Tunable absorption and emission wavelength in conjugated microporous polymers by copolymerization†
Jessica
Brandt
a,
Johannes
Schmidt
b,
Arne
Thomas
b,
Jan Dirk
Epping
b and
Jens
Weber
*a
aMax Planck Institute of Colloids and Interfaces,Dept. of Colloid Chemistry, D-14424, Potsdam, Germany. E-mail: jens.weber@mpikg.mpg.de; Fax: +49 (0)331 5679502; Tel: +49 (0)331 5679569
bDepartment of Chemistry, Functional Materials, Technische Universität Berlin, Englische Str. 20, D-10587, Berlin, Germany
First published on 8th July 2011
Abstract
Conjugated, microporous polymers based on a spirobifluorene core were synthesized by Suzuki polycondensation of the tetrabromospirobifluorene with benzene diboronic acid and/or thiophene diboronic acid. The optical properties (absorption and emission spectra) and the gas sorption properties (N2 and CO2) are analyzed and discussed.
Recently, a lot of interest was generated by the possibility to synthesize conjugated, microporous polymers (CMPs).1–4 Such polymers combine the specific features of conjugated polymers (photo- and electroluminescence etc.) with those of microporous materials (high surface area and pore volume). Consequently, a number of potential applications have been proposed and suggested, e.g.light harvesting.5 The concept presented by Jiang and coworkers investigated the possibility of using a CMP as an antenna for light. The energy could be transferred to a coumarin derivative, which showed brilliant emission with enhanced intensity. As the CMP acts as an antenna, its optical properties determine the light-harvesting process. Hence, it would be of interest to tune the range of operation by tuning the optical properties of the CMP. In analogy to linear conjugated polymers this might be achieved by copolymerization techniques to obtain polymers with tunable properties.
Typically, most CMPs are synthesized by metal-catalyzed coupling reactions of two distinct monomers, typically a multifunctional cross-linker and a bifunctional connector. Only very few studies have been presented thus far where different linkers have been used in copolymerizations and their ratio was varied in a continuous fashion.6,7 Typically, the impact of the different comonomers on the observable porosity was investigated and only limited interest was spent to the optical properties.
Here we present the synthesis of spirobifluorene-based CMPs by Suzuki polycondensation of 2,2′,7,7′-tetrabromo-9,9′-spirobifluorene (Sp, 1) with 1,4-benzene diboronic acid (Ph, 2) and 2,5-thiophene diboronic acid (Th, 3). Spirobifluorene was shown to be a suitable monomer for the synthesis of microporous polymersvia the “intrinsic microporosity” (PIM) concept.3,8–12
The ratio between (2) and (3) was varied over a broad range. The monomers were reacted such that an equimolar balance of the functional groups was achieved. For example, one equivalent of (1) was reacted with 2 equivalents of (2) to yield the material named SpPh2; one equivalent of (1) was reacted with 1 equivalent of (2) and with 1 equivalent of (3) to yield the material named SpPh1Th1 and so on (see Fig. 1 and Table 1).
| Entry | SBET [m2g−1] | Vp, N2a) [cm3 g−1] | SCO2 [m2 g−1] | Vp, CO2 [cm3 g−1] | band gap [eV] |
|---|---|---|---|---|---|
| a N2pore volume determined from total uptake at p/p0 = 0.1. | |||||
| SpPh2 | 800 | 0.27 | 640 | 0.22 | 2.86 |
| SpPh1.75Th0.25 | 870 | 0.33 | 650 | 0.21 | 2.59 |
| SpPh1.25Th0.75 | 601 | 0.23 | 520 | 0.15 | 2.55 |
| SpPh1Th1 | 560 | 0.2 | 490 | 0.18 | 2.53 |
| SpTh2 | 467 | 0.18 | 488 | 0.16 | 2.53 |
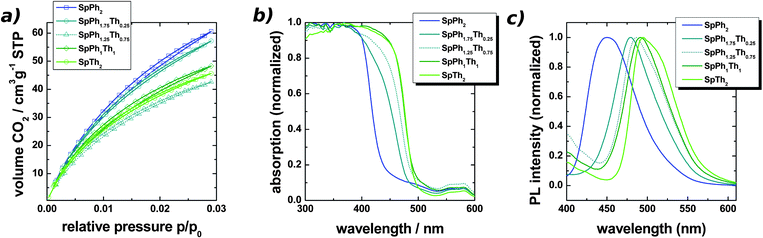 | ||
| Fig. 1 (a) CO2 sorption isotherms obtained at 273 K, (b) solid state UV/Vis absorption spectra (note: the weak absorption feature between 520–600 nm is due to the instrument setup), (c) PL spectra of the networks, (λexcitation = 350 nm). | ||
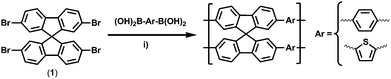 | ||
| Scheme 1 Synthesis of spirobifluorene based CMPs, (i) Pd(PPh3)4, DMF, K2CO3, 150 °C, 24h. | ||
The synthetic protocol followed the one suggested by Jiang and coworkers, i.e. using a DMF/water mixture as solvent, potassium carbonate as base and tetrakis(triphenyle phosphine) palladium(0) as catalyst.5 It is worth noting that this procedure yielded microporous conjugated spirobifluorene networks without the need of microwave irradiation and is superior to the routine we used formerly.3
The chemical identity of the materials was analyzed by Elemental Analysis, FTIR and solid state CP/MAS 13C-NMR spectroscopy (See ESI). IR spectroscopy revealed that there was indeed a true polycondensation. For instance, the band at 810 cm−1 (which can be ascribed to CH bending vibrations originating from phenyl rings having two 2 neighboured hydrogen atoms, i.e. the phenyl ring connecting two spirobifluorenes) decreases in intensity with increasing thiophene content. On the other hand, a new band arises at ∼798 cm−1 at increasing thiophene content, which is most probable due to CH bending from the thiophene ring.
Analysis of the C–Br region (∼1060 cm−1) revealed that the polymerization did not result in a large number of bromine end-groups, as the characteristic band at 1060 cm−1 is hardly observed.
Solid state CP/MAS 13C-NMR spectroscopy of the materials did also support the successful copolymerization. With increasing thiophene content an increase of the peaks at ∼120, 135 and 150 ppm was observed. The observed spectra were in qualitative agreement with those reported by Schmidt et al. for spirobifluorene-bisthiophene based materials.8
Finally, we determined the C S−1 ratio by elemental analyis. A very good agreement between the calculated ratio and the found ratio was observed (Fig. S5, ESI†).
The porosity of the materials was analyzed by means of nitrogen and carbon dioxide sorption (Fig 2a and S6). It was shown previously, that CO2 sorption can be an important additional tool for the characterization of microporous polymers, as it is only sensitive to micropores.9 Analysis of the gas sorption isotherms was made on the basis of the BET model and DFT and GCMC approaches.
We found with increasing thiophene content a decrease of the apparent BET specific surface areas from 800–870 m2g−1 (SpPh2) to 470 m2g−1 (SpTh2). This trend was also observed by CO2 sorption, which gave specific surface areas ranging from 640 m2g−1 to 490 m2g−1 (NLDFT model). The same trend was observed for the pore volumes. Table 1 summarizes the porosity data.
The difference in specific surface area might be due to a changed connection geometry,6 or due to slightly less effective cross-linking in the case of thiophene bisboronic acid. Typically, lower cross-linking leads to slightly higher flexibility and consequently to a decrease in porosity by elastic pore closure. Based on spectroscopic investigations, this scenario is not dominant, but can also not be excluded. Finally, it is also possible that there are changes in finer details of phase separation, when switching from benzene bisboronic acid to thiophene bisboronic acid. It was shown previously, that the details of phase separation can have severe impact on the observable porosity.13
For some materials a strong swelling upon N2 sorption was observed, which was manifested by a linear increase of the adsorbed volume in the intermediate pressure regime together with a pronounced hysteresis upon desorption (see ESI†). This points to the fact that the pores have some dynamic character and might have different sizes in the dry state and the (solvent) swollen state, respectively.9 Comparing the pore volumes determined by CO2 and N2 sorption, it can be observed that N2 analysis results in larger pore volumes. As CO2 can only probe subnanometer sized pores (R < 0.6 nm), this gives evidence for the presence of some larger pores together with ultramicropores, which seem to make up the major porosity. The observed swelling finally indicates that the pores get even larger when filled with solvent. In summary, all materials showed significant permanent microporosity.
The optical properties of the polymers were analyzed by solid state UV/Vis spectroscopy and photoluminescence (PL) measurements (Fig. 1b and 1c). From the absorption edge of the UV/Vis spectra it was possible to estimate the band gap energies (Table 1). The blue fluorescent SpPh2 has a band gap of approx. 2.86 eV, upon introduction of thiophene this can be lowered to ca. 2.5 eV (SpTh2).
Even the introduction of a low degree of thiophene units (SpPh1.75Th0.25) leads to a significant redshift of the absorption spectrum. Finally, the absorption spectra of SpPh1Th1 and SpTh2 are almost identical. As the thiophene units are the low-energy places, i.e. they will trap the excitons, they will dominate the absorption spectra already at low concentrations.14 At higher concentrations, they completely dominate the observed spectrum.
The PL spectra of the thiophene free material SpPh2 shows maximum emission at λ = 450 nm, which is slightly blueshifted to comparable networks.3 As those have however surface areas which are lower by a factor of two, it is possible that finer details of the network morphology can have influences on the emission wavelength. Upon introduction of 0.25 eq. thiophene (SpPh1.75Th0.25), we observed a redshift of the spectrum by ∼ 30 nm. A further increase of the thiophene content results in an additional redshift and the maximum emission of SpTh2 is found at λ = 495 nm. The polymer emits greenish, which is in agreement with reports on related spirobifluorene-thiophene polymers.15 The PL curve of SpTh2 does also show a shoulder at ca. 513 nm. This effect could only be observed for SpTh2 and the origin of the shoulder is not perfectly clear. It could either be due to interchain interaction between thiophene units or due to the vibronic structure.
Finally, we were interested, whether the pores are accessible and could be loaded with active materials. As a proof of principle, we loaded the SpTh2 network with Rhodamine 6G (R6G). At high loadings (see ESI† for details), the resulting polymer powder had an orange colour. Upon irradiation (λexcitation = 350 nm), dark green-yellow fluorescence was observed by eye, although this was not very strong. The PL spectrum showed a new shoulder at around 560 nm, while the shape of the peak at 490 (combined with the shoulder at 513 nm) changed significantly. At low loadings, the polymer powder had a bright greenish-yellow colour. Upon irradiation, the resulting PL spectrum was in between that of the parent network and the one with high loading of R6G, i.e the intensity of the new shoulder at 560 nm was less intense compared to high R6G loadings. This is in accordance with reports by Jiang et al., where the PL intensity of the dye loaded in the network also increased with increasing concentration.5 Interestingly, the maximum of the most intense emission peak is slightly blue-shifted upon R6G introduction. Also, we observe a change in the shape of the broad emission peak at 500 nm and the shoulder at 513 nm becomes clearer. The photophysical details of the network-dye interactions, i.e. whether re-absorption is active or whether the network acts as antenna is not yet clarified, but will be subject to further studies.
In summary, we have presented the possibility of band gap tuning of CMPs by simple copolymerization of tetrabromospirobifluorene with various diboronic acids. The PL emission maximum could be shifted by ca. 50 nm by replacing benzene diboronic acid with thiophene diboronic acid, while maintaining permanent microporosity. The pores are accessible for guests as exemplarily demonstrated by loading with Rhodamine 6G. Future research targets the use of these materials in sensing applications, making use of the optical activity and the pronounced porosity.
Acknowledgements
We acknowledge helpful discussions with C. Dosche (U Potsdam). Financial support from the Max Planck Society is acknowledged.Notes and references
- A. Thomas, Angew. Chem., Int. Ed., 2010, 49, 8328–8344 CrossRef CAS
.
- A. I. Cooper, Adv. Mater., 2009, 21, 1291–1295 CrossRef CAS
- J. Weber and A. Thomas, J. Am. Chem. Soc., 2008, 130, 6334–6335 CrossRef CAS
- A. Thomas, P. Kuhn, J. Weber, M.-M. Titirici and M. Antonietti, Macromol. Rapid Commun., 2009, 30, 221–236 CrossRef CAS
- L. Chen, Y. Honsho, S. Seki and D. Jiang, J. Am. Chem. Soc., 2010, 132, 6742–6748 CrossRef CAS
- J. Schmidt, M. Werner and A. Thomas, Macromolecules, 2009, 42, 4426–4429 CrossRef CAS
- J.-X. Jiang, F. Su, A. Trewin, C. D. Wood, H. Niu, J. T. A. Jones, Y. Z. Khimyak and A. I. Cooper, J. Am. Chem. Soc., 2008, 130, 7710–7720 CrossRef CAS
- J. Schmidt, J. Weber, J. Epping, M. Antonietti and A. Thomas, Adv. Mater., 2009, 21, 702–705 CrossRef CAS
- J. Weber, J. Schmidt, A. Thomas and W. Böhlmann, Langmuir, 2010, 26, 15650–15656 CrossRef CAS
- J. Weber, M. Antonietti and A. Thomas, Macromolecules, 2008, 41, 2880–2885 CrossRef CAS
- J. Weber, O. Su, M. Antonietti and A. Thomas, Macromol. Rapid Commun., 2007, 28, 1871–1876 CrossRef CAS
- N. B. McKeown and P. M. Budd, Macromolecules, 2010, 43, 5163–5176 CrossRef CAS
- R. Dawson, A. Laybourn, Y. Z. Khimyak, D. J. Adams and A. I. Cooper, Macromolecules, 2010, 43, 8524–8530 CrossRef CAS
- A. C. Grimsdale, K. Leok Chan, R. E. Martin, P. G. Jokisz and A. B. Holmes, Chem. Rev., 2009, 109, 897–1091 CrossRef CAS
- K. Takagi, M. Momiyama, J. Ohta, Y. Yuki, S.-ichi Matsuoka and M. Suzuki, Macromolecules, 2007, 40, 8807–8811 CrossRef CAS
Footnote |
| † Electronic Supplementary Information (ESI) available: Materials and methods, experimental procedures, analytical data (FTIR, NMR, EA, gas sorption data). See DOI: 10.1039/c1py00217a/ |
| This journal is © The Royal Society of Chemistry 2011 |