 The by-product in the present cycloaddition reaction of ECH was 3-chloro-1,2-propylene glycol, as shown by GC. Therefore, the main side reaction was the ring opening of cyclic carbonate caused by the trace of water at higher temperature. The optimal temperature for the reaction is 140 °C from a practical standpoint.
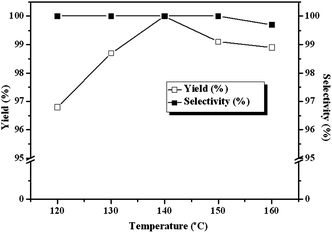 |
| | Fig. 4 Dependence of the carbonate yields and selectivities on reaction temperature. Reaction conditions: ECH 3 ml, catalyst (entry 5 in Table 2) 0.1 g, 3 MPa CO2, 3 h. | |
Due to the influence of diffusion on the reaction kinetics in the mass transfer accompanied, there are optimum ranges of CO2 pressure for the maximum conversion of ECH in the cycloaddition reactions of CO2 to epoxide.21 In order to understand the influence of pressure on the reactivity of CLPN catalyst, the pressure of CO2 is varied between 1 and 5 MPa. The results are described in Fig. 5. Yield and selectivity of cyclic carbonate increase as CO2 pressure increases up to 3.0 MPa. It is because the solubility of CO2 in the epoxides increases with pressure increasing, which favors the cycloaddition reaction considering that CO2 was a reactant. However, ECH conversion decreased when CO2 pressure is above 3.0 MPa. At the higher pressures, more ECH are extracted into the CO2-rich gas phase and the concentration of ECH in the liquid phase decreases, which reduces the reaction rate. In addition, too high CO2 pressure may inhibit the interaction between ECH and the catalyst, and causes a low concentration of ECH in the vicinity of the catalyst due to the so-called dilution effect.22 As a result, there exists an optimum CO2 pressure for the conversion of ECH and the favorable pressure is 3 MPa in the following experiments. At the same time, a lower pressure was more feasible in the industrial applications.
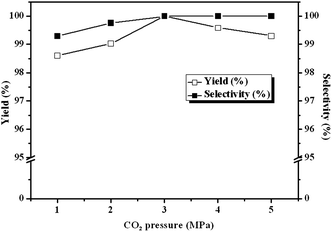 |
| | Fig. 5 Dependence of carbonate yields and selectivity on CO2 pressure. Reaction conditions: ECH 3 ml, catalyst (entry 5 in Table 2) 0.1 g, 140 °C. | |
Fig. 6 depicts the influence of reaction time on the yield and selective of cyclic carbonate. The reaction proceeds rapidly in the first 3 h, and as high as 100% ECH conversion could be achieved in 3 h at 140 °C and 3 MPa CO2, which demonstrated the high catalytic performance of the CLPNs. The high activity of CLPNs may be owing to the nanostructure which could afford larger specific surface area and be dispersed easily in the reaction system. Prolonging reaction time more than 3 h gives a little decrease in the yield of cyclic carbonate indicating 3 h is optimal. It is because long reaction time will result in the ring opening of the cyclic carbonate to propylene glycol by water. The by-product was also examined by GC technique.
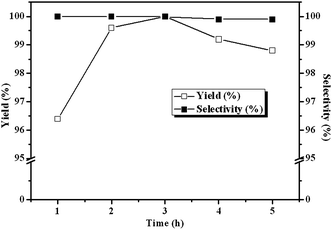 |
| | Fig. 6 Dependence of the carbonate yields on reaction time. Reaction conditions: ECH 3 ml, catalyst (entry 5 in Table 2) 0.1 g, 3 MPa CO2, 140 °C. | |
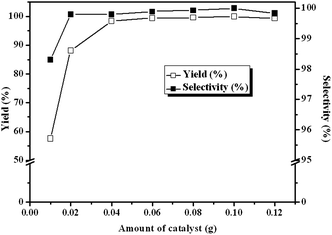
The influence of catalyst amount on the reaction was also evaluated under the identical reaction conditions. As presented in Fig. 7, the yield of cyclic carbonate markedly increases from 57.6% to 98.4% with catalyst from 0.01 g to 0.04 g. Notably, when the catalyst amount further increased to 0.1 g, ECH can be converted completely with 100% selectivity within 3 h. However, the yield decreases slightly when the amount of catalyst is more than 0.1 g, which may be because that the excess catalyst cannot be well dispersed in the reaction mixture and the mass transfer between the active sites and reactants limited.19,23
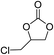
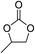
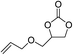
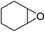
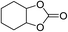
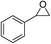
The results above indicated that CLPNs are an effective catalyst for the cycloaddition of ECH with CO2 in solvent-free conditions. In order to survey the applicability of various epoxides to this process, we also examined the cycloaddition reactions of CO2 with other epoxides at 140 °C and 3 MPa CO2. The results are summarized in Table 5. It is found that CLPNs can be used to prepare a variety of cyclic carbonates with high yield and excellent selectivity. All these cyclic carbonates have been characterized by 1H NMR (as shown in Fig. S6–S10†). Among the epoxides surveyed, propylene oxide is an active substrate as well as ECH. The possible reason for the fastest reaction rate of ECH may be due to the presence of the electron-withdrawing chloromethyl group, which results in easy ring opening of ECH by nucleophilic attack.24 The results also indicate that styrene oxide and allyl glycidyl ether are lower active substrates due to the steric hindrance and electronic effects. In addition, double bond in allyl glycidyl ether is stable in the process which illuminates that the reaction is a good method to prepare functional monomer with double bond. Due to the higher hindrance originated from the two rings, cyclohexene oxide exhibits the lowest activity.
| Substrate |
Product |
Reaction time (h) |
Yield (%) |
Selectivity (%) |
|
Reaction condition: substrate 3 ml, catalyst (entry 5 in Table 2) 0.1 g, 3 MPa CO2, 140 °C.
|
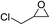
|
|
3 |
100 |
100 |
|
|
3 |
99.6 |
100 |

|
|
6 |
93.4 |
100 |
|
|
10 |
89.3 |
99.4 |
|

|
6 |
88.3 |
100 |
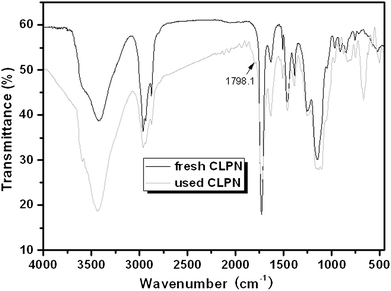
It is well known that the constancy of the activity and reusability of a catalyst are the most important factors that determine whether it can find practical application in industry. In order to test these characteristics of the novel catalyst, a series of catalytic cycles for the cycloaddition reaction of CO2 with ECH were run under the optimized reaction conditions. In each run, CLPN catalyst could be separated just by filtration after pouring the reaction mixture into toluene (as shown in Fig. S3†). As shown in Fig. 8, CLPNs could be reused for the cycloaddition reaction in six runs with less than 1.5% loss of its catalytic activity. The results indicate that CLPNs are very stable and reusable catalysts. The loss of catalytic activity may be due to the residual cyclic carbonate in the CLPNs. Cyclic carbonate is often used as a polar aprotic solvent and it is very difficult to remove it from the catalyst completely due to its high boiling point (above 200 °C). The residual cyclic carbonate in the CLPNs was confirmed by FT-IR and TGA techniques. As shown in Fig. 9 and 10, the peak at 1798.1 cm−1 in the curve of used catalyst was ascribed to the stretching vibration of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group in cyclic carbonate. Moreover, TGA curves indicated that the weight loss of the used catalyst emerged a little earlier which was due to the residual cyclic carbonate. Both of the results proved that there was residual cyclic carbonate which may result in loss of catalytic activity. The morphology of used CLPNs was also observed using SEM and TEM (as shown in Fig. S11†). It could be seen that used CLPNs remained intact, although some were aggregated.
O group in cyclic carbonate. Moreover, TGA curves indicated that the weight loss of the used catalyst emerged a little earlier which was due to the residual cyclic carbonate. Both of the results proved that there was residual cyclic carbonate which may result in loss of catalytic activity. The morphology of used CLPNs was also observed using SEM and TEM (as shown in Fig. S11†). It could be seen that used CLPNs remained intact, although some were aggregated.
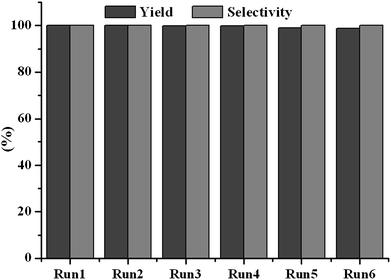 |
| | Fig. 8 The reusability of CLPN catalyst. Reaction condition: ECH 3 ml, catalyst (entry 5 in Table 2) 0.1 g, 3 MPa CO2, 140 °C. | |
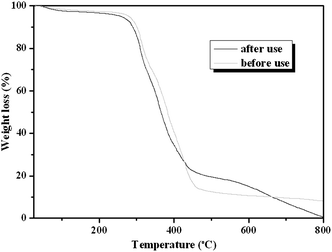 |
| | Fig. 10
TGA curves of fresh and used CLPNs. | |
CLPNs can be explored as highly effective catalysts for CO2 cycloaddition with epoxides, which may result from their unique nature, as well as the mechanism of CO2 cycloaddition. It has been reported that the active species in the cycloaddition reaction catalyzed by IL-based catalysts are the cations.25 As discussed above, the quaternary phosphonium salts are mainly on the surface of CLPNs, and suggested to be the active species for the reaction. This proposal is analogous to that of the quaternary phosphonium and ammonium salts catalysts in the same reaction.25 The proposed mechanism involves the activation of epoxide by the quaternary phosphonium cation, the ring opening of the epoxidesvia nucleophilic attack of chlorine at the less-hindered carbon, producing an oxy anion species, and the insertion of CO2 into the P–O bond. Subsequent cyclizationvia an intermolecular nucleophilic attack leads to the cyclic carbonate and the regeneration of the catalyst. It can be drawn that the role of CLPN in this system was both as the reaction site and a reusable supporter.
Conclusions
In summary, we have developed a very convenient one-step synthesis to cross-linked polymeric nanoparticles by conventional radical copolymerization of the cross-linker EGDMA and phosphorous ionic liquid without the addition of stabilizers or surfactants. The results indicated that solvent and the molar ratio of EGDMA to PIL had significant influence on the formation of CLPNs in this strategy, and the size of the CLPNs could be facilely tuned by the feed ratio of EGDMA and PIL. Moreover, the CLPNs exhibited excellent catalytic activity, selectivity, and stability because they were nanometre-sized and could be dispersed very well both in the substrates and the products. The CLPNs could also be easily separated by filtration from the products and reused six times without loss of catalytic activity and selectivity obviously. In addition, due to their good electrical properties, CLPNs have potential applications in polymer solar cells,26 and nanocomposites for sensor, optical and electronic device applications.27 It is expected that the one-step synthesis of CLPNs will attract great interest in the field of polymeric nanoparticle preparation and applications. Furthermore, the highly active and selective nanocatalyst provides a promising strategy to solve the problem of the greenhouse effect mainly by CO2.
Acknowledgements
This work was financially supported by National Natural Science Foundation of China (No. 20804031), Natural Science Foundation of Gansu Province (No. 2007GS03612), the Key Proj. Chin. Mini. Edu. China (No. 207120), NCET and Foundation of Key Laboratory of Polymer Materials of Gansu Province. We also thank Prof. Ming Jiang, Prof. Ping Yao and Prof. Daoyong Chen (Department of Macromolecular Science, Fudan University) for the DLS, AFM, and TEM measurements.
References
-
(a) J. M. Lu, F. Yan and J. Texterc, Prog. Polym. Sci., 2009, 34, 431–448 CrossRef CAS;
(b) V. I. Parvulescu and C. Hardacre, Chem. Rev., 2007, 107, 2615–2665 CrossRef CAS;
(c) P. Kubisa, Prog. Polym. Sci., 2004, 29, 3–12 CrossRef CAS;
(d) P. Kubisa, Prog. Polym. Sci., 2009, 34, 1333–1347 CrossRef CAS;
(e) T. Ueki and M. Watanabe, Macromolecules, 2008, 41, 3739–3749 CrossRef CAS.
-
(a) T. Welton, Chem. Rev., 1999, 99, 2071–2084 CrossRef CAS;
(b) J. Dupont, R. Souza and P. Suarez, Chem. Rev., 2002, 102, 3667–3692 CrossRef CAS;
(c) S. Ahrens, A. Peritz and T. Strassner, Angew. Chem., Int. Ed., 2009, 48, 7908–7910 CrossRef CAS;
(d) X. Zheng, S. Luo, L. Zhang and J. Cheng, Green Chem., 2009, 11, 455–458 RSC;
(e) H. Weingartner, Angew. Chem., Int. Ed., 2008, 47, 654–670 CrossRef;
(f) R. Giernoth, Angew. Chem., Int. Ed., 2010, 49, 2834–2839 CAS.
-
(a) H. Kawanami, A. Sasaki, K. Matsui and Y. Ikushima, Chem. Commun., 2003, 896–897 RSC;
(b) M. North, R. Pasquale and C. Young, Green Chem., 2010, 12, 1514–1539 RSC;
(c) H. Z. Yang, Y. L. Gu, Y. Q. Deng and F. Shi, Chem. Commun., 2002, 274–275 RSC;
(d) J. He, T. Wu, Z. Zhang, K. Ding, B. Han, Y. Xie, T. Jiang and Z. Liu, Chem.–Eur. J., 2007, 13, 6992–6997 CrossRef CAS;
(e) F. Shi, Q. Zhang, Y. He and Y. Deng, J. Am. Chem. Soc., 2005, 127, 4182–4183 CrossRef CAS;
(f) S. Liang, H. Liu, T. Jiang, J. Song, G. Yang and B. Han, Chem. Commun., 2011, 47, 2131–2133 RSC.
- W. L. Wong, P. H. Chan, Z. H. Y. Zhou, K. H. Lee, K. C. Cheung and K. Y. Wong, ChemSusChem, 2008, 1, 67–70 CrossRef CAS.
-
(a) Y. Xie, Z. Zhang, T. Jiang, J. He, B. Han, T. Wu and K. Ding, Angew. Chem., Int. Ed., 2007, 46, 7255–7258 CrossRef CAS;
(b) T. Sakai, Y. Tsutsumi and T. Ema, Green Chem., 2008, 10, 337–341 RSC.
-
(a) R. Savic, L. B. Luo, A. Eisenberg and D. Maysinger, Science, 2003, 300, 615–618 CrossRef CAS;
(b) S. B. Darling, Prog. Polym. Sci., 2007, 32, 1152–1204 CrossRef CAS;
(c) N. A. Lynda, A. J. Meuler and M. A. Hillmyer, Prog. Polym. Sci., 2008, 33, 875–893 CrossRef;
(d) K. Kataoka, A. Harada and Y. Nagasaki, Adv. Drug Delivery Rev., 2001, 47, 113–131 CrossRef CAS;
(e) K. Landfester, Angew. Chem., Int. Ed., 2009, 48, 4488–4507 CrossRef CAS;
(f) T. Higuchi, A. Tajima, K. Motoyoshi, H. Yabu and M. Shimomura, Angew. Chem., Int. Ed., 2009, 48, 5125–5128 CrossRef CAS;
(g) W. Meier, Chem. Soc. Rev., 2000, 29, 295–303 RSC.
-
(a) W. Li, K. Min, K. Matyjaszewski, F. Stoffelbach and B. Charleux, Macromolecules, 2008, 41, 6387–6392 CrossRef CAS;
(b) K. Min, H. Gao and K. Matyjaszewski, J. Am. Chem. Soc., 2006, 128, 10521–10526 CrossRef CAS;
(c)
M. Jiang, A. Eisenberg, G. J. Liu and X. Zhang, Macromolecular Self-assembly, Science Press, Beijing, 2006 Search PubMed;
(d) P. B. Zetterlund, Y. Kagawa and M. Okubo, Chem. Rev., 2008, 108, 3747–3794 CrossRef CAS;
(e) W. M. Wan and C. Y. Pan, Macromolecules, 2007, 40, 8897–8905 CrossRef CAS;
(f) J. Yuan and M. Antonietti, Macromolecules, 2011, 44, 744–750 CrossRef CAS.
-
(a) F. Checot, S. Lecommandoux, Y. Gnanou and H. Klok, Angew. Chem., Int. Ed., 2002, 41, 1339–1343 CrossRef CAS;
(b) Q. Zhang, E. Remsen and K. Wooley, J. Am. Chem. Soc., 2000, 122, 3642–3651 CrossRef CAS;
(c) Y. Kakizawa, A. Harada and K. Kataoka, J. Am. Chem. Soc., 1999, 121, 11247–11248 CrossRef CAS.
-
(a) T. He, D. J. Adams, M. F. Butler, C. T. Yeoh, A. I. Cooper and S. P. Rannard, Angew. Chem., Int. Ed., 2007, 46, 9243–9247 CrossRef CAS;
(b) H. Peng, D. Chen and M. Jiang, Macromolecules, 2005, 38, 3550–3553 CrossRef CAS;
(c) D. Chen and M. Jiang, Acc. Chem. Res., 2005, 38, 494–502 CrossRef CAS;
(d) L. Nie, S. Liu, W. Shen, D. Chen and M. Jiang, Angew. Chem., Int. Ed., 2007, 46, 6321–6324 CrossRef;
(e) J. Yuan and M. Antonietti, Macromolecules, 2011, 44, 744–750 CrossRef CAS.
- S. Sugihara, K. Sugihara, S. P. Armes, H. Ahmad and A. L. Lewis, Macromolecules, 2010, 43, 6321–6329 CrossRef CAS.
-
(a) A. M. I. Ali, P. Pareek, L. Sewell, A. Schmid, S. Fujii, S. P. Armes and I. M. Shirley, Soft Matter, 2007, 3, 1003–1013 RSC;
(b) D. Horak, F. Svec and J. M. J. Frechet, J. Polym. Sci., Part A: Polym. Chem., 1995, 33, 2961–2968 CrossRef CAS.
-
(a) K. Min, H. Gao, J. A. Yoon, W. Wu, T. Kowalewski and K. Matyjaszewski, Macromolecules, 2009, 42, 1597–1603 CrossRef CAS;
(b) G. Zheng, Q. Zheng and C. Pan, Macromol. Chem. Phys., 2006, 207, 216–223 CrossRef CAS;
(c) S. Liu and S. Armes, J. Am. Chem. Soc., 2001, 123, 9910–9911 CrossRef CAS.
- Y. B. Xiong, H. Wang, R. M. Wang, Y. F. Yan, B. Zheng and Y. P. Wang, Chem. Commun., 2010, 46, 3399–3401 RSC.
-
Y. B. Xiong, Y. J. Wang, H. Wang, R. M. Wang and Z. P. Cui, J. Appl. Polym. Sci., Accepted, Manuscript No. APP-2010-09-2927.R1 Search PubMed.
- S. Kawaguchi and K. Ito, Adv. Polym. Sci., 2005, 175, 299–328 CAS.
- M. Fineman and S. D. Ross, J. Polym. Sci., 1950, 5, 259–262 CrossRef CAS.
- H. Shinoda, P. J. Miller and K. Matyjaszewski, Macromolecules, 2001, 34, 3186–3194 CrossRef CAS.
- J. Sun, W. G. Cheng, W. Fan, Y. H. Wang, Z. Y. Meng and S. J. Zhang, Catal. Today, 2009, 148, 361–367 CrossRef CAS.
- L. F. Xiao, F. W. Li, J. J. Peng and C. G. Xia, J. Mol. Catal. A: Chem., 2006, 253, 265–269 CrossRef CAS.
- J. Q. Wang, X. D. Yue, F. Cai and L. N. He, Catal. Commun., 2007, 8, 167–172.
- J. Sun, W. G. Cheng, W. Fan, Y. H. Wang, Z. Y. Meng and S. J. Zhang, Catal. Today, 2009, 148, 361–367 CrossRef CAS.
- J. Q. Wang, D. L. Kong, J. Y. Chen, F. Cai and L. N. He, J. Mol. Catal. A: Chem., 2006, 249, 143–148 CrossRef CAS.
- Y. X. Zhou, S. Q. Hu, X. M. Ma, S. G. Liang, T. Jiang and B. X. Han, J. Mol. Catal. A: Chem., 2008, 284, 52–57 CrossRef CAS.
- W. L. Wong, K. C. Cheung, P. H. Chan, Z. Y. Zhou, K. H. Lee and K. Y. Wong, Chem. Commun., 2007, 2175–2177 RSC.
-
(a) Y. Du, J. Q. Wang, J. Y. Chen, F. Cai, J. S. Tian, D. L. Kong and L. N. He, Tetrahedron Lett., 2006, 47, 1271–1275 CrossRef CAS;
(b) E. H. Lee, J. Y. Ahn, M. M. Dharman, D. W. Park, S. W. Park and I. Kim, J. Mol. Catal. A: Chem., 2008, 131, 130–134 CAS.
- X. Chen, J. Yang, J. Liu, K. Manga and F. Zhu, Appl. Phys. Lett., 2009, 95, 133305–133308 CrossRef.
- A. N. Shipway, E. Katz and I. Willner, ChemPhysChem, 2000, 1, 18–52 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1py00201e |
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here.